Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2010; 1:209-222. doi:10.7150/jca.1.209 This volume Cite
Review
The Therapeutic Value of Monoclonal Antibodies Directed Against Immunogenic Tumor Glycoproteins
Myron Arlen ![]() , Philip Arlen, Al Tsang, XuePing Wang, Rishab Gupta
, Philip Arlen, Al Tsang, XuePing Wang, Rishab Gupta
Dept of Surgery North Shore Univ. Hosp. Division of Surgical Oncology, Manhasset NY, USA; Med. Univ of South Carolina and Neogenix Oncology Inc. Great Neck NY, USA
Received 2010-10-8; Accepted 2010-11-2; Published 2010-11-3
Citation:
Arlen M, Arlen P, Tsang A, Wang X, Gupta R. The Therapeutic Value of Monoclonal Antibodies Directed Against Immunogenic Tumor Glycoproteins. J Cancer 2010; 1:209-222. doi:10.7150/jca.1.209. https://www.jcancer.org/v01p0209.htm
Other stylesAbstract
Monoclonal antibodies developed against immunogenic proteins (Tumor Specific Antigens/TSA's) that are expressed in human cancers, display a unique behavioral pattern. They appear to serve in a dual role. This includes the early recognition of these immunogenic membrane proteins that can serve as diagnostic markers, and the targeting of such markers for the destruction of the tumor, primarily thru ADCC.
The monoclonals (mAbs) that we have developed against specific immunogenic tumor membrane proteins have been studied in detail. These tumor proteins, when first defined, were referred to as tumor associated antigens. With the ability of the mAbs to demonstrate therapeutic antitumor activity in those patients with relatively advanced malignancies, the term tumor specific was introduced. Monoclonals that we were able to develop from tumor specific proteins derived from colon and pancreas cancer were found capable of targeting those tumors to induce apoptosis. We were also able to define immunogenic membrane proteins from lung (squamous and adenoCa) as well as prostate neoplasms. Monoclonals developed from these tumor antigens are in the initial phases of investigation with regard to their specificity and antitumor activity.
Mabs capable of targeting the malignancies noted above were produced following immunization of BALBc mice with the Tumor Specific Antigens. The hybridomas that were screened and found to express the antibodies of interest appeared for the most part as IgG2a's. It became apparent after a short period of time that stability of the Fab CDR loops as well as the therapeutic efficacy of the hybridoma mAbs could be lost. Stability was achieved by chimerization and or humanization. The resulting mAbs were found to switch their isotypes to an IgG1 subsequent to chimerization and or humanization, when expressed in CHO cells. The monoclonals, so produced, were not only more efficient in controlling tumor growth but minimized the development of a HAMA response.
Because of 1) the specificity of this group of monoclonal antibodies in targeting well defined immunogenic proteins that were expressed on the tumor cell membrane,2) their lack of cross reactivity to normal tissue, 3) relatively low toxicity when delivered intravenously, 4) rapid targeting of tumor cell populations (4-6 hrs in vitro) and their 5) ability to destroy xenograft transplants (in vivo) within days of delivery, these mAbs were felt to be ideal for possible use in the treatment of patients with recurrent and or metastatic tumors.
Initial clinical studies have been planned for following the filing of an IND. It is required by FDA that the potential effects of tumor control and toxicity be defined using the naked antibodies produced under GMP conditions, In those situations where patients with recurrent malignancies are to be studied we have come to realize that a number of factors can influence the response to monoclonal therapy. This includes the amount of shed antigen in the serum at the time of treatment that could initiate immune complex formation as well as the shedding of inhibitory material into the serum possibly effecting an immune response. As such we plan to eventually employ the therapeutic mAbs in combination with chemotherapy as a means of enhancing the immunogenicity of the tumor system being treated and to possibly weaken the malignant growth for easier destruction by the mAb. We will also look at the combination of mAbs with immunostimulants such as GMCSF and IL-2 (fusion proteins) and eventual conjugation of the mAbs with alpha and possibly B-emitters to help in targeting bystander cells. The present paper reviews the potential therapeutic value of such mAbs in the treatment of recurrent malignancies, especially those having failed chemotherapy in established clinical trials.
Keywords: Monoclonal antibodies, hybridomas, chimeric antibodies, ADCC, apoptosis, tumor specific antigens (TSA).
1. Introduction
Based on our experience with the use of commercial monoclonal antibodies (mAbs) employed in therapeutic situations for managing metastatic malignancies, especially those targeting growth factors, these monoclonals appear to have played a limited role in controlling the spread of established tumors. Those growth factors presently being targeted by mAbs such as Erbitux, Vectibix and Avastin, however, are not only seen in the malignant state but are also expressed on the surface of many normal tissues, i.e. skin, bowel. Growth factors are essentially none immunogenic, do not shed from the cells that express them and are not recognized by the host. The mAbs, that have therefore been commercially developed and directed against such growth factors, appear to have limited therapeutic value and do demonstrate occasional adverse responses when delivered intravenously.
The newer classes of mAbs that we are developing are, as noted above, directed against immunogenic tumor specific antigens (TSA's) present on the surface of tumor cells and absent in normal adjacent tissue. Shedding of these TSA antigens into the serum does occur, but to a limited degree. This is due to the low level of TSA expression within the cytoplasm and secondarily to what is chaperoned to the cell membrane where shedding occurs into the serum as potential tumor markers. In contrast, epidermal growth factors which are present in normal as well as malignant cells, are not immunogenic and do not shed into serum. The host does recognize shed TSA, tending to produce circulating monoclonals (mAb1) with low levels of ADCC (Antibody Dependent Cell Cytotoxicity), immune complexes as well as circulating anti-TSA antibodies (mAb2 anti-idiotypes). A consequence of this phenomenon is a low level of humoral and cell mediated immunity at the level of what we term “tumor surveillance”. The existence of circulating mAbs, anti-TSA Abs and cytotoxic T-cells are not present at a significant level however, to induce continued apoptosis of developing tumor. They do help in minimizing the rate of tumor expression early in the clinical course of disease. Without therapeutic intervention tumor spread does occur. By raising the titer of circulating antibodies to those levels needed to reach threshold therapeutic levels by IV administration, even without directly enhancing levels of cell mediated cytotoxicity, we can anticipate obtaining a desired clinical response.
The mAbs that we have developed via immunization of BALBc mice and which have derived from functional hybridomas and secondarily thru expression in the chimeric version in CHO, appear to represent a new and potentially effective group of IgG's. They offer an alternative approach for treating metastatic malignancies alone and in combination with other antitumor agents. These same mAbs, expressed as chimeric or humanized mAbs, when given intravenously in high therapeutic doses, can initiate tumor destruction within hours of IV administration. They circulate with a half life exceeding 10 days and show a localization index of better than 10:1, where the concentration of the mAb fixed to the tumor is tenfold or greater than what remains in circulation.
In the face of progressive and metastatic disease that has failed to respond to chemotherapy, few approaches are available to reverse or control the disease process. Specific active immunotherapy with vaccines from which the above mAbs are derived, is not a viable option in such clinical situations. The optimum immunologic response to such vaccines (inducing cell mediated and humoral immunity) occurs during a 4-6 month period of time following completion of immunization (4). This interval is too long to anticipate seeing a therapeutic response in patients with advanced disease. The existence of a more advanced malignant state usually has progressed in terms of tumor volume during this period of time and has been shown to have expressed a number of factors capable of suppressing the immune response (immunological escape) (5). Passive immunization with a therapeutic monoclonal antibody which works within hours of delivery becomes the method of choice in such instances.
Following our early vaccine studies in high risk tumor patients using TSA vaccines, the end results appeared to demonstrate a marked enhancement in survival approximating a three-fold increase over controls during a five to seven year period of time. A significant finding seen among the long term survivors in the above mentioned clinical trials was a high serum titer of IgG1 directed against the tumor antigen. These elevated IgG levels lasted from 5 to 10 or more years and paralled the diminution in a delayed cutaneous hypersensitivity response to skin test dose of antigen. In those patients having received TSA immunization and who had failed therapy showing evidence of recurrence of tumor within a year of treatment, it became apparent that there was an innate inability to produce and maintain high levels of the anti-tumor IgG. This set the stage for looking into the development of the antitumor IgG's as potential monoclonal therapeutic agents to be used in patients with advanced, recurrent and possibly metastatic tumor (6).
Development of hybridomas following inoculation of BALB/c mice with defined human TSA's i.e those immunized with colon or lung TSA, confirmed that the resulting murine mAb's were capable of inducing marked destruction of specific tumors both in vivo and in vitro. Chimerization of these murine mAbs demonstrated an enhancement in antitumor effect because of the presence of the human Fc portion of the IgG1 molecule which contains NK cell receptors.
Those studies that followed, were designed to characterize the possible effectiveness of our TSA derived mAb's as potential therapeutic agents in patients with advanced disease, especially in those having failed chemotherapy and who then demonstrated progression of disease. Of importance was the need to define the nature of the mechanisms associated with destruction of existing tumor other than that of ADCC, if such existed. Such studies had been accomplished in defining the mechanisms for Rituxan control of CD-20 expressing cells such as lymphomas (7).
2. Methods
A.TSA Isolation and characterization
In the past, we have isolated and characterized several human tumor specific membrane antigens (TSA's) defining a number of human solid tumor malignancies. Because of the low levels of antigen expression in any one patient's tumor, pooled allogeneic membrane proteins were employed in evaluating the threshold level of antigen necessary to optimize cellular and humoral antitumor responses. Among the tumors studied, were those derived lung, colon/pancreas, prostate and ovarian cancers. The specific TSA's that were studied (4), were isolated from human tumor membranes obtained following surgical resection. They were then characterized by separation into membrane proteins of different molecular weight by Sephadex-200 and further defined after separation by discontinuous polyacrilamide gel electrophoresis. The nearly purified antigen preparation was then delivered as a vaccine to patients with Stage II and III malignancies having undergone resection, in an attempt to minimize the incidence of recurrence (IRB and FDA approved). Each of these immunogens were studied and found capable of turning on both cellular and humoral immune responses in patients manifesting the specific malignancy. Further study of the antigens indicated that each were oncofetal in origin and were not usually found to be expressed in normal tissue. In fetal tissue they appeared to allow maturation of associated tissues prior to birth. At birth it was felt that re:methylation of the functional fetal gene resulted in its silencing and thus effects of the oncofetal protein were negated. Only after the silenced gene was found to be reactivated in the adult host, probably as a result of carcinogenic reactivation or viral transfection, does one see the tumor oncofetal protein expressed in the adult, but this time in a mutated form. A number of attempts have been made in the past to define tumor antigens in various malignancies including colon cancer. Those antigens that have been isolated and characterized by other groups have failed to define the immunogens. This appears to be due to the extremely low levels of expression in the individual malignancy and the need for skin testing with the isolated pooled antigens to see if DHR is present in the antigen preparation of question. Catimel et. al. described methods for purification and characterization of restricted antigens in normal and transformed human colonic epithelium (8).
In the malignant state, such as in colon and pancreatic cancer, the tumor variant of the oncofetal protein becomes expressed and defined within the cytoplasm of the tumor cell to serve as a diagnostic marker as well as a therapeutic target. This tumor protein (TSA) then appears to be chaperoned to the tumor membrane where it is shed into the serum. The isolation of such immunogenic tumor associated antigens, while still highly expressed in the tumor membrane, was achieved through a process of membrane sonication of multiple specimens, gel electrophoresis and HPLC. TSA activity was confirmed by evaluating the delayed cutaneous hypersensitivity response to an intradermal test dose of antigen. The DHR skin test which was approved by the hospital IRB allowed patients with the corresponding tumor, patients with different malignancies and normals to be tested to define specificity as well as to rule our cross reactivity. This procedure is no longer allowed due to the potential of introducing a virus from one of the donors into the pooled antigemn preparation. Following this we were able to define an elevation in an associated circulating serum IgG1 response that is capable of targeting the TSA membrane antigen in patients free of disease, five or more years following surgery. This latter response has been noted to persist for periods lasting up to 10 years post immunization in those patients that have remained free from recurrent tumor. Because of the potential problem of utilizing pooled allogeneic material to isolate TSA, the FDA has restricted tumor use to the patient's own tumor, preparing personalized vaccines. During the studies of TSA specificity, it was noted that the immune response required somewhere between 500 and 1000 µgs plus an adjuvant to result in full immunization. It is rare however to find more than 100 µg in a patient's own tumor preparation so that self vaccines that have added heat shock protein, dendritic cells or various viral preparations to allow full immunization to occur, have usually been ineffective.
Each of the monoclonals that have been developed in our labs to date, appear to demonstrate a high level of ADCC as long as one can identify the presence of the target protein in tumor sections derived from both fresh and from paraffin blocks using immunoperoxidase staining (immunohistochemistry).
B. Hybridoma development
Antigens which demonstrated delayed cutaneous hypersensitivity and which proved to have therapeutic efficacy in clinical trials, exhibiting cellular and humoral immunity, were used to inoculate mice for the production of hybridomas. BALB/c mice were immunized by IP injections of 50ug of colon carcinoma TSA emulsified in complete Freund's adjuvant, 0.1ml x3. After 10 days the mice received an IV booster injection of the same amount of TSA in 0.9% NaCl solution. Mice were sacrificed three days later. Spleen cells were used for cell fusion. This fusion was performed by incubation of 5x107 mouse spleen cells with 107 sp2/0-Ag14 myeloma cells in 40% PEG. An enzyme linked antibody assay (ELISA) was used for the detection of clones producing monoclonal antibodies against the specific TSA's.
C. Method used to determine antibody dependent cell cytotoxicity (ADCC )
A 4 hr. 51Cr or more recently the 111In release assay was used to measure antibody dependent cell cytotoxicity. The target cells, whether colon carcinoma, pancreatic carcinoma or lung squamous cancer cell lines, were obtained from the American Type Culture Collection (ATCC) in Rockville MD. Target cells were labeled with 200 µCi sodium 51chromate or the equivalent in radiolabelled indium. In 0.2 ml. fetal calf serum for 1 hour. Target cells, (1x104 in 50 µl) were added to 96 U-bottom wells assay plates containing effector mononuclear cells. Effector to target cell ratios' of 100, 50, and 25 were assayed in the presence of the mAb of interest compared to normal IgG at mAb concentrations of 2.5- 5.O µg/well. The plates were incubated for 4 hrs. at 370C. in a humidified atmosphere containing 5% CO2. Supernatants were harvested for gamma counting using Skatron Harvester Frames. Experiments were carried out in triplicate. Specific lysis was calculated using the following formula. It appeared that 5.0 ug/well of the mAb produced optimum results and that the 100:1 E:T ratio produced the highest range of tumor destruction over a 4-6 hr. period of time. This of course set the stage for eventual employment of a humanized or chimerized monoclonal IgG's since one had the opportunity to see clinical responses in patients with recurrent disease where standard forms of therapy had failed.
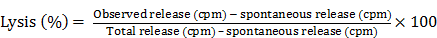
Spontaneous release was determined by measuring the radioactivity released from target cells incubated in medium alone. Total releasable radioactivity was obtained after treatment with 2.5% Triton X-100. Another method of tabulating data is in terms of “lytic units”. Such units can be calculated where one lytic unit is defined as the number of cells required to lyse 15% of a population of 5 × 103 target cells in a 6-h assay. Lytic unit values are then expressed as mean ± standard error of the mean.
The MTT assay that is occasionally employed is a similar laboratory test for antibody cytotoxicity using a standard colorimetric assay (an assay which measures changes in color) for measuring cellular proliferation (cell growth).The amount of yellow MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) oxidised to purple formazan (see figure) is measured spectrophotometrically. This oxidation takes place only when mitochondrial reductase enzymes are active, and thus conversion is directly related to the number of viable cells. The production of purple formazan in cells treated with an agent is measured relative to the production in control cells, and a dose-response curve can be generated. We feel that the radioactive release assay measuring cytotoxicity, has proven to be more reliable as well as accurate. We have found the sensitivity for the MTT assay not to be as reliable as when we measure isotope release as an indication of cell death, and as such are now employing the 111In release assay routinely. As newer non isotopic assays are developed we do however plan to compare results with both procedures.
D. Measurement of Apoptosis by Annexin V binding
While the major attack by mAb therapy occurs thru ADCC we have defined that other mechanisms are involved including an apoptotic event. He, apoptosis represents programmed cell death wherein the process is characterized by a number of morphological and biochemical features including shrinkage of cytoplasm, condensation and margination of nuclear chromatin, fragmentation of DNA into 120-180 base pair segments, and finally blebing of the plasma membrane and formation of apoptotic bodies, which are rapidly removed by neighboring phagocytes. Different changes taking place on the surface of apoptotic cells such as the expression of thrombospondin binding sites, loss of sialic acid residues and exposure of a phospholipid-like phosphatidylserine (PS) have been previously described (9). Phospholipids are asymmmetrically distributed between inner and outer leaflets of the plasma membrane with phosphatidylcholine and sphingomyelin exposed on the external leaflet of the lipid bilayer, and phosphatidylserine predominantly observed on the inner surface facing the cytosol (9).
3. Results
In spite of the poor response of recurrent tumors to the use of chemotherapy, we now believe that it is possible to develop an efficient therapeutic approach to the management of many of these malignancies by employing specific mAb's directed against an array of immunogenic proteins expressed on their cell surfaces (10,11). For the particular group of antibodies that we have developed to date as well as for others being developed, all appear capable of initiating destruction of existing tumor by the process of antibody dependent cell cytotoxicity (ADCC). Prior to considering the therapeutic potential of such antibodies, we analyzed their capability for accurately quantifying the intensity of their staining capabilities, and the % of reactive cells expressing the relevant cell surface target antigen. The binding of the mAbs have varied with the tumor population studied, but combinations of mAbs have shown that tumor recognition can be optimized to offer a superior response. This procedure has also offered a mean density distribution of the antigen so that we are able to predict the number of binding sites available (12). In this manner we are evaluating the procedure for defining antibody therapy; one that can be checked for optimum response with each monoclonal or group of monoclonals studied. When we evaluated ADCC as an in-vitro assay for tumor cytotoxicity, we employed our protein derived mAb's, demonstrating that these mAbs were associated with a 50-60% or better rate of tumor cell destruction. This process was found to occur in-vitro, over a 4-6 hr. period of time.
The rate of destruction of tumor cells that we used in this assay was defined as noted above, by a chromium or indium release assay, wherein the antibody specific to a tumor marker expresses its efficacy in the presence of human effector cells. We have noted that with those antibodies having a low destructive rate, especially under 10%, the potential for effecting a clinical response in patients is minimal. This is probably due to the rapid rate of tumor cell proliferation in culture, requiring destructive (apoptotic) rates in excess of a 30% kill in the in-vitro assay before clinical evidence of destruction of tumor is seen. Methods for regulation of ADCC by improving effector cell function are being further evaluated (13).
In an analysis of the ability of a carbohydrate monoclonal antibody such as CA 17.1A to induce apoptosis in colon cancer cells, we compared that mAb to one that was derived from an immunogenic glycoprotein (TSA) of colon Ca origin proven to have efficacy in clinical trials. Monoclonal antibody 17.1A was originally employed in a detailed study of that mAb targeting residual colon tumor cells that might remain following surgery for Dukes C2 carcinoma suggesting a marked improvement in the incidence of recurrence when this carbohydrate monoclonal followed resection of disease. This monoclonal (Reitmueller) (19) had originally been used in clinical trials in Germany suggesting that a clinical response could be anticipated when the antibody was added as an adjuvant to the surgery of Dukes C2 colon carcinoma. A major trial utilizing this monoclonal antibody now referred to as Panorex, was run by Glaxo, but prior to initiation of the study, an ADCC study was performed. The results are seen in Fig. 1.
Fig 1
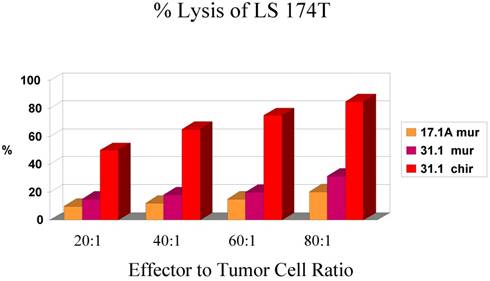
Demonstrates the effect of chimerization on the function of mAb 31.1 and the need for an effective E:T ratio.
Needing a backup monoclonal to compare the effectiveness ofa tumor kill from mAb 17.1A, Tsang utilized mAb 31.1 that had been developed by Tsang and Arlen (6). The mAb had been derived from the immongenic TSA for colon cancer. Noting a low level antitumor response for the 17.1A mAb, that was below 20% tumor kill every 6 hours and especially when compared with the results from chimerized mAb 31.1, it appeared that 17.1A would not deliver the therapeutic response that was hoped for. Fig.1 clearly demonstrates the enhanced effectiveness of a chimerized immunogenic protein derived mAb. In a similar approach to the evaluation of the carbohydrate mAbs such as Ca19.9 (pancreatic Ca), PSA (prostate Ca) and Ca125 (ovary), all demonstrate minimal if any therapeutic effect when delivered intravenously to patients with metastatic malignancy
Having identified three separate tumor specific antigens (TSA's) expressed in colon and pancreas cancers, we went on to produce monoclonals for each target protein via hybridoma production. Following this, the heavy and light chains for each mAb were defined and transfected into CHO cell lines as noted above for production of the chimeric versions of these mAbs that is mAbs NPC-1, 31.1 and 16C3.
With plans for studying the tumor effect of Panorex in patients having undergone Dukes C2 colon resection, requests were made for in-vitro data to be obtained. ADCC as such was carried out by Tsang and mechanisms of action studied (14). A control for comparing Panorex, mAb 17.1A, to a monoclonal that shown activity in previous studies, that is mAb 31.1 as mentioned above, was employed. The data from this study is illustrated in Fig.1. It was apparent that tumor destruction by a carbohydrate monoclonal was not that efficient even when compared to a murine mAb derived from a tumor associated immunogenic protein. The use of the chimeric version of the mAb 31.1 clearly illustrated the benefit of employing this class of mAb's that were optimized to deliver the highest rate of tumor kill. At this point mechanisms by which the mAbs functioned to control tumor were studied in detail, Tsang et al. (14).
A number of studies were performed with the mAb 31.1 to evaluate potential efficacy. Cell flow cytometric data, followed by ADCC and Annexin V binding were used to lead up to animal studies for evaluating efficacy. Of interest as the study progressed. was that when the target proteins were evaluated as to time of expression, it was found that those cells undergoing genotypic transformation could be shown to express the target protein 3-4 months prior to the phenotypic appearance of malignancy. This made the proteins being evaluated to be ideal tumor markers when expressed, and paved the way for the development of early diagnostic tests by IHC as well as in ELISA serum assays.
The rationale for testing various effector to tumor cell ratios in the ADCC study reflects that the true effector cell presence in a population of human peripheral blood mono nuclear cells (PMNC), is defined by a specific subset within the effector cell group, those expressing CD16 surface marker. A proper number of such cells must be present in order to induce the release of products initiating cell cytotoxicity. It appears that the primary effector cell responsible for inducing the ADCC response is the NK cell. When this cell population, containing ligands for the human Fc receptor is delivered with an effective monoclonal antibody to the tumor cell surface expressing the proper TSA, there is an apparent release of NO which initiates the cytotoxic process In the in-vitro model one finds that the effector cell to tumor cell ratio must be maintained at 80:1 to 100:1 for optimum efficacy.
Fig. 1 denotes that as the ratio of E:T decreases, the effective response is halved. It is also apparent that when the antibody is chimerized, allowing proper exposure to the human Fc receptors, there is an increase in response which results in an improved destruction of the tumor population.
Tsang et. al. (14) evaluated the use of hrIL-6 as a means of enhancing the ADCC response. PMNC (polymorphonuclear cells) were preincubated in 100-400 u/ml. of the lymphokine. The effect was to significantly enhance the activity of ADCC as long as the proper mAb was employed; one that defined the tumor immunogenic protein. Depletion of the CD16 population of PMNC suggested that these were the responsible cells for the IL-6 augmentation and as such the NK cells were the prime candidates for the mAbs to secondarily induce tumor cell destruction, rather than that of a direct effect of the mAb on the tumor.
In evaluating the specificity of the ADCC response to several tumor types and to controls, our prime candidate at the time was chimeric mAb 31.1. The other mAbs being studied against colon and pancreas cancer are being prepared for chimerization in a high expression vector to attempt to achieve production levels above 1000mg./L Chimeric 31.1 was the only potential antibody available for study and to compare to CA 19.9, a carbohydrate monoclonal antibody showing activity in pancreatic neoplasms as well as some colorectal lesions. UPC-10, a myeloma antibody was used as the control. One can see relatively good function of mAb 31.1 against colon and pancreas when compared to CA19.9. UPC-10 represented the negative control, Tables 1 and 2.
Table 1
In-111 labeled target cells, antibodies used at 5 ug/ml, human PBMC used as effector cells, 4 hour incubation at 370C before harvest.
| Target | Effector:Target Ratio | % Specific ADCC Activity (± SEM) | ||
|---|---|---|---|---|
| mAb 31.1chi | CA 19-9 | UPC-10 negative cntl | ||
| SW1463 (colorectal adeno) | 100:1 | 51.0 ± 1.3 | 2.0 ± 1.2 | 3.4 ± 0.8 |
| 50:1 | 36.0 ± 1.0 | 1.9 ± 0.1 | 1.1 ± 0.8 | |
| 25:1 | 24.4 ± 1.7 | 1.6 ± 0.3 | 1.3 ± 0.2 | |
| AsPC-1 (pancreas adeno) | 100:1 | 30.3 ± 1.3 | 19.5 ± 0.4 | 3.2 ± 0.9 |
| 50:1 | 19.3 ± 1.3 | 1.4 ± 0.4 | 0.6 ± 0.5 | |
| 25:1 | 12.7 ± 0.1 | 3.3 ± 1.1 | -0.6 ± 1.9 | |
| H441 (lung epithelial ) | 100:1 | 5.0 ±0.4 | 0.6 ± 3.0 | 3.0 ±1.2 |
| 50:1 | 5.2 ± 3.6 | 1.8 ± 0.6 | -0.5 ± 0.4 | |
| 25:1 | 6.7 ± 0.6 | 1.0 ± 0.2 | 0.1 ± 0.5 | |
| MCF-7 (breast adeno) | 100:1 | 2.4 ± 0.3 | 2.5 ± 0.5 | 2.6 ± 0.8 |
| 50:1 | 2.4 ± 0.5 | 1.7 ± 0.1 | 1.8 ± 0.3 | |
| 25:1 | 2.8 ± 0.4 | 2.5 ± 0.2 | 1.7 ± 0.1 | |
Table 2
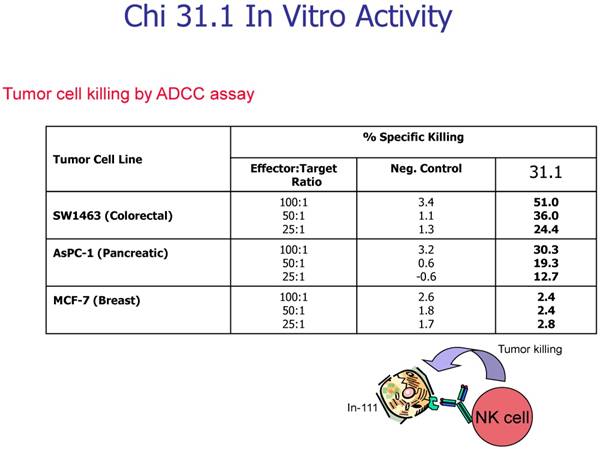
Chimeric 31.1 in a secondary study indicates the strong ADCC activity of mAb 31.1 against colon and pancreatic Ca.
Regardless of the array of different mechanisms involved in initiating tumor cell death thru intravenous administration of an effective IgG, the appropriate monoclonal antibody required is best delivered in the chimeric or humanized form. Here the presence of the human Fc component of the specific IgG employed always contains the NK receptors which optimizes the transfer of this cytotoxic cell to the surface of the tumor. At that point NO can be released at the site on the membrane where the antibody binds resulting in the phenomenon of ADCC. It is also suspected that in those instances where tumor dormancy occurs, that the NK cell may release nitric oxide synthase which potentially suppresses kRas function to initiate the process of dormancy.
In order to evaluate the potential for a clinical response initiated by those mAb's that we were interested in and which were derived from immunogenic proteins, an in-vivo murine model was designed. We chose a therapeutic situation to evaluate the effect of the antibody on fully established tumors in mice. Control animals (nude mice) were primed by injecting 2 million of either human colon adenocarcinoma or pancreatic adenocarcinoma cells (obtained from ATCC) into the hind legs of the animals. Each animal (7per group) were studied for the appearance and progression of tumor cell growth. At 10 days following innoculation, clinical tumor masses appeared from 2-3 cm in diameter and progressed so that within 10 to 15 days each animal was limping and having difficulty in functioning. At the end of following week each animal appeared pre terminal. The experiment was then repeated, to test the ability of the monoclonals to control tumor growth by indicating regression of the established tumor mass.
Groups were divided to receive the 2 million cancer cells noted above, by subcutaneous injections in the leg. On the 10th day of the experiment one group was given 400µgs of non specific human IgG with human effector cells intraperitoneally.
This was performed as a negative control to rule out the ability of nonspecific IgG in the presence of effector cells to elicit an apoptotic effect on the growing human tumor. A second injection of antibody was given on day 11. The second group that received tumor cells was given the therapeutic antibody, mAb 31.1 without the potential benefit of effector cells. This second arm was employed to evaluate if any additional mechanism other than NK cell activity induced tumor destruction. The third group received both intraperitoneal mAb plus human effector cells. It became readily apparent that without the proper mAb, non specific human IgG in conjunction with human effector cells had no response on tumor growth and the masses appearing on the animals' upper leg continued progressing. Therapeutic monoclonals given without effector cells did have some capability of controlling growth of tumor suggesting that a “second”, but lesser mechanism than ADCC might be coming into play. We suspect that many of these therapeutic mAbs secondarily act as TRAIL ligands initiating apoptosis defined by annexin V binding. When the more detailed animal study was performed evaluating mAb 31,1 alone and in combination with effector cells further information was gained on the ability of these TSA monoclonals to initiate tumor destruction thru a number of different means including its primary function in ADCC, Figs. 2,3,4.
Realizing that one mechanism (ADCC), might not completely explain the ability of protein derived monoclonals to control human tumor growth, especially after seeing some weak response in the absence of effector cells, we examined the effect of this group of mAbs on the induction of apoptosis as defined by annexin V binding. At the time of cell death, just prior to DNA destruction by the apoptosome, phosphoinosityl serine is transported to the surface of the tumor cell membrane where it binds to annexin V tagged with a fluorescent signal.
Fig 2
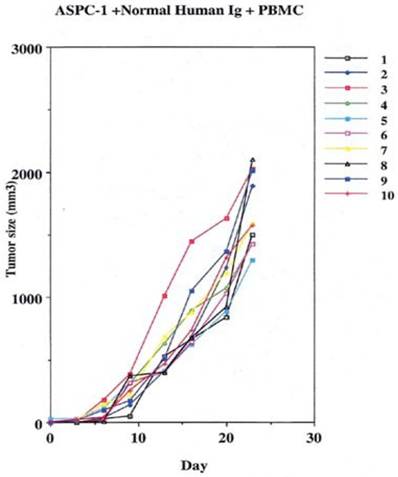
Control arm using normal IgG to replace the therapeutic effect demonstrating that the addition of effector cells can alter the growth of tumor cells.
Fig 3
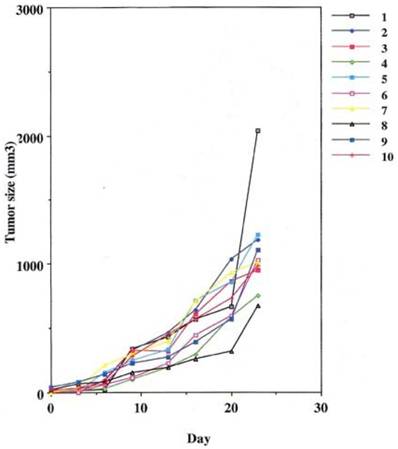
Some control of tumor growth is seen when a potentially therapeutic antibody 31.1 is delivered in the absence of human effector cells.
Fig 4
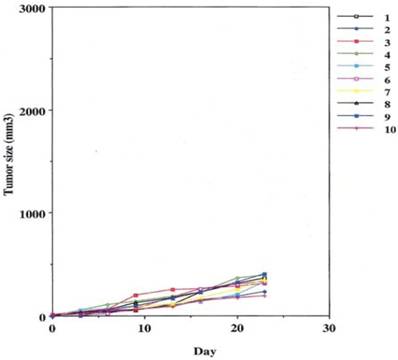
Defines the ability of an effective human IgG1 (TSA derived 31.1 given in combination with human effector cells to control growth of human cancers.
The results of the shift in phosphoinosityl serine to the outer cell membrane are seen in Figs.5 and 6 - where treated and untreated pancreatic cancer cells were compared for the degree of annexin V binding.
Fig 5
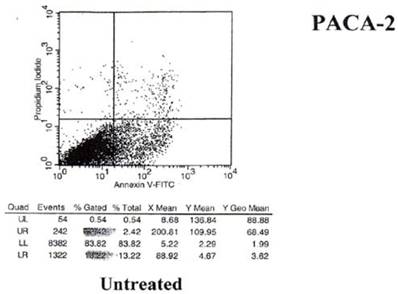
Results of annexin V binding on untreated cells.
Fig 6
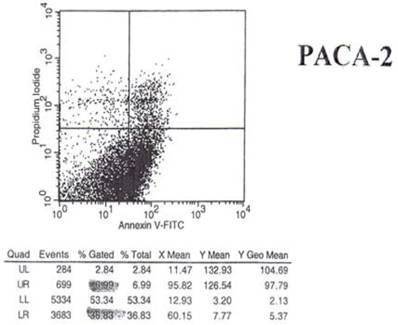
Effect of mAb 31.1 treatment on Annexin V binding of pancreatic cancer cells.
Examination of the 2 scattergrams indicates that in the 4 quadrants illustrated by Figs 5 and 6, that the left lower quadrant represents the population of live cells. The right lower quadrant expresses the incidence of early apoptotic cells before and after induction by mAb therapy and the right upper quadrant, the later apoptotic or necrotic cells that begin to appear. As such, the degree of binding based on shift to the right quadrant (lower and upper) of the scattergram indicates that the % of annexin + cells in the untreated group was 18.22%. This was found to have increased to 36.83% of the cells undergoing apoptosis in the treated group. This possibly explains the shift we see in the animal model curve, where the mAb alone was used without effector cells being present.
Additional possibilities arise as to other mechanisms by which therapeutic mAbs effect tumor growth. We evaluated the effect of this group of TSA mAb's on VEGF expression and were able to demonstrate a significant diminution in the level of these surface growth factors similar to what is seen with Herceptin (Table 3).
Table 3
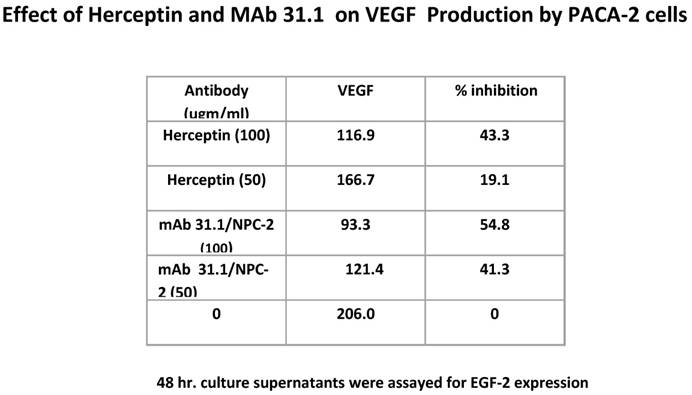
Antibody suppression of VEGF by both 31.1 and Herceptin.
4. Discussion
The ability to employ passive immunization, that is, mAb therapy, alone or in combination with chemotherapy has become a valid approach for managing patients with malignancies that have failed standard chemotherapy. While most of those antibodies used in the treatment of malignancies are presently targeting growth factors, we believe that antibodies, IgG1's, directed against immunogenic proteins expressed on the tumor cell surface membrane, offer a greater potential for effective tumor control in the metastatic setting.
Liu et al. (15) noted the presence of tumor associated antigens on the cell surface of malignant lesions as characteristic of many cancers. They reported that antibodies to these TSA's could be developed and that it might be possible to use such antibodies for targeting the specific tumors. They used replacement of the mouse constant C domain regions with the corresponding human equivalent (human Fc) to create chimeric mAbs. Antibodies obtained by this approach retained specificity for antigen but were felt to be less immunogenic if they were given to patients.
In general, the present approach we are employing and that is required for developing such antibodies for therapeutic use in patients requires that the IgG be humanized or human in structure. In addition the expression of the monoclonal should be produced in a mammalian cell line such as CHO (dhfr -) and for commercial value, the antibody should be expressed at over 1000 mg/L of bioreactor fluid that contains no fetal calf serum.
The mouse mAb L6 (IgG2aK) that Liu's group studied binds to a carbohydrate antigen that is present at the surface of cells from such human carcinomas as lung, breast, colon, and ovary. L6 was found to mediate CDC (complement-mediated cytotoxicity) with human complement or ADCC with human effector cells. cDNA's encoding the immunoglobulin genes for L6 were isolated. Restriction enzyme recognition sites were created in the cDNA sequences at the V/C junction by in-vitro mutagenesis using oligodeoxy- ribonucleotides. The chimeric antibody they produced was then compared with the mouse L6 for effector function. It was founds to bind to tumor cells as well as the mouse L6 antibody but was more active in ADCC, killing a greater number of target cells at a concentration lower by a factor of 100. The chimeric mAb that was produced was never developed for clinical testing. Other mAb's such as A33 were also tested clinically but did not reach the therapeutic effect felt necessary for commercialization.
We realized the need for the chimerization of our monoclonal antibodies, with the concept that if the human anti-mouse antibody (HAMA) responses were high with the murine hybridoma version, humanization might be indicated both for enhancement in response and for minimization of the HAMA response. In a single patient study with the chimeric version of our monoclonal 31.1 directed against hepatic metastasis from pancreatic cancer, the HAMA response became minimally elevated in contrast to pretreatment levels. After 2 doses of the chimeric mAb at 25 mg and 50 mg IV the serum marker for Ca 19.9 fell from 3000U to 300U. The HAMA prior to therapy was 5 nanogms/ml prior to administration of the mAb and rose to 7 nanogms/L 2 weeks after the second and final dose was given. Additional mAb was not available for further study. We are presently preparing a new version of our mAbs derived from the original immunogens used in clinical trials. It is being cloned into a vector with a strong promoter capable of delivering the anticipated 1000 mg/L or more, needed for production as a GMP product.
Where the need to minimize the HAMA response is important, it can be accomplished by modifiying the structure of the mAbs utilizing the backbone structure of the human FAb in the humanization process leaving only the murine CDR loops. This modificatyion in structure results in reduction in HAMA response however it may not be as important as the therapeutic efficacy of the antibody being employed in its chimeric form.
A secondary host response contributing to the potential efficacy of the mAb may be seen in the production of an anti-idiotype response to the therapeutic monoclonal (16). As part of the hypothesis of an antiidiotype network, immunization with a given TSA generates production of antibodies mAb1. In turn, Ab1 will initiate production of a series of anitidiotype antibodies termed Ab2 which express the internal image of the TSA. Some of the Ab2 molecules can effectively mimic the 3 dimensional structure of the TSA. These particular antibodies can induce a specific immune response similar to the original TSA. This particular antiidiotype antibody, the Ab2, structurally represents an exogenous peptide that can be endocytosed by the APC's and degraded into 14-25 mer peptides to be presented by class II antigens to activate CD4 helper cells. Cross presentation to CD8+ T cells also occur. We are now in the process of developing the procedures to define and quantify the appearance of the anti id response to the clinical delivery of our NPC-1 and 31.1 chimeric monoclonals. When entering clinical trials we plan to measure levels of anti-idiotypes appearing in the serum in response to mAb's NPC-1, 31.1, and 16C3. If enhancement in immune responses are noted in relation to MHC Calss 1, it will be important to determine if mAb2 does mimic the mirror image of the antigen used in the development of our monoclonals and if Ab2 in of itself has the immunogenic capabilities to generate cytotoxic T-cells. The failure of previous attempts to produce effective antiidiotypic monoclonals resided in the fact that the monoclonals being studied were carbohydrate in origin. As such any anti idiotype response would theoretically mimic the carbohydrate moiety of the antigen.
The first monoclonal antibody technology was introduced in 1985 with the introduction of OKT3 (17). This antibody was developed against an epitope on the epsilon unit within the CD3 complex. It proved to be a potent immunosuppressive agent, capable of minimizing allograft rejection in heart, lung and kidney transplant. In 1989 Meldstedt (18) at the Karolinska Institute tested the first potential antitumor antibody Ca 17.1A for its activity in metastatic colon Ca. He noted regression of metastatic lung lesions, but no prolongation in survival. As such the monoclonal was discontinued in therapeutic trials. Reitmuller (19) later suggested (1994) that marked improvement in the survival of Dukes C colon cancer could be achieved using monoclonal 17.1A in the adjuvant rather than therapeutic setting. This finding however, was not confirmed in more extensive clinical trials carried out in the US using 17.1A under the name Panorex as described previously. We had anticipated minimal anti-tumor activity for Panorex since it was carbohydrate in origin, and as with other carbohydrate mAbs, apoptosis if present, was usually minimal. On completion of a detailed 5 year clinical study employing Panorex in patients with Dukes C carcinoma, no clinical improvement was found. As such this mAb was removed as potential clinical reagent for use in locally advanced, recurrent and possible metastatic colon cancer.
Most therapeutic mAbs with human backbones are of the IgG1 isotype which effectively mediate the Fc domain based function, including compliment fixation and ADCC. Other isotypes such as the IgG2's have been used when the mAb was designed to act simply thru its antigen binding capabilities rather than have the inherent capability of working to induce apoptosis. Examples of the latter category include the anti-EGF monoclonals, Cetuximab(Erbitux)(20), Herceptin(21) and Bevacizumab(Avastin)(22) which are murine/human chimeric antibodies. We have found that by humanizing the Fc portion of our antibodies, we almost double the ability to destroy tumor growth, probably reflecting the presence of the proper ligands on the human Fc for ADCC as well as the initiation of complement fixation. While we understand that those mAbs that address the issue of a human epidermal growth factor have some effect on clinical response, these mAb's eventually lose control of tumor growth with subsequent progression ofdisease as a result of the tumor finding another metabolic pathway for its growth and development. Where Avastin was believed to function through diminution in the vascular supply to the tumor, it is now thought that this agent may affect microcirculation within the tumor making the introduction of chemotherapeutic agents in combination with the antibody more effective in tumor destruction.
Whereas tumor growth in the case of pancreatic cancer having recurred, has an average response to the chemotherapeutic agent Gemcitabine of 7 months, few agents have improved on this. Tarceva when added to a gemcitabine regimen does appear to have a slight advantage of 14 or so days. When Avastin was added to the Gemcitabine protocol for recurrent pancreatic cancer there was no evidence that any efficacy could be determined. We do however believe, that those monoclonals that have been derived from an immunogenic target in pancreatic and or colon cancer such as TSA NPC-31.1, when employed alone or in combination with chemotherapy, may show a survival advantage in those recurrent malignancies having failed standard chemotherapy.
In the initial chimerization of our protein derived mAbs, we chose a vector which allowed insertion of both heavy and light chain genes with transfection to CHO cells for optimum production. The plasmid vector pRc/CMV that we employed was designed with the intent of achieving high expression of our chimeric monoclonal antibodies. The vector had a cloning site which accepted the heavy and light chain genes, inserting them downstream from the human CMV. The vector allowed antibody to be produced at levels greater than 100mg/L in bioreactor media, so that therapeutic doses of 250-500mg, could be delivered.
We do believe that with mAbs demonstrating minimal HAMA and high levels of ADCC, that doses of 200mg. to 400mg.delivered every two weeks I.V. could be effective in controlling metastatic cancer. At the present time we have chosen a newer vector which allows similar insertion of heavy and light chain genes, but has a potential for production in excess of 1000 mg/L of bioreactor fluid. Both plasmid vectors carry a dhfr expression unit driven by an enhancer-deficient SV40 early promoter. The vector is inserted into the CHO-D-SFM (dihydrofolate reductase (dhfr)-deficient Chinese hamster ovary) cells in near serum-free medium supplemented with 1 μg/ml of MTX. At the end of the production, cells have to be adapted to serum free media before final purification of the antibody can occur.
ADCC as we have described above, appears to be one of the important mechanisms for tumor cell lysis of unconjugated mAb's. Many of these antibodies having high affinity and specificity do not elicit a strong lytic response. These monoclonals contain a conserved N-linked glycosylation site in each of the CH2 domains of the constant region. The sites are normally occupied by complex oligosaccharide structures which have an important effect on the activity of the mAbs. One of the qualitative differences in the oligosaccharide component between human circulating antibodies and those derived from recombinant antibody producing cell lines, is the presence of bisected oligosaccharides on the naturally occurring antibodies. These oligosaccharides are produced by a reaction catalyzed by N-acetylglucosaminyl transferase III (GnTIII) which essentially is not expressed in the commercially produced mAbs. The effect of GnTIII was assessed in terms of the potential enrichment of humanized therapeutic mAb's (23). The ADCC activity of three independent antibodies was substantially increased, ten to one hundred fold when the antibodies were derived from cell lines overexpressing GNTIII. Based on the final ADCC levels that we have defined with our mAbs, we are now evaluating the possibility that alteration in glycosylation patterns can further enhance ADCC to yield higher therapeutic responses.
The first therapeutic mAb to receive FDA approval, Rituximab, was for use in the low and intermediate grade lymphomas. This monoclonal product, developed at IDEC, is a naked antibody directed against the surface antigen, CD20. Many of the mechanisms of activity have been defined (5). This is of importance to our group since we are still defining the multiple methods for tumor destruction initiated by those monoclonals derived from immunogenic proteins expressed on the surface of the tumor cell. The CD20 antigen seen on all lymphocytes and most lymphoma cells proved to be a transmembrane protein of 297 amino acids. While not serving as a surface immunogen, the binding of CD20 by Rituxan did prove to initiate apoptosis. The intracellular portion of the CD20 antigen was shown to contain phosphorylation sequences for protein kinase C, calmodulin, and casein kinase -2. When CD20 is transfected into the cell, intracellular Ca++ increases. When Rituxan attacks or cross links to CD20 there is an increase in intracellular Ca++ and further phosphorylation of the inner chain of CD20. There is also an associated upregulation of C-myc and an upregulation of MHC-2 proteins.
Caspace 3 is activated in the above mechanism of Rituxin/CD20 binding to induce apoptosis in intermediate grade lymphoma. This has resulted in one of the more active approaches to the treatment of a hematologic malignancy. CD20 has a 95% expression in B cell malignancies explaining the high incidence for potential cases to be treated.
In those cases where commercial monoclonals target surface growth factors such as epidermal growth factors (Erbitux, Herceptin), the incidence of expression of these growth factors for many of the patients is in the 20% range, explaining the low number of patients with colon and breast cancer that can be subjected to effective treatment.
One year survival with Rituxan in lymphoma has been shown to be 69% and for 2 year 67%. When delivered with chemotherapy, the survival results have increased. This stands in contrast to the commercial growth factor mAb's noted above, such as Erbitux, Herceptin and Avastin where, as noted above, enhanced survival is defined in months even when given in combination with chemotherapy (2). We anticipate that the protein based mAb's that we have been developing should demonstrate responses in solid tumor malignancies similar to what is seen for lymphoma patients, when Rituxan is employed. With further analysis of our protein based mAbs we believe that a number of additional mechanisms will be defined in addition to those of ADCC, apoptosis as defined by annexin V binding and suppression of VEGF to minimize vascularity to the tumor. We believe that this class of TSA derived mAbs have the potential to further improve results expected to be obtained when the naked antibody is used. When given in combination with immune stimulants and then chemotherapy is used for additional treatment, one can anticipate a further improvement in survival.
Mechanisms defining the action of NK cells are being resolved especially in terms of their relationship to the Fc component of the IgG as noted above. This pertains to receptors that are essential for initiating the ADCC process and are potent regulators of the ADCC. Inhibitory Fc receptors have been found to modulate in-vivo cytoxicity against tumor targets (24).
The inhibitory FcγRIIB molecule is an important part of the activator suppressor system. The FcγRIII receptor enhances the activity of the effector cell defining the associated ligand on the IgG. The antitumor activities of therapeutic antibodies include blocking, signaling pathways, activation of apoptosis, effector cell mediated cytotoxicity, and engagement of Fc γ receptors on effector cells. It's been shown that Herceptin (Trastuzumab) and Rituxan (Rituximab) engage both activation with Fc γ III, as well as inhibition with the Fc γ IIB antibody receptor on myeloid cells. Negative signals can down-regulate T, B, monocytes, dendritic cells as well as NK cells functions. These signals belong to numerous subfamilies but we focus on the inhibitory receptors of the B7 family. However other negative regulators are also important such as the FcRIIB, CD22 and others.
Human Natural Killer Cells express only the A (transmembrane) iso form of CD16. This facilitates activation of the NK cell. ADCC has been viewed as a mechanism to directly induce a variable degree of immediate tumor destruction. This process looks to antigen presentation and induction of tumor directed T cell responses.
With regard to complement fixation cytotoxicity, the IgM's have the most effective method for induction of cell death. However because of the size of the molecule, it has little therapeutic effect since it is difficult for this IgG to penetrate those vascular structures needed to reach the tumor. This is why the proper antibody for therapy must not only have tumor killing capability, and induce apoptosis, but be capable of reaching the target cells with ease. These tasks can be accomplished more readily with the IgG1's and 3's. Isotyping those clinical antibodies that we have seen to be potent in the in-vitro as well as in-vivo situation still indicate that the IgG1's have the greatest versatility to enhance immune reactivity.
5. Conclusion
What appears unique to those immunogenic TSA protein derived antitumor mAb's, in comparison to those directed against surface epidermal growth factors (EGF-1, EGF-2,VEGF), is their specificity to the malignancy only as well as high rate of tumor kill in a relatively short period of time. This obviates the need for long clinical trials attempting to illustrate an improvement in the interval to progression of the metastatic disease process.
The apoptotic effect which occurs in 6-8 hrs when these proteins derived mAbs are employed via intravenous administration is defined by ADCC mediated by the recruitment of NK, CD-16 cells. There is also a suggestion of inhibition in the expression of VEGF and that a degree of apoptosis seen in the absence of NK cells may be mediated by TRAIL ligation. This same group of TAA derived antibodies appears to demonstrate an ability to define target antigens as diagnostic molecular markers, months before the phenotypic expression of malignancy becomes apparent. A requirement for employment in the metastatic situation is defined by the need to demonstrate that the target immunogenic antigen is expressed in the tumor and specifically on the cell surface membrane where it eventually sheds into the serum. We have been able to illustrate that tumor antigen (TSA) is routinely shed into serum as a potential diagnostic marker and as such are developing a serum ELISA to use not only for monitoring clinical responses but as a potential diagnostic serum test.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Liu W.M, Fowler D.W, Smith P, Dalgleish A.G. Pre-Treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumors by promoting adaptive immune responses. Br. J. Cancer. 2010;102:115-123
2. Ramakrishnan R, Assudani D, Nagaraj S, Hunter T, Cho H. et al. Chemotherapy enhances tumor cell susceptibility to CTL-mediated killing during Cancer Immunotherapy in mice. J Clin Invest. 2010;120:1111-1124
3. Qu CF, Song EY, Li Y, Rizvi SMA, Raja C, Smith R, Morgenstern A, Apostolidis C, Allen BJ. Preclinical study of 213Bi labeled PAI2 for the control of micrometastatic pancreatic cancer. Clinical & Experimental Metastasis. 2005;22:575-586
4. Hollinshead A, Elias G, Arlen M. et al. Specific Active Im-munotherapy in patients with adenocarcinoma of the colon utilizing tumor associated antigens (TAA). Cancer. 1985;56:480-489
5. Arlen M. Escape Mechanisms employed by tumor cells to allow for growth, invasion and metastasis; In Chemoimmunosensitization of Resistant Tumor Cells to Cell Death by Apoptosis (text). Bonavida. 2006:243-261
6. Arlen M, Tsang K.W. The Nature of the Monoclonal Antibodies derived from Immunogenic membrane antigen of colon carcinoma origin. J. Tumor Marker Oncology. 1990;5:313-319
7. Jazirehi A.R, Bonavida B. Cellular and Molecular Signal Trans-duction Pathways Modulated by Rituximab in Non Hodgkins Lymphoma. Implications in Chemotherapy Sensitization. Oncogene. 2005;24:2121-2143
8. Catimel B. et.al Purification and characterization of a novel restricted antigen expressed by normal and transformed human colonic epithelium. J. Biol. Chem. 1999;271:25664-25670
9. Morrone S. Annexin V. Cytometry CD Rom Series V.4. USA: Purdue Cytometry. 1998
10. Francis RJ, Sharma SK, Springer C. et al. A phase I trial of anti-body directed enzyme prodrug therapy (ADEPT) in patients with advanced colorectal carcinoma or other CEA producing tumors. Br. J. Cancer. 2002;87:600-607
11. Schuster M, Umana P, Ferrara C, Brunker P, Gerdes C. et al. Improved Effector Functions of a Therapeutic Monoclonal Lewis Y-Specific Antibody by Glycoform Engineering. Cancer Research. 2005;65:7934-7941
12. Durrant L.G, Robbins R.A, Baldwin R.W. Flow Cytometric Screening of Monoclonal Antibodies for Drug or Toxin Targeting to Human Cancer. J. Nat Cancer Institute. 1989;81:688-695
13. Tang Y, Lou J, Alpaugh RK, Robinson MK, Marks JD, Weiner LM. Regulation of Antibody-Dependent Cellular Cytotoxicity by IgG Intrinsic and Apparent Affinity for Target Antigen. J. Immunol. 2007;179:2815-2832
14. Tsang K.Y, Finch M.D, Primus F.J, Schlom J. Human re-combinant interleudin-6 enhances antibody-dependent cellular cytotoxicity of human tumor cells mediated by human peripheral blood mononuclear cells. Cancer Immunol Immunother. 1991;34:9-16
15. Liu A.Y, Robinson R.R, Hellstrom K.E, Murray E.D, Chang C.P, Hellestrom I. Chimeric mouse-human IgG1 antibody that can mediate lysis of cancer cells. Proc. Nat. Acad. Sci. USA. 1987;84:3439-3443
16. Fagerberg J, Hjelm A.L, Ragnhammar P, Frodin J.E, Wigzell H, Mellstedt H. Tumor regression in monoclonal antigody treated patients correlates with the presence of anti-idiotype reactive T- lymphocytes. Cancer Research. 1995;55:1824-1827
17. Krotkiewski H, Gronbergl G, Krotkiewska B, Nilssonll B, Svensson S. The Carbohydrate Structures of a Mouse Monoclonal IgG Antibody OKT3. J. Biol. Chem. 1990;265:2019-2021
18. Mellstedt H, Frodin JE, Massucci G. Clinical status of monoc-lonal antibodies in the treatment of colorectal carcinoma. Oncology. 1989;3:25-31
19. Reitmuller G. et al. Randomized trial of mAb for adjuvant therapy of resected Dukes'C colorectal cancinoma. The Lancet. 1994;343:1177-1183
20. Wu X, Fan Z, Masui H, Rosen N. Apoptosis induced by an anti-epidermal growth factor receptor monoclonal antibody in a human. J Clin Invest. 1995;95(4):1897-1905
21. Romond EH, Perez EA, Bryant J. et al. Trastuzumab plus adjuvant chemotherapy for operable HER2+ breast cancer. N Engl J Med. 2005;353:1673-1684
22. Muhsin M, Graham J, Kirkpatrick P. Bevacizumab. Nature Reviews Drug Discovery. 2004;3:995-996
23. Lipscomb M.L, Palomarea L.A, Hernandez V, Ramirez O.T, Kompala D.S. Effect of Production Method and Gene Amplification on the Glycosylation Pattern of a Secreted Reporter Protein in CHO Cells. Biotechnol. Prog. 2005;21:40-49
24. Clynes R.A, Tower T Dresta G, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nature Med. 2000;6:443-446
Author contact
![]() Corresponding author: Email: myronarlencom
Corresponding author: Email: myronarlencom
Citation styles
APA
Arlen, M., Arlen, P., Tsang, A., Wang, X., Gupta, R. (2010). The Therapeutic Value of Monoclonal Antibodies Directed Against Immunogenic Tumor Glycoproteins. Journal of Cancer, 1, 209-222. https://doi.org/10.7150/jca.1.209.
ACS
Arlen, M.; Arlen, P.; Tsang, A.; Wang, X.; Gupta, R. The Therapeutic Value of Monoclonal Antibodies Directed Against Immunogenic Tumor Glycoproteins. J. Cancer 2010, 1, 209-222. DOI: 10.7150/jca.1.209.
NLM
Arlen M, Arlen P, Tsang A, Wang X, Gupta R. The Therapeutic Value of Monoclonal Antibodies Directed Against Immunogenic Tumor Glycoproteins. J Cancer 2010; 1:209-222. doi:10.7150/jca.1.209. https://www.jcancer.org/v01p0209.htm
CSE
Arlen M, Arlen P, Tsang A, Wang X, Gupta R. 2010. The Therapeutic Value of Monoclonal Antibodies Directed Against Immunogenic Tumor Glycoproteins. J Cancer. 1:209-222.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.