Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2011; 2:271-279. doi:10.7150/jca.2.271 This volume Cite
Research Paper
Differential Genetic Expression in Large Versus Small Clear Cell Renal Cell Carcinoma: Results from Microarray Analysis
Gennady Bratslavsky1# ![]() , Thomas Sanford 1#, Ramaprasad Srinivasan1, Olga Aprelikova2, Jack Liu 1, Martha Quezado 3, Maria Merino3, W. Marston Linehan1
, Thomas Sanford 1#, Ramaprasad Srinivasan1, Olga Aprelikova2, Jack Liu 1, Martha Quezado 3, Maria Merino3, W. Marston Linehan1
1. Urologic Oncology Branch, National Cancer Institute, Bethesda, MD, USA
2. Cell and Cancer Biology Branch, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA
3. Surgical Pathology Branch, National Cancer Institute, National Institutes of Health, Bethesda MD, USA
# Equal contribution
Received 2011-4-21; Accepted 2011-5-12; Published 2011-5-12
Citation:
Bratslavsky G, Sanford T, Srinivasan R, Aprelikova O, Liu J, Quezado M, Merino M, Linehan WM. Differential Genetic Expression in Large Versus Small Clear Cell Renal Cell Carcinoma: Results from Microarray Analysis. J Cancer 2011; 2:271-279. doi:10.7150/jca.2.271. https://www.jcancer.org/v02p0271.htm
Other stylesAbstract
Purpose: Tumor growth and progression requires multiple steps and genetic alterations. The molecular events that occur as tumors increase in size are unknown. Patients with von Hippel-Lindau (VHL) provide a unique opportunity to study molecular alterations during tumor growth as these patients develop multiple bilateral renal tumors. To better characterize biologic events associated with tumor growth, we evaluated the alterations in gene expression in large versus small renal tumors removed from the same kidney of the same individuals.
Experimental Design: We reviewed pathology reports from patients who underwent partial nephrectomies at the National Cancer Institute for multiple tumors. We identified 11 patients who fulfilled the following inclusion criteria: 1) The patient must have had a surgical resection of more than one solid tumor from the same kidney during the same operation; 2) Among the solid tumors at least one must have been greater than 3 cm in the largest dimension and at least one less than 2 cm; 3) the nuclear Furhman grade for both larger and smaller solid tumors was identical; 4) a portion of each tumor was procured and snap frozen after surgical removal; 5) Hematoxylin and eosin staining of the frozen sample confirmed clear cell carcinoma to be present in at least 80% of the section.
Affymetrix platform and protocol for gene expression arrays were used. RNA from the frozen large and small tumor samples was extracted using Trizol-Chlorophorm method. The RNA was then reverse transcribed, labeled, fragmented, and hybridized on to an Affymetrix U133 Plus 2.0 array that contains 54,000 probe sets representing 24,568 genes. Analysis included unsupervised clustering and chromosomal analysis. The paired t-test was performed to compare gene expression levels in small and large tumors. P<0.01 was considered statistically significant.
Results: Gene expression profiles were assessed for 22 tumors (11 patients). Upon unsupervised clustering the pairs with larger tumor volume difference clustered separately from pairs with smaller volume difference. Chromosomal analysis revealed few consistent changes other than reduced expression of chromosome 3p25 among all tumors. Paired t-test showed 860 differentially expressed genes in the T1b vs T1a group, a number far greater than expected due to chance alone. When analyzed by gene function, most differences were observed in genes involved in DNA replication and in cytokine signaling.
Conclusions: This study demonstrates that as tumors increase in size there is an increasing difference in gene expression. Unsupervised clustering analysis confirms that as the volume difference increases there are a distinct set of genes that are regulated either as a response to a tumor's growth or as an early event that causes the tumor to grow. While we did not observe chromosomal instability, we did note differences in expression of individual transcripts as tumors grew larger.
Keywords: Renal cell carcinoma, tumor, size, microarray, kidney
Introduction and Objectives
During the last two decades, the molecular events that lead to the formation of renal cell carcinoma have been elucidated, particularly for the clear cell type (ccRCC). Most clear cell renal tumors form as the result of dysregulation of the von Hippel-Lindau tumor (VHL) suppressor gene with loss-of-function somatic mutations and epigenetic silencing that may be found in up to 90% of sporadic ccRCC (1). While the inciting genetic events that lead to the formation of ccRCC have been worked out in great detail, the events that give primary tumors the ability to grow and potentially gain aggressiveness remain poorly understood.
There is a strong relationship between the size of the primary tumor and likelyhood of metastasis. Frank et al demonstrated that estimated cancer specific survival rates for tumors >7cm were 15-25% higher than for tumors that were less than <7cm following nephrectomy (2). In a separate study from the NCI of a cohort of patients with inherited germline mutations of the VHL gene, there have been no cases of metastasis of tumors less than 3 cm (3). To explore the genetic events involved in the development and progression of ccRCC as tumors grow larger, we surveyed the global gene expression profiles of tumors of different sizes in patients with confirmed germline VHL gene mutations. By using tumors of different sizes removed from the same patient, we aimed to study the differences in genetic profiles associated with increase in tumor size.
Materials/Methods
Patient selection
Pathology reports from patients who underwent partial nephrectomies for multiple renal tumors at the National Cancer Institute were reviewed. Eleven patients fulfilled the following inclusion criteria: 1) The patient must have had a surgical resection of more than one solid tumor from the same kidney during the same operation; 2) Among the solid tumors at least one must have been greater than 3 cm in the largest dimension while the other removed solid tumor was less than 2 cm; 3) the nuclear Furhman grade for both larger and smaller solid tumors was identical; 4) a portion of each tumor was procured and snap frozen after surgical removal; 5) Hematoxylin and eosin staining of the frozen sample confirmed clear cell carcinoma to be present in at least 80% of the section. Original tumor volumes were calculated using the formula for an ellipsoid (4/3πr3). All patients had confirmed mutations in the VHL tumor suppressor gene. All patients were enrolled in an IRB approved NIH intramural research protocol.
A summary of the tumor sizes for the eleven patients included in this study can be seen in Table 1. Four pairs have at least 1 tumor >4cm (used to compare T1a vs T1b), and seven pairs have two tumors <4cm (used to compare T1a vs T1a).
An example of the multifocal renal tumors in a patient with VHL disease can be seen in the bivalved nephrectomy specimen in Figure 1 demonstrating multiple tumors of different sizes throughout the kidney.
RNA Isolation
Fifteen 20μm thick frozen sections from each tissue sample were homogenized in TRIZOL reagent (Invitrogen Life Techonolgies, Carlsbad, CA USA). Total RNA was extracted using a standard chloroform protocol followed by purification with the Qiagen RNeasy Mini Kit (QIAGEN Inc, Valencia, CA USA). RNA integrity was evaluated by using RNA 6000 Nano LabChips on an Agilent 2100 Bioanalyzer (Agilent Technologies, Foster City, CA USA). RNA purity was assessed by the ratio of spectrophotometric absorbance at 260 and 280 nm (A260/280nm) using NanoDrop ND-1000 (NanoDrop Inc, Wilmington, DE USA). All chips were prepared according to the manufacturer's instructions. Total RNA degradation was evaluated by reviewing the electropherograms and the RNA integrity number (RIN): only samples with preserved 18S and 28S peaks and RIN values greater than 7 were selected for gene expression analysis.
Table 1
Tumor sizes for the patients
| Size | Tumor size (cm) | Size Difference (cm) | Tumor volume (cc3) | Volume Difference (cc3) | |
|---|---|---|---|---|---|
| Patient 1 | large | 6 | 4.7 | 84.8 | 84.0 |
| small | 1.3 | 0.8 | |||
| Patient 2 | large | 5.5 | 3.7 | 25.9 | 24.5 |
| small | 1.8 | 1.4 | |||
| Patient 3 | large | 5.2 | 4 | 34.3 | 31.9 |
| small | 1.2 | 2.4 | |||
| Patient 4 | large | 4.5 | 2.5 | 24.7 | 22.0 |
| small | 2 | 2.7 | |||
| Patient 5 | large | 3.5 | 1.7 | 11.9 | 11.4 |
| small | 1.8 | 0.5 | |||
| Patient 6 | large | 3.5 | 2 | 9.2 | 8.9 |
| small | 1.5 | 0.3 | |||
| Patient 7 | large | 3.4 | 2.1 | 15.9 | 15.3 |
| small | 1.3 | 0.6 | |||
| Patient 8 | large | 3.1 | 1.1 | 11.8 | 10.3 |
| small | 2 | 1.5 | |||
| Patient 9 | large | 3 | 1 | 4.7 | 2.3 |
| small | 2 | 2.4 | |||
| Patient 10 | large | 3 | 1.6 | 7.9 | 6.8 |
| small | 1.4 | 1.1 | |||
| Patient 11 | large | 3 | 1.8 | 14.1 | 13.8 |
| small | 1.2 | 0.3 |
Figure 1
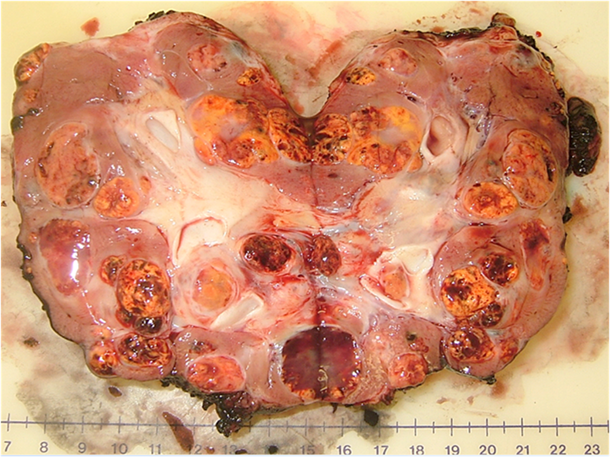
Multiple renal tumors with a single renal unit: bivalved nephrectomy specimen from a patient with a germline mutation in the VHL gene.
Figure 2
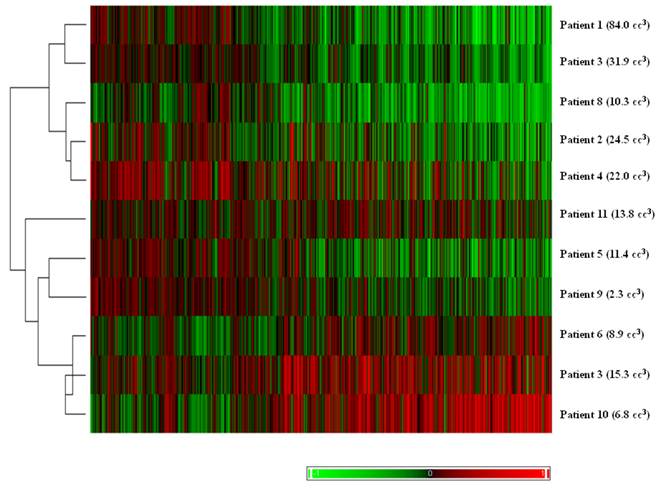
Hierarchical clustering of tumor pairs based on differences in size. The volume difference between larger and smaller tumor is listed in parenthesis for each patient.
Microarray hybridization and Image Acquisition
Messenger RNA expression levels were measured using the GeneChip® HG-U133 plus 2.0 platform following the manufacturer's protocol (Affymetrix Inc, Santa Clara, CA USA). Briefly, double-stranded cDNA was synthesized from 11µg of total RNA from each tumor sample using GeneChip Two-Cycle cDNA Target Synthesis Kit (Affymetrix Inc, Santa Clara, CA USA). After one round of amplifications, in vitro transcription was performed using IVT Labeling Kit (Affymetrix Inc, Santa Clara, CA USA) to synthesize biotin-labeled cRNA. An aliquot of 15ug labeled cRNA was fragmented and subsequently hybridized for 16 hours at 45C in a hybridization oven to a HG-U133 plus 2.0 olgionucleotide array. Arrays were washed and scanned with an Affymetrix GeneChip 3000 Scanner.
Statistical Analysis
Raw data (.CEL) files were imported into BRB array tools (4). The data were normalized using the RMA algorithm (5). Unsupervised clustering was performed using hierarchical clustering (1-correlation) based on the differences in size between large and small tumors. The data were then separated into two groups for supervised analysis: the first consisted of pairs of tumors that were both T1a (<4cm), and the second group had one T1a tumor and one T1b (between 4 and 7 cm) to evaluate the accepted size cut offs of AJCC staging. Differences in gene expression were analyzed using a paired t-test with random variance model set at 1000 permutations. Significant P-value was set at 0.01. BRB array tools was used to analyze which categories are over-represented relative to the prevalence of Gene Ontology categories on the array (4).
Data were then imported into the R environment for further analysis (www.r-project.org). Chromosomal abnormalities were assessed with a technique known as comparative genomic microarray analysis. This method has been shown to have good correlation with more traditional CGH techniques (6). For a common reference pool, a pool of normal renal parenchyma from the gene expression omnibus database (http://www.ncbi.nlm.nih.gov/geo/) was used (GSE7392).
Results
The results of unsupervised clustering based on the volume difference of the pairs can be seen in Figure 2. With one exception, the pairs with larger differences in volume clustered separately from pairs with smaller differences.
A schematic of the results of the paired t-test are observed in figure 3. The Affymetrix HGU133 plus 2.0 has 54,675 probe sets. The threshold for significance was set at 0.01. Thus, we would expect approximately 550 false positives. In the paired analysis where both tumors were less than 4cm, there were 458 genes were differentially expressed, less than would be expected by chance. When tumors greater than 4cm were compared to tumors less than 4cm in the same patient, there were 890 differentially expressed genes identified, which is greater than the expected false positives. When we examined the gene ontology of the significant gene changes, we found that transcripts involved in cytokine production and DNA synthesis to be over-represented (Table 2).
The first part of the cGMA chromosomal analysis was an examination of the expression pattern for chromosome 3. Since all the patients had a confirmed VHL mutation, as expected the level of expression was decreased in chromosome 3p25 in all samples when compared to normal renal parenchyma (Figure 4). Interestingly, there is was also a decrease in gene expression near chromosome 3p21. Full chromosomal analysis revealed that the expression profiles within each pair appeared similar. No consistent differences in expression on chromosomal level between large and small tumors were observed. (Figure 5).
Table 2
Gene ontology
| GO ID | GO Term | Observed/Expected |
|---|---|---|
| GO:0032602 | chemokine production | 8.08 |
| GO:0042364 | water-soluble vitamin biosynthetic process | 6.79 |
| GO:0051187 | cofactor catabolic process | 6.57 |
| GO:0034381 | lipoprotein particle clearance | 6.47 |
| GO:0009110 | vitamin biosynthetic process | 5.5 |
| GO:0006306 | DNA methylation | 5.39 |
| GO:0006305 | DNA alkylation | 5.39 |
| GO:0007031 | peroxisome organization | 4.85 |
| GO:0031047 | gene silencing by RNA | 4.53 |
| GO:0006304 | DNA modification | 4.04 |
| GO:0030336 | negative regulation of cell migration | 3.99 |
| GO:0001935 | endothelial cell proliferation | 3.93 |
| GO:0016458 | gene silencing | 3.85 |
| GO:0043542 | endothelial cell migration | 3.83 |
| GO:0009062 | fatty acid catabolic process | 3.83 |
| GO:0045765 | regulation of angiogenesis | 3.7 |
| GO:0051271 | negative regulation of cell motion | 3.64 |
| GO:0043414 | biopolymer methylation | 3.45 |
| GO:0045762 | positive regulation of adenylate cyclase activity | 3.38 |
| GO:0019362 | pyridine nucleotide metabolic process | 3.31 |
| GO:0022618 | ribonucleoprotein complex assembly | 3.17 |
| GO:0006733 | oxidoreduction coenzyme metabolic process | 3.17 |
| GO:0031281 | positive regulation of cyclase activity | 3.16 |
| GO:0051349 | positive regulation of lyase activity | 3.1 |
Figure 3
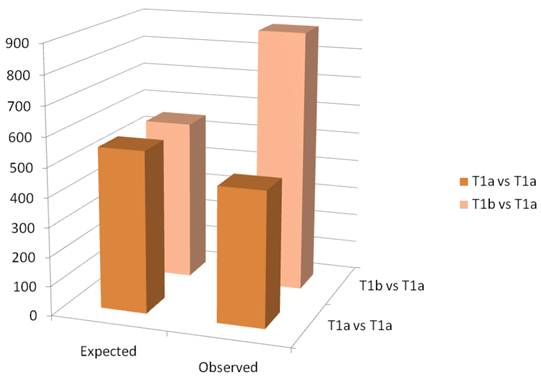
Column graph of the observed and expected gene expression changes based on chance alone. In the T1a vs T1b group, there are more changes than would be expected by chance.
Figure 4
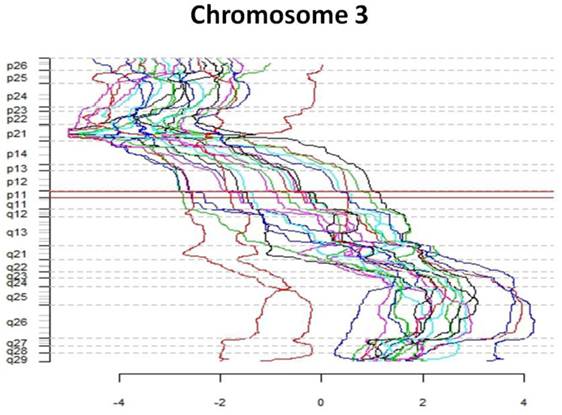
Comparative genome microarray analysis of chromosome 3. Note that each tumor sample has reduced expression of the short arm of chromosome 3, the site of the VHL gene
Figure 5
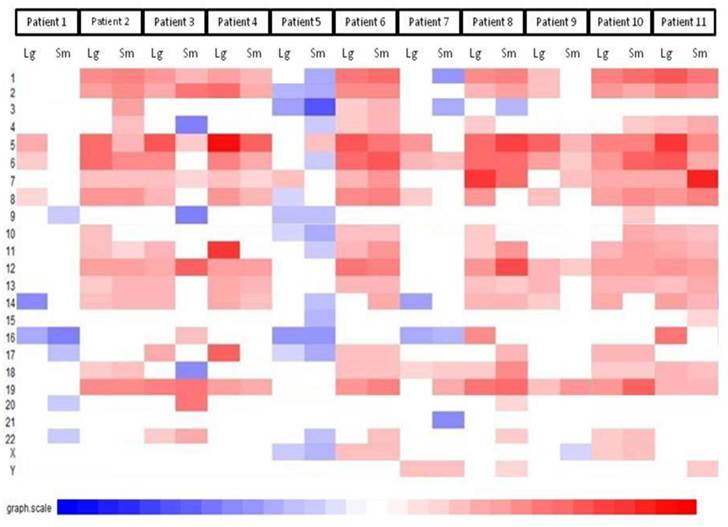
Comparative genomic microarray analysis - global chromosomal heat map. No consistent differences in expression on chromosomal level between large and small tumors were observed. Note that the changes between individuals are greater than changes between tumors of different size in a given individual.
Discussion
Renal tumors in patients with von Hippel Lindau syndrome result from biallelic inactivation of the VHL gene located on chromosome 3p25 with one allele inactivated via a germline mutation and the second due to a random somatic event (7). The protein product of the VHL gene (pVHL) is a key component of an ubiquitin ligase complex that marks the alpha subunit of hypoxia inducible factor (HIF) for degradation (8). The VHL protein serves a critical role in prevention of dysregulated HIF-alpha levels (9). Downregulation of HIF-a by pVHL has been shown to be both necessary and sufficient for renal tumor suppression. In the absence of pVHL patients are prone to form tumors of the retina, CNS, kidneys, pancreas, and epididymis (10).
Among the most severe manifestations of the VHL syndrome are the renal manifestations. These patients are at risk for forming multiple bilateral clear cell renal cell carcinomas - as many as 70 clinically lesions in one kidney have been documented to be removed (11). Despite multiple foci of disease that cumulatively represent substantial tumor burden, the renal tumors in patients with VHL do not attain the ability to metastasize until the largest tumor reaches 3cm (3). Similar to VHL tumors, size of sporadic renal masses is also one of the most important prognostic indicators in sporadic RCC. Frank et al estimated 5 year survival rates to be 97% for patients with T1a (<4cm) tumors, 87% for patients with T1b tumors (4-7 cm), and 71 % for patients with T2 disease (>7cm) (2). While the relationship between size and metastatic potential is well-established, molecular changes that are associated with tumor growth are poorly characterized.
In the present study we investigated the changes in gene expression profiles that occur with tumor growth by performing microarray analysis on small and large tumors from the same patient. Interestingly, unsupervised clustering revealed that the tumor pairs with larger volume differences clustered separately from tumor pairs with smaller volume differences. This suggests that the global transcriptome of larger tumors are more biologically dissimilar from smaller tumors within the same patient. Such observation is of potential clinical relevance as these tumors were not only resected from the same patient and the same kidney, they had the same germline mutation, and were exposed to the same environmental and host factors throughout their development. Therefore, the differences observed may be representative of biologically significant processes because the only difference between the tumors was their size.
We were also able to investigate the nature of the genetic changes on a chromosomal level between large and small tumors. CGMA was used to examine gene expression changes between tumor samples and a pool of normal renal parenchyma samples. Analysis of the gene expression from each chromosome revealed only one area that had decreased expression across all samples: 3p - the location of the VHL gene. These findings are also useful as confirmatory results of our analysis, demonstrating global loss of gene expression on the 3p, consistent with the VHL samples studied.
In addition to demonstration of 3p loss as a confirmation of our analyses, the study of cytogenetics may be particularly relevant in patients with VHL. Indeed, inactivation of pVHL was found to lead to defects in spindle orientation thus provoking chromosomal instability (12). Consistent with this theory, multiple chromosomal abnormalities have been reported in the literature for patients with both hereditary and sporadic ccRCC (13, 14). For example, Phillips et al performed cytogenetic studies on cell lines derived from vHL patients with multifocal RCC and found that higher stage tumors were associated with a greater degree of aneupolidy (14).
Notably, not all studies demonstrated genomic instability in RCC. A study of the sporadic ccRCC population failed to show an association between the degree of genomic instability and the clinical stage of renal tumors (13). Interestingly, another large scale study of 432 clear cell renal tumors supported the loss of chromosome 3p as a common event in the development of ccrCC (60%), but the prevalence of other chromosomal aberrations were much lower (14% for loss of chromosome 4p to 33% for gain of chromosome 5q). In that study, loss of chromosome 9 was the only independent predictor associated with survival (15). The lack of chromosomal changes between larger and smaller tumors in our study is inconsistent with the concept that chromosomal instability occurs in larger tumors. However, it should be noted that all our samples are derived from T1 tumors, which are associated with a very good prognosis. In contrast, the tumors studied by Phillips et al included a wider spectrum of disease, including tumors that had achieved the ability to metastasize (14). Therefore, chromosomal instability may be a late, rather than an early event, in tumor progression.
While consistent cytogenetic changes between large and small tumors were not observed, we did note significant changes in the expression of individual genes. For this analysis, our sample was split into two groups: groups where the larger tumor was less than 4 cm (T1a) and groups where the larger tumor was greater than 4cm (T1b). As Figure 3 demonstrates, the number of statistically significant genetic changes between large and small tumors was no greater than would be expected by random chance when both tumors were T1a. This finding further supports the strategy used at the urologic oncology branch at the NCI, when small tumors are surveilled until the largest tumor reaches 3 cm (Duffey at al). However, once the largest tumor reached T1b size (greater than 4cm), the number of statistically significant genetic changes between larger and smaller tumors becomes much greater than would be expected by random chance. This suggests that even in the absence of large cytogenetic changes, there are significant changes in gene expression as tumors grow larger. When Gene Ontology analysis was performed, it demonstrated that there was an over-representation of transcripts involved in DNA replication and in cytokine signaling, particularly IL-6. This may be clinically elevant, especially since cytokine therapy is one of the durable treatments used for advanced ccRCC.
This study represents one of the first attempts to perform microarrays on tumors derived from the same patient. Gieseg studied the gene expression profiles of smaller (200mg) and larger tumors (1000 mm) in subcutaneously grown tumors from cell lines, and found no difference in gene expression between large and small tumors (16). This is not unexpected as a cell line represents a clonal population of cells that has been selected for high proliferative ability. Perou et al examined breast cancer samples derived from primary tumor and lymph nodes in the same patient, and found that gene expression patterns in two tumor samples from the same individual were almost always more similar to each other than either was to any other sample (17). Similarly, Chen et al examined multiple tumors from patients with HCC (18). The investigators noted that the most important factor in clustering was the individual of origin, not the size of the tumor. They found genetic differences between clonally related tumors. Consistent with our findings, the differences were particularly evident in one patient with a large tumor (8cm), who had different chromosomal abnormalities than smaller tumors in the same patient (1cm and 2cm) (18).
The results of prior studies on tumors from the same patient corroborate our data. It appears that each individual has a very distinct pattern of transcribed genes when surveyed by gene expression microarray. Tumors have genetic aberrations, but continue to have a transcriptome that is more similar to its parental pattern than to another individual's transcriptome. Our data provide evidence of differences in gene expression between larger and small renal cell carcinoma tumors, although the number of differences may small in number in T1 renal tumors. It may not be until renal tumors are even larger that chromosome instability would be detectable.
Of note, our size criteria were chosen to reflect a clinically useful landmark. Tumors less than 4cm are also known as small renal masses. As the use of CT scans increases, the incidence of these small renal masses is rising, creating a new debate about the utilization of active surveillance in select patients (19). We found that there may be a biologic basis for this practice by demonstrating few genetic differences in T1a tumors. Consistent with our findings, recent work from MSKCC studying metastatic events of sporadic RCC, found the metastatic rate negligible in patients with renal masses less than 3cm (20). However, as the size of the tumors increases beyond 4 cm, there are ongoing changes in the transcriptome, possibly reflecting the potential change in biologic aggressiveness.
The present study has a few limitations. First, the samples analyzed were archived, and there is a possibility that some of the observed changes may be due to a length of storage and procurement conditions. Second, although all patients had VHL, the different germline mutations or the second hits of the normal allele may have also be a variable, determining the expression profile of the tumors studied. Third, the sample size is small (although it includes patients treated during a period of 5 years at the NCI). Finally, we recognize that the transcriptome analysis do not always translate into proteome and biologic significance. Despite these limitations, this is the first study in RCC that compares the expression levels of large and small tumors obtained from the same individual in the same setting. Additionally, his study may provide new biologic rationale for active surveillance of small renal masses, practiced for years at the National Cancer Institute.
Conclusion
Through analysis of genetic expression profiles between larger and smaller tumors in the same patient, we found that larger tumor volume differences progressively increased global transcriptome changes. Although few changes in gene expression were appreciated on a chromosomal level, on the level on the individual transcripts, there were an increasing number of changes between large and small tumors, especially when the larger tumor was greater than 4cm (T1b). This may provide a genetic rational behind active surveillance of small renal masses in select patients and further support practice of active surveillance at the Urologic Oncology Branch at the NCI.
Acknowledgements
We would like to thank Dr. Chandramouli Gadisetti for his help with statistical analysis.
Analyses were performed using BRB-Array Tools developed by Dr. Richard Simon and the BRB-Array Tools Development Team.
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research
Conflicts of Interest
The authors have no conflicts of interest to report
References
1. Nickerson ML, Jaeger E, Shi Y. et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res. 2008;14:4726-34
2. Frank I, Blute ML, Leibovich BC, Cheville JC, Lohse CM, Zincke H. Independent validation of the 2002 American Joint Committee on cancer primary tumor classification for renal cell carcinoma using a large, single institution cohort. J Urol. 2005;173:1889-92
3. Walther MM, Choyke PL, Glenn G. et al. Renal cancer in families with hereditary renal cancer: prospective analysis of a tumor size threshold for renal parenchymal sparing surgery. J Urol. 1999;161:1475-9
4. Simon R, Lam A, Li MC, Ngan M, Menenzes S, Zhao Y. Analysis of Gene Expression Data Using BRB-Array Tools. Cancer Inform. 2007;3:11-7
5. Irizarry RA, Wu Z, Jaffee HA. Comparison of Affymetrix GeneChip expression measures. Bioinformatics. 2006;22:789-94
6. Crawley JJ, Furge KA. Identification of frequent cytogenetic aberrations in hepatocellular carcinoma using gene-expression microarray data. Genome Biol. 2002;3:0075
7. Linehan WM, Bratslavsky G, Pinto PA. et al. Molecular diagnosis and therapy of kidney cancer. Annu Rev Med. 2010;61:329-43
8. Linehan WM, Walther MM, Zbar B. The genetic basis of cancer of the kidney. J Urol. 2003;170:2163-72
9. Lubensky IA, Gnarra JR, Bertheau P, Walther MM, Linehan WM, Zhuang Z. Allelic deletions of the VHL gene detected in multiple microscopic clear cell renal lesions in von Hippel-Lindau disease patients. Am J Pathol. 1996;149:2089-94
10. Kaelin WGJr. The von Hippel-Lindau tumor suppressor protein and clear cell renal carcinoma. Clin Cancer Res. 2007;13:680s-4s
11. Fadahunsi AT, Sanford T, Linehan WM, Pinto PA, Bratslavsky G. Feasibility and outcomes of partial nephrectomy for resection of at least 20 tumors in a single renal unit. J Urol. 2011;185:49-53
12. Thoma CR, Toso A, Gutbrodt KL. et al. VHL loss causes spindle misorientation and chromosome instability. Nat Cell Biol. 2009;11:994-1001
13. Thrash-Bingham CA, Salazar H, Freed JJ, Greenberg RE, Tartof KD. Genomic alterations and instabilities in renal cell carcinomas and their relationship to tumor pathology. Cancer Res. 1995;55:6189-95
14. Phillips JL, Ghadimi BM, Wangsa D. et al. Molecular cytogenetic characterization of early and late renal cell carcinomas in von Hippel-Lindau disease. Genes Chromosomes Cancer. 2001;31:1-9
15. Klatte T, Rao PN, de Martino M. et al. Cytogenetic profile predicts prognosis of patients with clear cell renal cell carcinoma. J Clin Oncol. 2009;27:746-53
16. Gieseg MA, Man MZ, Gorski NA, Madore SJ, Kaldjian EP, Leopold WR. The influence of tumor size and environment on gene expression in commonly used human tumor lines. BMC Cancer. 2004;4:35
17. Perou CM, Sorlie T, Eisen MB. et al. Molecular portraits of human breast tumours. Nature. 2000;406:747-52
18. Chen X, Cheung ST, So S. et al. Gene expression patterns in human liver cancers. Mol Biol Cell. 2002;13:1929-39
19. Jewett MA, Zuniga A. Renal tumor natural history: the rationale and role for active surveillance. Urol Clin North Am. 2008;35:627-34
20. Thompson RH, Hill JR, Babayev Y. et al. Metastatic renal cell carcinoma risk according to tumor size. J Urol. 2009;182:41-5
Author contact
![]() Corresponding author: Gennady Bratslavsky, M.D., Urologic Oncology Branch, National Cancer Institute, National Institutes of Health, Building 10, Room 1-5940, Bethesda, Maryland 20892-1414 USA. Phone: (301)496-6353; Fax: (301) 480-5626; E-mail: bratslagnih.gov or bratslagedu
Corresponding author: Gennady Bratslavsky, M.D., Urologic Oncology Branch, National Cancer Institute, National Institutes of Health, Building 10, Room 1-5940, Bethesda, Maryland 20892-1414 USA. Phone: (301)496-6353; Fax: (301) 480-5626; E-mail: bratslagnih.gov or bratslagedu
Citation styles
APA
Bratslavsky, G., Sanford, T., Srinivasan, R., Aprelikova, O., Liu, J., Quezado, M., Merino, M., Linehan, W.M. (2011). Differential Genetic Expression in Large Versus Small Clear Cell Renal Cell Carcinoma: Results from Microarray Analysis. Journal of Cancer, 2, 271-279. https://doi.org/10.7150/jca.2.271.
ACS
Bratslavsky, G.; Sanford, T.; Srinivasan, R.; Aprelikova, O.; Liu, J.; Quezado, M.; Merino, M.; Linehan, W.M. Differential Genetic Expression in Large Versus Small Clear Cell Renal Cell Carcinoma: Results from Microarray Analysis. J. Cancer 2011, 2, 271-279. DOI: 10.7150/jca.2.271.
NLM
Bratslavsky G, Sanford T, Srinivasan R, Aprelikova O, Liu J, Quezado M, Merino M, Linehan WM. Differential Genetic Expression in Large Versus Small Clear Cell Renal Cell Carcinoma: Results from Microarray Analysis. J Cancer 2011; 2:271-279. doi:10.7150/jca.2.271. https://www.jcancer.org/v02p0271.htm
CSE
Bratslavsky G, Sanford T, Srinivasan R, Aprelikova O, Liu J, Quezado M, Merino M, Linehan WM. 2011. Differential Genetic Expression in Large Versus Small Clear Cell Renal Cell Carcinoma: Results from Microarray Analysis. J Cancer. 2:271-279.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.