Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2010; 1:108-111. doi:10.7150/jca.1.108 This volume Cite
Case Report
Adrenocortical Carcinoma and Synchronous Malignancies
Marlon A. Guerrero1 ![]() , Electron Kebebew2
, Electron Kebebew2
1. University of Arizona, Department of Surgery, 1501 N. Campbell Ave., Room 4327D, Tucson AZ, 85724-5131, USA
2. National Cancer Institute, Building 10 - Hatfield CRC, Room 4-5952, 10 Center Drive, MSC 1201, Bethesda, MD 20892-1201, USA
Published 2010-8-12
Citation:
Guerrero MA, Kebebew E. Adrenocortical Carcinoma and Synchronous Malignancies. J Cancer 2010; 1:108-111. doi:10.7150/jca.1.108. https://www.jcancer.org/v01p0108.htm
Other stylesAbstract
Objective: Adrenocortical carcinoma (ACC) is an aggressive tumor that accounts for 0.02% of all reported cancers. ACC commonly arises in a sporadic manner, but may also manifest as part of a familial syndrome. Regardless of the setting, ACC rarely arises concurrent with other malignant tumors.
Methods: In this report we describe a 32-year-old woman who on work-up for abnormal vaginal bleeding was diagnosed with synchronous uterine adenocarcinoma, ovarian adenocarcinoma and ACC. We also provide a literature review of the past 20 years to identify other patients with ACC and synchronous malignant tumors, and those with familial syndromes associated with an increased risk of developing ACC.
Results and Conclusions: To our knowledge this is the first report of a patient with synchronous malignant tumors of the uterus, ovary and adrenal gland. Review of the literature revealed only 5 other cases in which a patient had concurrent ACC and malignant tumors in other organs.
Keywords: Adrenocortical carcinoma, Ovarian cancer, Uterine cancer, Synchronous malignancies, and Hereditary syndrome
Introduction
Adrenocortical carcinoma is a rare tumor with an estimated incidence of 1-2 per million per year. (1) It affects women nearly 60% more often than men and accounts for only 0.02% of all reported cancers. (2) These tumors arise with a peak bimodal age distribution during the first and fifth decades of life and 2-10% can be bilateral. (2-4)
The clinical presentation varies, ranging from vague, non-specific abdominal or back pain to overt hormonal syndromes when the tumors become biochemically active. Between 57 and 67% of patients present with either local invasion or widely metastatic disease. (4, 5) The most common hormone secreted in ACC is cortisol, resulting in Cushing's syndrome. Steroid overproduction is present in about 60% of patients with ACC. (6) In children, virilizing tumors are the most common functional adrenocortical tumors and account for 20-30% of ACC. Despite our surgical, medical and chemotherapeutic advances, the overall prognosis of patients with ACC is dismal, with a 5-year survival of less than 35%. (7)
Most ACCs arise sporadically; however, ACC does develop as part of a constellation of tumors in inherited familial cancer syndromes. The most commonly reported include the Li-Fraumeni Syndrome (8), Beckwith-Wiedemann Syndrome (BWS), Gardner Syndrome, and Multiple Endocrine Neoplasia, type 1. (9) Each syndrome is associated with a unique germline mutation. The commonality, however, is their inherit risk of developing ACC. Despite the risk of developing ACC in an inherited fashion, the risk of developing synchronous ACC with other malignant tumors is exceedingly rare. We report a patient who was diagnosed with synchronous ovarian cancer, uterine cancer and ACC.
Description of Case
A 32-year-old woman with a history of vaginal bleeding presented to a referring hospital with edema of the left lower extremity. A computer tomography (CT) scan of the pelvis showed a left pelvic mass and a deep venous thrombosis. She underwent a uterine biopsy which revealed a grade I endometrial adenocarcinoma. A staging CT scan demonstrated a heterogeneous central and left pelvic mass measuring 16.8 x 14.8 cm and a 14.5 x 10.7 cm left adrenal mass. (Figure 1) Before she was referred to our institution, a biopsy of the adrenal mass was done to exclude hormonal overproduction and interpreted as consistent with an ACC. Magnetic resonance imaging further delineated the pelvic mass as two separate masses: a 2.3 cm uterine mass and a 15.5 x 15.3 cm left adnexal mass. (Figure 2)
Figure 1
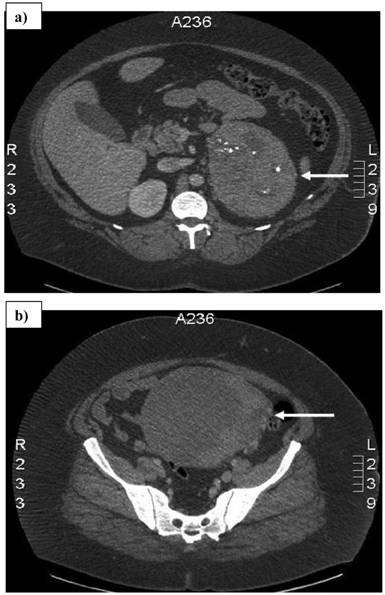
Computed tomography scan showing a) 14.5 x 10.7 cm left adrenal mass with areas of necrosis and internal calcifications, and b) 16.8 x 14.8 cm pelvic mass. Arrows denote the tumors.
Figure 2
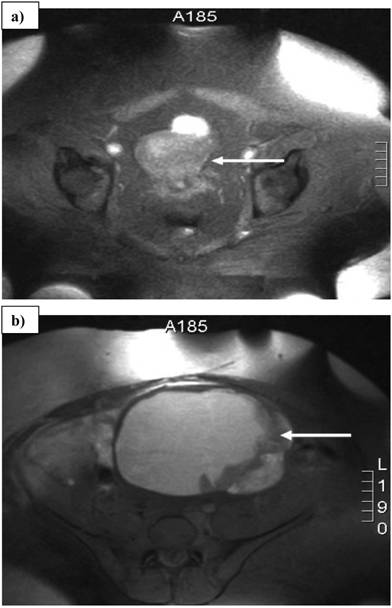
Magnetic resonance imaging scan showing a) 2.3 cm uterine mass and b) 15.5 x 15.3 cm left adnexal mass. Arrows denote the tumors.
Before surgery at our institution, an inferior vena cava filter was placed and hormonal activity was excluded from the adrenal tumor. The patient underwent an exploratory laparotomy, total abdominal hysterectomy, bilateral salpingo-oophorectomy, pelvic lymph node dissection, omentectomy, abdominal washing and left adrenalectomy via a midline incision. Final pathologic examination revealed a stage I endometrioid adenocarcinoma, stage I adenocarcinoma of the ovary and a stage II adrenocortical carcinoma. The adrenal tumor displayed 5 of 9 Weiss criteria (10) for malignancy: abundant necrosis, high mitotic count, atypical mitoses, nuclear pleomorphism and a diffuse architecture. The tumor did not exhibit the remaining criteria of non-clear cytoplasm, vascular invasion, capsular invasion or sinusoidal invasion. The patient received postoperative chemotherapy consisting of 6 cycles of paclitaxel and carboplatin. Mitotane was not given. She remains free of recurrence at 8 months after surgery.
Literature Review
The vast majority of ACCs arise sporadically, but ACC can develop in association with familial tumor syndromes. Patients with these inherited syndromes have an increased risk of developing ACC, but the diagnosis of synchronous ACC with other cancers in these families has not to our knowledge been described. The following syndromes are associated with ACC:
Li- Fraumeni Syndrome
Li-Fraumeni Syndrome (LFS) is an autosomal dominant disorder caused by a germ-line mutation in the TP53 gene located on chromosome region17q13.1. A mutation in this gene results in the inability of p53 to initiate cell growth arrest and DNA repair. (3) The resulting cascade of events leads to the formation of a myriad of tumors including soft tissue sarcomas, osteosarcomas, breast cancer, brain tumors, and leukemia.(9) ACC develops in approximately 3-4% of patients with LFS, most commonly in those under 20 years of age. (3)
Beckwith-Wiedemann Syndrome
The Beckwith-Wiedemann Syndrome (BWS) results from an epigenetic dysfunction of the chromosomal region 11p15. This syndrome arises most often sporadically, but 15% of cases are familial. (3) The genes located in this region are the insulin-like growth factor 2 (IGF-2), cyclin-dependent kinase inhibitor 1C (CDKN1C, p57kip2), and H19. (3) Abnormalities in these genes result from parental imprinting that causes a gain in the paternal allele and loss of the maternal allele. This leads to the overgrowth sequela that is characteristic of BWS. Patients suffer from macrosomia, exomphalos, macroglossia, abdominal wall defects, ear anomalies, renal abnormalities, and cleft palate. Patients also develop nephroblastoma, hepatoblastoma, rhabdomyosarcoma and nesideoblastosis. Additionally, ACC arises in 5% of patients with BWS. (3)
Gardner Syndrome
Gardner syndrome (11) is a disorder inherited in an autosomal dominant fashion that results in an inherit risk of developing colorectal cancer. This syndrome results from mutations in the APC gene in the chromosomal region 5q21. (3) The manifestation of this syndrome was first described in the early 1950's as a combination of gastrointestinal polyps, osteomas and soft tissue tumors. (12) This syndrome is characterized by epidermal cysts, gastric and duodenal polyps, desmoid tumors, periampullary cancer, and about 2% of women with this syndrome develop a crifiborm type of papillary thyroid cancer. (13) Adrenocortical carcinoma also affects individuals with Gardner's syndrome. Few cases have been described in the literature since the original report by Marshall (14), so the precise prevalence is unknown.
MEN1 Syndrome
Multiple Endocrine Neoplasia 1 is a syndrome resulting from mutation in the MEN1 gene located in the chromosomal region 11q13. (9) The resulting mutation leads to a defect in the MENIN protein, leading to the formation of pituitary tumors, parathyroid tumors and pancreatic neuroendocrine tumors, as well as adrenal tumors, multiple lipomas and angiomas. Adrenocortical tumors develop in 55% of people affected with this syndrome; the majority being adenomas and less commonly ACC. (3)
Synchronous Tumors
The development of synchronous tumors in patients diagnosed with ACC occurs even more infrequently and is evidenced by the scarcity of reports in the English language literature. A Pubmed literature using the key terms “adrenocortical carcinoma and synchronous tumors”, “adrenocortical carcinoma and synchronous cancer”, “adrenal cancer and malignancies”, “adrenal cancer and ovarian cancer”, “ACC and synchronous cancer” and “ACC and malignancies” search revealed only five patients with synchronous tumors. The first case, described in 1995, was of a patient with cancer of the rectum, breast and adrenal gland. (15) ACC has also been reported to occur synchronously with stage IIIB testicular seminoma (16), ganglioneuroblastoma (17), osteosarcoma (18), and renal cell carcinoma (19). (Table 1)
Synchronous tumors appear to develop independent of familial syndromes. The genetic abnormalities in these syndromes and subsequent development of metachronous tumors raise the suspicion for a genetic component. Two patients showed a mutation of the p53 gene; one showed a single base substitution in codon 248 (17) and the other had a mutation at codon 273. (18) Our patient underwent molecular testing for possible Lynch syndrome, given the high association of endometrial cancer with this syndrome, but she was not found to have microsatellite instability. Additionally, the patient was found to not have a mutation of the p53 gene.
Conclusion
Although patients with familial syndromes are at higher risk of developing ACC, reports in the literature of synchronous ACC and other malignant tumors are scant. To date, only 5 cases have been reported of synchronous malignant tumors in patients with ACC. There appears to be no association between the histological types of cancers that arise synchronously since all 5 cases had tumors of different origin.
To our knowledge, this is the first report of synchronous cancers arising in the adrenal gland, uterus and ovary. This is the only patient who has been diagnosed with synchronous tumors from the 50 (2%) patients in our ACC tumor registry. Furthermore, no patients were diagnosed with metachronous tumors arising within a year of ACC diagnosis. This denotes not only the rarity of ACC, but also the rarity of developing synchronous cancers.
Table 1
Reports of patients with ACC and synchronous malignancies
| Published Study | Patient Age | Synchronous Tumor | ACC Stage | Duration of recurrence free disease |
|---|---|---|---|---|
| Eisinger, et al., 1983 (15) | n/a | Breast, Rectal | n/a | n/a |
| Inoue, et al., 1998 (16) | 57y | Testicular seminoma | II | 42 months |
| Pivnick, et al., 1998 (17) | 18m | Ganglioneuroblastoma | IV (Metastases to lung) | 17 months |
| Khayat, et al., 2004 (18) | 7y | Osteosarcoma | IV (Renal invasion) | Died from osteosarcoma metastases |
| Jani, et al., 2008 (19) | 53y | Renal cell carcinoma | III | 24 months |
| Guerrero, et al. | 32y | Ovarian and Uterine carcinoma | II | 8 months |
Acknowledgements
We would like to thank Pamela Derish for her help in editing this manuscript.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Schteingart DE, Doherty GM, Gauger PG. et al. Management of patients with adrenal cancer: recommendations of an international consensus conference. Endocr Relat Cancer. 2005;12:667-680
2. Kuruba R, Gallagher SF. Current management of adrenal tumors. Curr Opin Oncol. 2008;20:34-46
3. Soon PS, McDonald KL, Robinson BG, Sidhu SB. Molecular markers and the pathogenesis of adrenocortical cancer. Oncologist. 2008;13:548-561
4. McNicol AM. A diagnostic approach to adrenal cortical lesions. Endocr Pathol. 2008;19:241-251
5. Kebebew E, Reiff E, Duh QY, Clark OH, Clark OH, McMillan A. Extent of disease at presentation and outcome for adrenocortical carcinoma: have we made progress? World J Surg. 2006;30:872-878
6. Allolio B, Fassnacht M. Clinical review: Adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab. 2006;91:2027-2037
7. Patalano A, Brancato V, Mantero F. Adrenocortical cancer treatment. Horm Res. 2009;71(Suppl 1):99-104
8. Gonzalez KD, Noltner KA, Buzin CH. et al. Beyond Li Fraumeni Syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol. 2009;27:1250-1256
9. Li FP, Fraumeni JF, Mulvihill JJ. et al. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988;48:5358-5362
10. Weiss LM, Medeiros LJ, Vickery AL. Pathologic features of prognostic significance in adrenocortical carcinoma. Am J Surg Pathol. 1989;13:202-206
11. Groussin L, Bonardel G, Silvéra S. et al. 18F-FDG PET for the diagnosis of adrenocortical tumors: a prospective study in 77 operated patients. J Clin Endocrinol Metab. 2009;94:1713-1722
12. Gardner EJ, Plenk HP. Hereditary pattern for multiple osteomas in a family group. Am J Hum Genet. 1952;4:31-36
13. Jagelman DG. Extracolonic manifestations of familial polyposis coli. Cancer Genet Cytogenet. 1987;27:319-325
14. Marshall WH, Martin FI, Mackay IR. Gardner's syndrome with adrenal carcinoma. Australas Ann Med. 1967;16:242-244
15. Eisinger F, Aubert L, Zanlucca S. et al. [Synchronous cancers. A case with three localizations: rectal cancer associated with familial polyposis, breast cancer and adrenal gland cancer]. Presse Med. 1995;24:1446
16. Inoue T, Terai A, Terachi T, Souma T, Yoshida O. Synchronous testicular seminoma and adrenocortical carcinoma: a case report. Int J Urol. 1998;5:615-617
17. Pivnick EK, Furman WL, Velagaleti GV, Jenkins JJ, Chase NA, Ribeiro RC. Simultaneous adrenocortical carcinoma and ganglioneuroblastoma in a child with Turner syndrome and germline p53 mutation. J Med Genet. 1998;35:328-332
18. Khayat CM, Johnston DL. Rhabdomyosarcoma, osteosarcoma, and adrenocortical carcinoma in a child with a germline p53 mutation. Pediatr Blood Cancer. 2004;43:683-686
19. Jani P, Nasr AL, Demellawy DE. Synchronous renal cell carcinoma and adrenocortical carcinoma: a rare case report and clinicopathologic approach. Can J Urol. 2008;15:4016-4019
Author contact
![]() Corresponding author: Phone/Fax numbers: (520) 626-6788/(520) 626-7785; Email: mguerreroarizona.edu
Corresponding author: Phone/Fax numbers: (520) 626-6788/(520) 626-7785; Email: mguerreroarizona.edu
Citation styles
APA
Guerrero, M.A., Kebebew, E. (2010). Adrenocortical Carcinoma and Synchronous Malignancies. Journal of Cancer, 1, 108-111. https://doi.org/10.7150/jca.1.108.
ACS
Guerrero, M.A.; Kebebew, E. Adrenocortical Carcinoma and Synchronous Malignancies. J. Cancer 2010, 1, 108-111. DOI: 10.7150/jca.1.108.
NLM
Guerrero MA, Kebebew E. Adrenocortical Carcinoma and Synchronous Malignancies. J Cancer 2010; 1:108-111. doi:10.7150/jca.1.108. https://www.jcancer.org/v01p0108.htm
CSE
Guerrero MA, Kebebew E. 2010. Adrenocortical Carcinoma and Synchronous Malignancies. J Cancer. 1:108-111.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.