Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2011; 2:333-338. doi:10.7150/jca.2.333 This volume Cite
Review
Immunological Effects of Multikinase Inhibitors for Kidney Cancer: A Clue for Integration with Cellular Therapies?
Camillo Porta1,2 ![]() , Chiara Paglino1,2, Ilaria Imarisio1,2, Carlo Ganini1, Paolo Pedrazzoli1
, Chiara Paglino1,2, Ilaria Imarisio1,2, Carlo Ganini1, Paolo Pedrazzoli1
1. Oncologia Medica, Fondazione I.R.C.C.S. Policlinico San Matteo, Pavia, Italy.
2. Gruppo Italiano di Oncologia Nefrologica - G.I.O.N. (Italian Nephro-Oncology Group).
Received 2011-4-27; Accepted 2011-5-28; Published 2011-6-2
Citation:
Porta C, Paglino C, Imarisio I, Ganini C, Pedrazzoli P. Immunological Effects of Multikinase Inhibitors for Kidney Cancer: A Clue for Integration with Cellular Therapies?. J Cancer 2011; 2:333-338. doi:10.7150/jca.2.333. https://www.jcancer.org/v02p0333.htm
Other stylesAbstract
The multikinase inhibitors Sunitinib and Sorafenib not only inhibit angiogenesis and tumor growth, but also have the potential of interacting with the function of the immune system.
Presently available data seem to suggest that Sorafenib may exert immune suppressive effects, whilst the effects of Sunitinib are not so clear, being immune stimulatory in the vast majority - but not all - the studies reported.
Trials of combination of these multikinase inhibitors with different types of immune manipulation - and cellular therapies in particular - should be rationally designed taking into account all these complex effects, which ultimately deserve further insights.
Keywords: immunology, kidney cancer, multikinase inhibitors, cellular therapies.
INTRODUCTION
Renal cell carcinoma (RCC) is the most common malignancy of the kidney and accounts for approximately 3% of all adult malignancies and 2% of all deaths from neoplasms.
The only treatment with curative intent for patients with localized RCC is radical surgical resection of the tumor; however, 20-30% of patients present with synchronous metastases at diagnosis, while up to 30-35% of more patients, initially radically resected, will eventually develop metastases over time [1].
Overexpression of P-glycoprotein (P-gp) encoded by the multidrug resistance 1 (MDR1) gene or multidrug resistance-associated protein (MRP) (or both), as well as decreased expression of DNA topoisomerase II is responsible for expression of the multidrug resistance (MDR) phenotype in the vast majority of RCCs [2], so that conventional chemotherapy is largely ineffective in this tumor, which is also resistant to radiations, radiotherapy being only useful for palliation of bone or cerebral metastases [3].
The above peculiarities of this neoplasm, together with the recognition of the frequent presence of several immunologic dysfunctions in RCC [4], even in the absence of metastases [5], have rendered this tumor a privileged field for the development and clinical application of immunotherapy.
Indeed, until 2005, immunotherapy has been the treatment of choice for advanced RCC patients, even though the generic term 'immunotherapy' comprises a vast array of different therapeutic approaches (including cellular therapies), unfortunately too often evaluated only within small, non-randomized, phase II studies.
Starting from 2005 onward, six molecularly targeted drugs (i.e., the three multikinase inhibitors Sorafenib, Sunitinib and Pazopanib, the anti-VEGF monoclonal antibody Bevacizumab - used in combination with Interferon-α - and the two mTOR inhibitors Temsirolimus and Everolimus), proved to be able, within large randomized trials, to change the natural history of advanced RCC [6-11], thus establishing new standards for both first- and even subsequent-lines of treatment (Table 1).
Table 1
A proposed algorithm of treatment for advanced RCC (in bold are reported the options supported by at least one randomized, controlled, phase III, clinical trial).
| 1st line | ||
| MSKCC* good and intermediate risk | Sunitinib | |
| Bevacizumab + Interferon-a | ||
| Pazopanib | ||
| Sorafenib (selected patients) | ||
| High-dose i.v. IL-2 (?) | ||
| MSKCC* poor risk | Temsirolimus | |
| Sunitinib | ||
| 2nd line | ||
| Post-cytokines | Sorafenib | |
| Pazopanib | ||
| Sunitinib | ||
| Post-one multi-kinase inhibitor | Everolimus | |
| a second multi-kinase inhibitor | ||
| 3rd line | ||
| Post-two multi-kinase inhibitors | Everolimus | |
| Post-Everolimus | Clinical trial | |
| Sorafenib (?) |
*MSKCC (or Motzer's) score takes into consideration the following parameters: ECOG Performance Status, hemoglobin levels, LHD levels, corrected calcium levels, and prior history of nephrectomy - or interval between diagnosis and treatment start; 0 risk factors = good risk; 1-2 risk factors = intermediate risk; >2 risk factors = poor risk.
Despite such an abundance of active agents (a scenario that has been brilliantly defined as the 'embarassment of the riches') [12], cure is still out of our sights, progression-free survival (PFS) for almost all these agents rarely exceeding eleven months.
To further improve the results obtained so far, the combination of molecularly targeted agents with immunotherapy is considered as a promising investigational therapeutic approach, even though - to date - only Interferon-α and Interleukin-2 (IL-2) have been successfully combined with the multikinase inhibitor Sorafenib [13-17].
However, it is important to highlight that all the above molecularly targeted drugs inhibit different key signal transduction pathways that, not only are implicated in the complex processes of angiogenesis, tumor growth, metabolism and survival, but also play many other physiological roles.
In particular, if these pathways are relevant also for the immune system - and they indeed are relevant - what does really happen from an immunological viewpoint when these pathways are pharmacologically inhibited? These actions may further impair the already compromised antitumor activity of the immune system? And, more than any other issue, could we exploit all these informations to more rationally design the next generation of clinical trials, including both molecularly targeted agents and immunotherapy (including cellular therapies)? [18]
IMMUNOLOGICAL EFFECTS OF SORAFENIB TOSYLATE
Sorafenib tosylate (a.k.a. BAY 43-9006, Nexavar™) is an oral, small molecule inhibitor of several tyrosine protein kinases (e.g., VEGFR-2 and -3, PDGFR-β, Flt-3 and c-Kit), heavily involved in tumor angiogenesis, as well as of the serine-threonine kinase Raf, a key protein within the RAF/MEK/ERK signaling pathway [19].
Sorafenib and Dendritic cells (DCs)
An elegant paper addressed the different effects of Sorafenib (and Sunitinib) on DCs phenotype and function [20].
Exposure to pharmacologic concentrations of Sorafenib induced, on myeloid-derived DCs, a pronounced down-regulation of CD1a, i.e., a glycoprotein structurally related to the Major Histocompatibility Complex (MHC), of the co-stimulatory molecule B7.2 (or CD86), as well as of the Dendritic Cell Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN or CD209), a C-type lectin that mediates the adhesion of DCs to T cells by stabilizing the DC/T cells' contact zone [20]. In vivo, treatment with Sorafenib significantly reduced the induction of antigen-specific T cells in C57BL/6 mice immunized with the OVA257-264 peptide [20], a class I (Kb)-restricted peptide epitope of OVA, an octameric peptide from ovalbumin presented by the class I MHC molecule, H-2Kb.
Furthermore, Sorafenib-treated DCs displayed impaired migratory capacity, through a reduced expression of the chemokine-CC motif receptor 7 (CCR7), the receptor for CCL19/MIP-3β, a system that regulates the migration of mature DCs from peripheral tissues to local lymph nodes across a CCL19/MIP-3β gradient [21].
Pre-treatment with Sorafenib reduced the capacity of TLR4 ligand-activated antigen-presenting cells (APCs) to stimulate lymphocyte proliferation, and lowered DC cytokine secretion [20].
Sorafenib (but not Sunitinib) induced apoptosis of DCs, and attenuated primary CD8+ T-cell response in a mouse model in vivo [20].
As a whole, Sorafenib - but not Sunitinib - proved able to inhibit DCs' function, cytokine production, and ability to migrate and stimulate T-cell responses [20]. Notably, all these immunosuppressive effect were mediated via Sorafenib-induced inhibition, not only of the MAPK pathway, but also of the PI3K and NFκB signaling pathways [20], a relatively unexpected finding since Sorafenib is not known to directly inhibit the two latter pathways.
Sorafenib, Natural Killer (NK) cells and CD4+/CD25high T-cells
In another experimental study it has been demonstrated that cytokine production and antitumor cytotoxicity of resting and IL-2-activated peripheral blood mononuclear cells (PBMCs) were inhibited by pharmacological concentrations of Sorafenib; furthermore, pharmacological concentrations of Sorafenib proved able to impair granule mobilization of NK cells among PBMCs, and inhibit NK cells reactivity [22]. Once again, these immunosuppressive effects were mediated via Sorafenib-induced inhibition of both the MAPK pathway, as well as of the PI3K pathway [22].
Sorafenib proved also able to induce apoptosis of CD4+/CD25high T cells if administered prior to antigenic stimulation, whilst this effect was prevented by the administration of IL-2 [23].
Sorafenib tosylate and the immune system: conclusions
From the above evidence, it is clear that Sorafenib has potentially immunosuppressive effects (Figure 1).
Figure 1
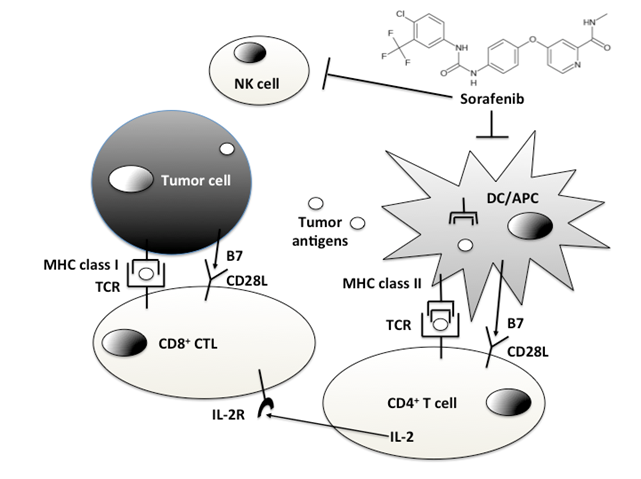
- The putative immune-suppressive effects of Sorafenib are exerted on Dendritic Cells (DCs)/Antigen Presenting Cells (APCs) and Natural Killer (NK) cells.
At this point, we should ask ourselves at least a couple of questions: since Sorafenib is highly active, in man, against several tumors (not only kidney cancer, but also HCC and thyroid cancer) [24-26], which - if any - is the real impact of such drug-induced immune suppression? And then, is it really possible that a drug such as Sorafenib, we suppose to know quite well, could really interfere with pathways such as the PI3K, or the NFκB ones, as it appears from the previously cited experimental papers?
To date, the latter question has no clear-cut answers, more biological studies being needed to address this point.
As far as the first answer, it is clear that the putative immune suppressant properties of Sorafenib have no impact at all on its anti-angiogenic and antitumor activity; however, these immunological effects should be taken into account when (and if) Sorafenib will be administered together with other forms of immunotherapy.
IMMUNOLOGICAL EFFECTS OF SUNITINIB MALATE
Sunitinib malate (a.k.a. SU011248, Sutent™) is an oral, small molecule inhibitor of several tyrosine protein kinases involved in tumor angiogenesis (e.g., VEGFR1 through -3, PDGFR-α and -β, Flt-3, CSF-1) [27].
Sunitinib malate: anti-immune suppressive effects
Sunitinib was the first multikinase inhibitor whose immunological effects have been specifically studied in vivo.
After a single cycle of Sunitinb an increase in the percentage of Interferon-γ producing T-cells, a reduction in the percentage of IL-4 producing T-cells, and a diminished type-2 bias, was observed [28]; furthermore, immune suppressant CD4+/CD25high/Foxp3+ regulatory T-cells (Treg) proved to be down-regulated after Sunitinib treatment [28].
CD33+/HLA-DR- and CD15+/CD14- Myeloid-Derived Suppressor Cells (MDSC), which are usually elevated in RCC patients [29], declined in response to Sunitinib treatment, which suppresses bone marrow production of MDSC, while enhancing lymphoid cell proliferation [30].
Differently from Sorafenib, Sunitinib do not affect DCs or NK cells phenotype and function [20,22]; on the contrary, Sunitinib reduces the expression of immune suppressive cytokines and co-stimulatory molecules, such as IL-10, Foxp3, PD-1, CTLA4 and BAFF (B lymphocyte-activating factor), in tumor infiltrating lymphocytes (TILs) from Sunitinib-treated mice [31]. More importantly, the expression of negative costimulatory molecules CTLA4 and PD-1 in both CD4+ and CD8+ T cells, and PDL-1 expression on MDSC and plasmacytoid DCs, was also significantly decreased by Sunitinib treatment [31].
Furthermore, T cells from Sunitinib-treated mice exhibited stronger cytotoxic activity, and increased 2.5 fold the number of CD4+ and CD8+ cells within the TILs population [31]. Finally, Treg and MDSC are reduced by Sunitinib treatment in this mouse model [31].
Finally, Sunitinib restores DC number and normalizes myeloid lineage distribution in RCC patients; indeed, subsequent to Sunitinib treatment, an increase to high levels of myeloid DCs subset frequencies relative to other myeloid subsets, was specifically observed in patients experiencing tumor regression; moreover, high CD1c/BDCA-1+ myeloid DC frequencies were predictive for tumor regression and improved PFS [32].
Sunitinib: fellow or foe?
At this point, the whole picture appears to be clear: Sunitinib enhances antitumor immunity, while Sorafenib has immunosuppressive properties, isn't it?
Not really.
Indeed, differently from what has been reported above, another recent study suggested that Sunitinib may also have immune suppressive effects.
In this study, Sunitinib proved able to inhibit the proliferation of primary human T cells from normal healthy volunteers as well as from RCC and other cancer patients, an inhibition that was recoverable after drug withdrawal [33]. In addition, Sunitinib led to accumulation in G0/G1 phase of the cell cycle, inhibition of cytokine production, down-regulation of activation markers expression, and blockade of Zap-70 signaling in the T cells [33].
CONCLUSIONS
A relevant body of evidence suggests that the multikinase inhibitors Sunitinib and Sorafenib may heavily impact on the immune system.
This is not strange, since the signaling pathways inhibited by these drugs are relevant also for the immune system. For example, the Mitogen-Activated Protein Kinase (MAPK) pathway is necessary for the positive selection (but not negative selection) of immature, double-positive, thymocytes [34].
Furthermore, circulating VEGF, which may increase after multikinase inhibitor treatment [35], proved able to inhibit DCs; indeed, the ability of mature DCs to stimulate allogeneic T cell was dose-dependently inhibited by the addition of VEGF in an experimental system [36].
As a whole, presently available evidence seems to suggest that the multikinase inhibitor Sorafenib may exert immune suppressive effects, whilst the effects of Sunitinib are not so clear, being immune stimulatory in the vast majority - but not all - the studies reported.
For years immunotherapy was the only available treatment for advanced RCC [37], but the development of molecularly targeted agents relegated it to a corner; among different immune therapeutic approaches, cellular therapies held particular promise for the treatment of advanced RCC, since the early demonstration that non-myeloablative allogeneic stem-cell transplantation was able to induce sustained regression of metastatic RCC, and even cure a small percentage of these patients [38]. This effect was associated with the recognition of a peptide (i.e., CT-RCC-1) by RCC-specific CD8+ T cells; the genes encoding this antigen were found to be derived from human endogenous retrovirus type E (HERV) and were expressed in RCC cell lines and fresh RCC tissue but not in normal kidney or other tissues [39], a finding that opens the field to further research on the role of viruses in the processes of carcinogenesis [40].
Despite the fact that, differently from cellular therapy, molecularly targeted agents do not allow to cure any of these patients, it is clear that their absolute benefit (i.e., a disease control rate in the range of 75 to 80%) makes them the treatment of choice for advanced RCC.
However, a combination of these novel agents with cellular therapy makes sense and deserve future studies.
But any future combination between multikinase inhibitors with immunotherapy as a whole (and cellular therapy, in particular) should be rationally designed taking into account all the above complex immunological effects of the latter drugs, effects which ultimately deserve further insights.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Flanigan RC, Campbell SC, Clark JI, Picken MM. Metastatic renal cell carcinoma. Curr Treat Options Oncol. 2003;4:385-90
2. Naito S, Koga H, Yokomizo A. et al. Molecular analysis of mechanisms regulating drug sensitivity and the development of new chemotherapy strategies for genitourinary carcinomas. World J Surg. 2000;24:1183-6
3. Motzer RJ, Russo P. Systemic therapy for renal cell carcinoma. J Urol. 2000;163:408-17
4. Enquist E, Zambiano N, Zbar B. et al. Molecular mechanisms of immune dysfunction in renal cell carcinoma. In: (ed.) Bukowski RM, Novick AC. Renal cell carcinoma. Molecular biology, immunology and clinical management. Totowa, NJ (USA): Humana Press. 2000:63-78
5. Porta C, Bonomi L, Lillaz B. et al. Renal cell carcinoma-induced immunosuppression: an immunophenotypic study of lymphocyte subpopulations and of circulating dendritic cells in patients at first diagnosis. Anticancer Res. 2007;27:165-73
6. Escudier B, Eisen T, Stadler WM. et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125-34
7. Motzer RJ, Hutson TE, Tomczak P. et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115-24
8. Sternberg CN, Davis ID, Mardiak J. et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061-8
9. Escudier B, Pluzanska A, Koralewski P. et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet. 2007;370:2103-11
10. Hudes G, Carducci M, Tomczak P. et al. Temsirolimus, interferon-alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271-81
11. Motzer RJ, Escudier B, Oudard S. et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449-56
12. Vogelzang NJ. Treatment options in metastatic renal carcinoma: an embarrassment of riches. J Clin Oncol. 2006;2:1-3
13. Gollob JA, Rathmell WK, Richmond TM. et al. Phase II trial of sorafenib plus interferon alfa-2b as first- or second-line therapy in patients with metastatic renal cell cancer. J Clin Oncol. 2007;25:3288-95
14. Ryan CW, Goldman BH, Lara PN Jr. et al. Sorafenib with interferon alfa-2b as first-line treatment of advanced renal carcinoma: a phase II study of the Southwest Oncology Group. J Clin Oncol. 2007;25:3296-301
15. Jonasch E, Corn P, Pagliaro LC. et al. Upfront, randomized, phase 2 trial of sorafenib versus sorafenib and low-dose interferon alfa in patients with advanced renal cell carcinoma: clinical and biomarker analysis. Cancer. 2010;116:57-65
16. Bracarda S, Porta C, Boni C. et al. Randomized prospective phase II trial of two schedules of Sorafenib daily and Interferon-α2a (IFN) in metastatic renal cell carcinoma (MRCC) (RAPSODY): GOIRC Study 0681. Genitourinary Cancers Symposium. 2008:abs357
17. Procopio G, Verzoni E, Bracarda S. et al. Sorafenib with interleukin-2 vs sorafenib alone in metastatic renal cell carcinoma: the ROSORC trial. Br J Cancer. 2011;104:1256-61
18. Porta C, Imarisio I, Paglino C, Ferraris E, Tagliani F. Kinase inhibitors and the immune system: friend or foe? Oncol Rev. 2008;2(Suppl.1):S43-5
19. Wilhelm SM, Carter C, Tang L. et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099-109
20. Hipp MM, Hilf N, Walter S. et al. Sorafenib, but not sunitinib, affects function of dendritic cells and induction of primary immune responses. Blood. 2008;111:5610-20
21. Lebre MC, Burwell T, Vieira PL. et al. Differential expression of inflammatory chemokines by Th1- and Th2-cell promoting dendritic cells: a role for different mature dendritic cell populations in attracting appropriate effector cells to peripheral sites of inflammation. Immunol Cell Biol. 2005;83:525-35
22. Krusch M, Salih J, Schlicke M. et al. The kinase inhibitors sunitinib and sorafenib differentially affect NK cell antitumor reactivity in vitro. J Immunol. 2009;183:8286-94
23. Molhoek KR, McSkimming CC, Olson WC, Brautigan DL, Slingluff CL Jr. Apoptosis of CD4+CD25high T cells in response to Sirolimus requires activation of T cell receptor and is modulated by IL-2. Cancer Immunol Immunother. 2009;58:867-76
24. Porta C, Paglino C, Imarisio I, Ferraris E. Sorafenib tosylate in advanced kidney cancer: past, present and future. Anticancer Drugs. 2009;20:409-15
25. Keating GM, Santoro A. Sorafenib: a review of its use in advanced hepatocellular carcinoma. Drugs. 2009;69:223-40
26. Duntas LH, Bernardini R. Sorafenib: rays of hope in thyroid cancer. Thyroid. 2010;20:1351-8
27. Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol. 2007;25:884-96
28. Finke JH, Rini B, Ireland J. et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res. 2008;14:6674-82
29. Kusmartsev S, Su Z, Heiser A. et al. Reversal of myeloid cell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2008;14:8270-8
30. Ko JS, Zea AH, Rini BI. et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15:2148-57
31. Ozao-Choy J, Ma G, Kao J. et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69:2514-22
32. van Cruijsen H, van der Veldt AA, Vroling L. et al. Sunitinib-induced myeloid lineage redistribution in renal cell cancer patients: CD1c+ dendritic cell frequency predicts progression-free survival. Clin Cancer Res. 2008;14:5884-92
33. Gu Y, Zhao W, Meng F. et al. Sunitinib impairs the proliferation and function of human peripheral T cell and prevents T-cell-mediated immune response in mice. Clin Immunol. 2010;135:55-62
34. Alberola-Ila J, Hernández-Hoyos G. The Ras/MAPK cascade and the control of positive selection. Immunol Rev. 2003;191:79-96
35. Deprimo SE, Bello CL, Smeraglia J. et al. Circulating protein biomarkers of pharmacodynamic activity of sunitinib in patients with metastatic renal cell carcinoma: modulation of VEGF and VEGF-related proteins. J Transl Med. 2007;5:32
36. Mimura K, Kono K, Takahashi A, Kawaguchi Y, Fujii H. Vascular endothelial growth factor inhibits the function of human mature dendritic cells mediated by VEGF receptor-2. Cancer Immunol Immunother. 2007;56:761-70
37. Porta C, Paglino C, Imarisio I, Bonomi L. Cytokine-based immunotherapy for advanced kidney cancer: past results and future perspectives in the era of molecularly targeted agents. ScientificWorldJournal. 2007;7:837-49
38. Childs R, Chernoff A, Contentin N. et al. Regression of metastatic renal-cell carcinoma after non-myeloablative allogeneic peripheral-blood stem-cell transplantation. N Engl J Med. 2000;343:750-8
39. Takahashi Y, Harashima N, Kajigaya S. et al. Regression of human kidney cancer following allogeneic stem cell transplantation is associated with recognition of an HERV-E antigen by T cells. J Clin Invest. 2008;118:1099-109
40. Porta C. Allogeneic transplantation following non-myeloablative conditioning in renal carcinoma. New evidence of the immune mechanisms responsible for the activity of this form of immunotherapy and the pathogenetic role of endogenous retroviruses. Oncol Rev. 2008;2:1-3
Author contact
![]() Corresponding author: Camillo Porta, MD. Medical Oncology, I.R.C.C.S. San Matteo University Hospital Foundation, Piazzale C. Golgi, 19, I-27100 Pavia, Italy. Phone: +39-0382-501355 Fax: +39-0382-502442 E-mail: c.portapv.it
Corresponding author: Camillo Porta, MD. Medical Oncology, I.R.C.C.S. San Matteo University Hospital Foundation, Piazzale C. Golgi, 19, I-27100 Pavia, Italy. Phone: +39-0382-501355 Fax: +39-0382-502442 E-mail: c.portapv.it
Citation styles
APA
Porta, C., Paglino, C., Imarisio, I., Ganini, C., Pedrazzoli, P. (2011). Immunological Effects of Multikinase Inhibitors for Kidney Cancer: A Clue for Integration with Cellular Therapies?. Journal of Cancer, 2, 333-338. https://doi.org/10.7150/jca.2.333.
ACS
Porta, C.; Paglino, C.; Imarisio, I.; Ganini, C.; Pedrazzoli, P. Immunological Effects of Multikinase Inhibitors for Kidney Cancer: A Clue for Integration with Cellular Therapies?. J. Cancer 2011, 2, 333-338. DOI: 10.7150/jca.2.333.
NLM
Porta C, Paglino C, Imarisio I, Ganini C, Pedrazzoli P. Immunological Effects of Multikinase Inhibitors for Kidney Cancer: A Clue for Integration with Cellular Therapies?. J Cancer 2011; 2:333-338. doi:10.7150/jca.2.333. https://www.jcancer.org/v02p0333.htm
CSE
Porta C, Paglino C, Imarisio I, Ganini C, Pedrazzoli P. 2011. Immunological Effects of Multikinase Inhibitors for Kidney Cancer: A Clue for Integration with Cellular Therapies?. J Cancer. 2:333-338.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.