Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2011; 2:413-424. doi:10.7150/jca.2.413 This volume Cite
Commentary
Malignant Transformation and Stromal Invasion from Normal or Hyperplastic Tissues: True or False?
Yan-gao Man1, Michael Grinkemeyer2, Mina Izadjoo1, Alexander Stojadinovic3 ![]()
1. Diagnostic and Translational Research Center, Henry Jackson Foundation, Gaithersburg, MD, USA
2. Armed Forces Institute of Pathology, Washington DC, USA
3. Surgical Oncology, Walter-Reed Army Medical Center, Washington DC, USA
Received 2011-6-29; Accepted 2011-7-22; Published 2011-7-23
Citation:
Man Yg, Grinkemeyer M, Izadjoo M, Stojadinovic A. Malignant Transformation and Stromal Invasion from Normal or Hyperplastic Tissues: True or False?. J Cancer 2011; 2:413-424. doi:10.7150/jca.2.413. https://www.jcancer.org/v02p0413.htm
Other stylesAbstract
Carcinogenesis is believed to be a multi-step process, progressing sequentially from normal to hyperplastic, to in situ, and to invasive stages. A number of studies, however, have detected malignancy-associated alterations in normal or hyperplastic tissues. As the molecular profile and clinical features of these tissues have not been defined, the authors invited several well-recognized pathologist, oncologists, biologist, surgeons, and molecular biologist to offer their opinion on: (1) whether these tissues belong to a previously unrevealed malignant entity or focal alterations with no significant consequence? (2) whether these alterations are linked to early onset of cancer or cancer of unknown primary site, and (3) how to further define these lesions?
Keywords: Malignant transformation, Tumor invasion, Sick lobe, Tumor microenvironment, Tumor capsule
Introduction
It is a commonly held belief that carcinogenesis is a multi-step process, progressing sequentially from normal to hyperplastic, to in situ, and to invasive stages [1-3]. Progression from one stage to another is believed to result from increasing accumulation of hereditary mutations in major regulatory genes or uncorrected and acquired mutations in somatic genes [4-6]. It is estimated that an average of 16 years is needed for a cancer-initiating cell to develop into a 10-mm, clinically detectable tumor, as the averaged volume doubling time in most tumors is about 210-days during exponential growth [7-9].
A vast majority of the studies during the past have consistently shown that a number of genetic, biochemical, and morphologic alterations, including a loss of heterozygosity (LOH), aberrant expression of p53, c-erbB2, CA-125, disruptions of the tumor capsule, and stromal or vascular invasion of epithelial cells, are exclusively or almost exclusively seen in malignant lesions [10-14]. Consequently, these have been considered as malignancy-associated alterations (MAA) or malignant tumor signatures [10-14].
However, a number of studies have reported that: (1). The same pattern of LOH at several chromosome loci was detected in both breast cancer tissues and in adjacent morphologically normal lobules [15], (2). Some morphologically normal ductal intraepithelial neoplasia (flat type) shared the same LOH and monoclonality identified in adjacent in situ and invasive ductal carcinoma [16], (3). Some healthy men between 19-29 years old showed a spectrum of proliferative abnormalities, including atypical hyperplasia, prostatic intraepithelial neoplasia, or invasive cancer [17-19], and (4). Prostate tissues of certain healthy man or normal tissues adjacent to prostate cancer showed a DNA phenotype identical to the DNA structure of invasive prostate cancer [20-22].
More recent studies have further revealed that about 15% of human breast and prostate tumors harbor variable numbers of morphologically normal or hyperplastic ductal and acinar structures with malignancy-associated alterations, including aberrant expression of p53 and c-erbB2, focal disruptions of the tumor capsules, and morphological signs of stromal or vascular invasion [23-33]. These structures are distributed as clusters or lobules with a distinct boundary to adjacent counterparts. Microdissected cells from these clusters or lobules showed a substantially elevated frequency of genetic instabilities and expression of invasion-related genes [24].
These findings suggest that the linear model of tumor progression [1-3] may not apply to all cases, and that the morphological features of some tissues may not fully reflect their genetic and biochemical profiles. However, as only a few such cases have been reported, it is not clear whether these tissues represent a previously unrevealed malignant entity, or focal changes with no major consequences. In addition, the structural relationships of these tissues with their adjacent counterparts have yet to be revealed. Thus, our current study intends to expand our previous observations, to assess: (1) whether cells with malignancy-associated changes could originate from these structures, (2) whether these structures are in physical continuity with distinct invasive lesions, and (3) whether aberrant leukocyte infiltration correlates with malignancy-associated alterations within these structures.
Materials and Methods
Five cases harbored large normal human breast ductal or acinar clusters or lobules with malignancy-associated alterations were selected from our previous studies [23-33], in which all tissue samples were retrieved from files of the Armed Forces Institute of Pathology. Consecutive sections at 7-um thickness were prepared and placed sequentially on positive charged slides. For each set of 10 sections, the first 3-4 sections were used for H&E staining and immunohistochemistry. The remaining sections were used for different molecular assays.
To identify malignancy-associated alterations, two-technical approaches were used. First, the physical integrity of the capsule surrounding epithelial structures was examined with tumor capsule specific markers, smooth muscle actin (SMA; clone:1A4; Sigma, St. Louis, MO, USA) and/or collagen IV (clone: CIV22, Dako, Carpinteria, CA, USA). Immunostained sections were examined under high magnification to identify the absence or focal disruptions of the tumor capsule (defined as the absence of myoepithelial cells and/or the basement membrane that results in a gap greater than the combined size of at least 3-myoepithelial cells). Second, sections were double imunostained for SMA and p53 (clone: D07, Dako, Carpinteria, CA, USA) or c-erbB2 (clone:10A7; Novocastra, Newcastle, UK). To differentiate ductal from acinar cells and to assess the impact of aberrant leukocyte infiltration on physical integrity of the myoepithelial cell layers and adhesion molecules, sections were double immunostained for E-cadherin (clone: 36B5; Lab Vision, Fremont, CA,USA) and leukocyte common antigen (LCA, clone:2B11+ PD7/26, USA). To identify isolated epithelial cells within leukocyte aggregates, sections were double immunostained for LCA (which reacts with all hematopoietic cells, including lymphocytes) and cytokeratin (CK) AE1/AE3 (clone; AE1/AE3, Dako, Carpinteria, CA,USA) (which react with all epithelium derived cells). The biological presentation and tissue microenvironment were assessed with a panel of biomarkers, including ER, PR, Ki-67, CK5, CK19, CD31, D2-40, and others.
Immunostaining was carried out following manufactures' instruction. The secondary antibody, ABC detection, and DAB chromogen kits were obtained from Vector Laboratories (Burlingame, CA, USA). The AP red-chromogen kit was purchased from Zymad Laboratories (South San Francisco, CA, USA). To assess the specificity of the immunostaining, negative controls included (1) the substitution of the primary antibody with the same isotype or pre-immune serum of the antibody, and (2) the omission of the secondary antibody. The immunostaining procedure was repeated at least twice using the same protocol and the same conditions. A given cell was considered immunoreactive if distinct immunoreactivity was consistently seen in its cytoplasm, membrane, or nucleus, while all negative controls lacked distinct immunostaining.
Results
Each case harbored variable numbers of morphologically normal- or hyperplastic-appearing ductal or acinar cells, which were distributed as clusters or lobules with a distinct boundary to adjacent counterparts. The size of these clusters or lobules varied from about one hundred to several thousand cells/per profile, and extended from about 50 to over 400 sections (about 350 - 3,000-um). Compared to adjacent morphologically clear-cut normal or hyperplastic counterparts, these ductal and acinar clusters or lobules have the following unique profiles:
- They are indistinguishable from adjacent counterparts in H & E stained sections under low magnification, but under high magnification, they often show a high nuclear-cytoplasm ratio and substantially enlarged nucleoli.
- In sections immunostained for tumor capsules, the myoepithelial cell layer and basement membrane of these structures are generally discontinuous or focally disrupted, or even totally absent.
- They were exclusively associated with large leukocyte aggregates, which completely or partially surrounded these structures. Some leukocytes were directly attached to the myoepithelial cell layers. Within leukocyte aggregates, a significant number of isolated epithelial cells was seen.
- Neither distinct in situ carcinoma nor enlarged tumor nests (with over 50-cells/nest) were seen.
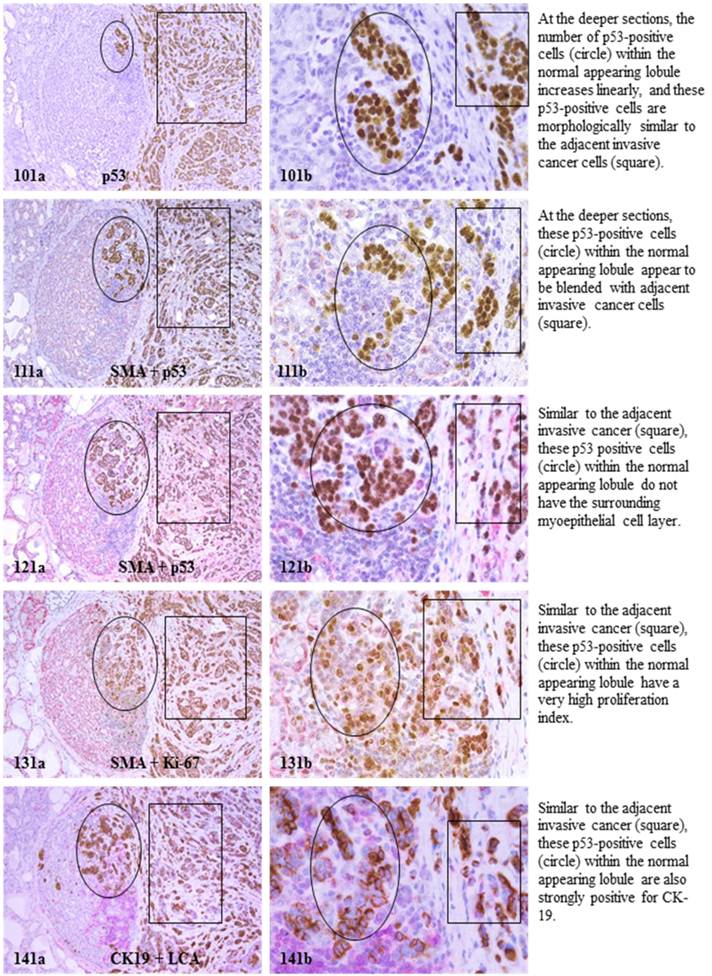
- In the superficial cuts, p53-positive cell was not detected. The number of p53-positive cells increased linearly in the deeper cuts, and eventually, the entire lobule was replaced by p53-positive cells.
- These p53-positive cells lacked E-cadherin expression with morphological features of lobular cells. These p53-positive cells also lacked the surrounding myoepithelial cell layer and the basement membrane, a typical feature of invasive cancers.
- These p53-positive cells were eventually blended with morphologically distinct invasive cancer cells.
- Leukocyte aggregates were exclusively or preferentially located at the junctions between these normal appearing structures and invasive lesion, and seemed to “flow” or “migrate” to different locations, correlating with the emergence of p53-positive cells.
The above features are depicted in the following two sets of consecutive sections. The first set elucidates malignancy-associated alterations in lobules distant from invasive lesion. The second set shows similar alterations in a normal-appearing lobule immediately adjacent to the invasive component. Each number at the figure sets represents the sequential number of the sections.
he above findings further suggest that lobular structures.
In summary, the above findings from consecutive sections indicate that:
- These clusters or lobules were not associated with morphologically distinct in situ or large tumor nests.
- Cells with malignancy-associated alterations (including strong p53-positivity, the absence of myoepithelial cell layers and the basement membrane, a high nuclear-cytoplasm ratio and substantially enlarged nucleoli, significantly elevated cell proliferation, morphological resemblance and physical continuity with invasive lesions, and disassociation from main structures) could originate from morphologically normal structures.
- The number of cells with malignancy-associated alterations linearly increased in consecutive sections, and these cells were eventually blended with morphologically distinct invasive lesions.
- Leukocyte aggregates were exclusively located at the junctions between these structures and invasive lesion, and seemed to “flow” or “migrate” to different locations, correlating with emergence of p53-positive cells. Together, these findings suggest that malignant transformation and stromal invasion could originate or emerge from morphologically normal structures.
Discussion
Based on these and previous findings, these morphologically normal- or hyperplastic-clusters and lobules seem to represent a previously unrevealed malignant entity that has acquired significant genetic abnormalities and could directly progress to invasive or metastatic breast lesions. This speculation is consistent with the “sick lobe” theory of breast carcinogenesis proposed by Tot at al [34, 35], which suggests that “The sick lobe carries some kind of genetic instability already from its initialization during the early embryonic life and is more sensitive to noxious influences than the other lobes within the same breast”.
The exact cause for the formation of these lobules is unknown, but could potentially result from diagnostic or therapeutic radiation exposure at the very early age [36,37], which may have resulted in significant genetic damages on a subset of stem cells within these lobules, especially those for myoepithelial cells. Myoepithelial cells are also very sensitive to certain chemicals. For example, exposure to lambdacarrageenan can specifically result in filament disassembly and loss of myoepithelial cells, whereas exposure to oxytocin could substantially enhance myoepithelial cell differentiation and proliferation in mouse breasts [38,39]. Damages to myoepithelial stem cells can direct impair the normal replenishment process, resulting in an aged or inactive myoepithelial cell population. As the myoepithelial cell layer is the sole source of several tumor suppressors, damages to this cell layer could lead to the loss of its paracrine inhibitory functions on tumor cell proliferation [40-42].
Figure 1
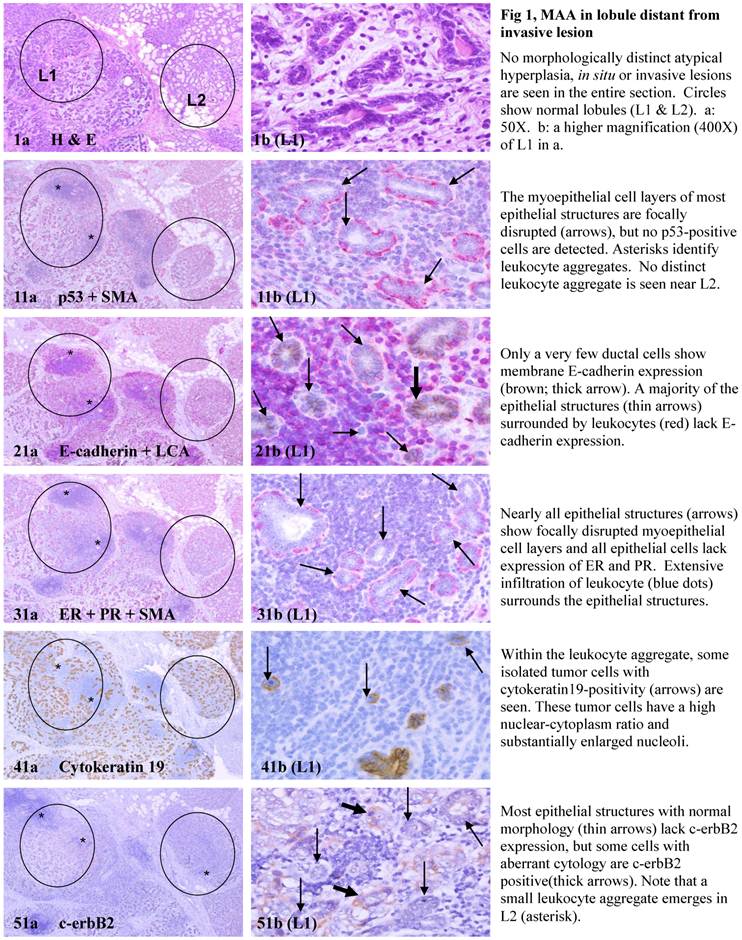
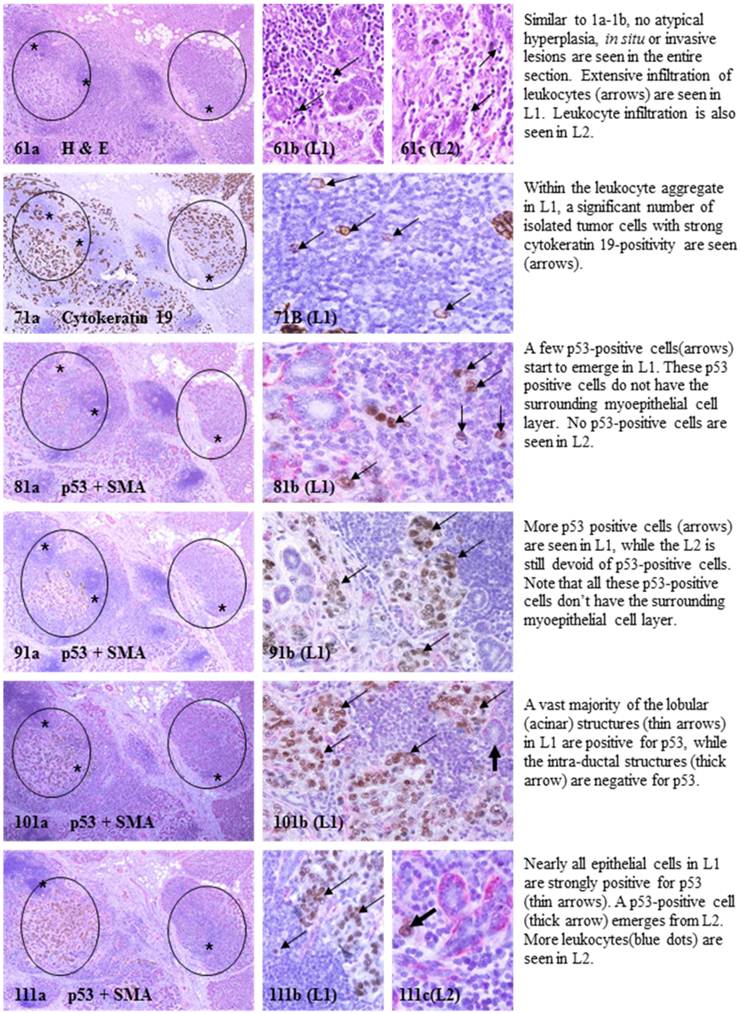
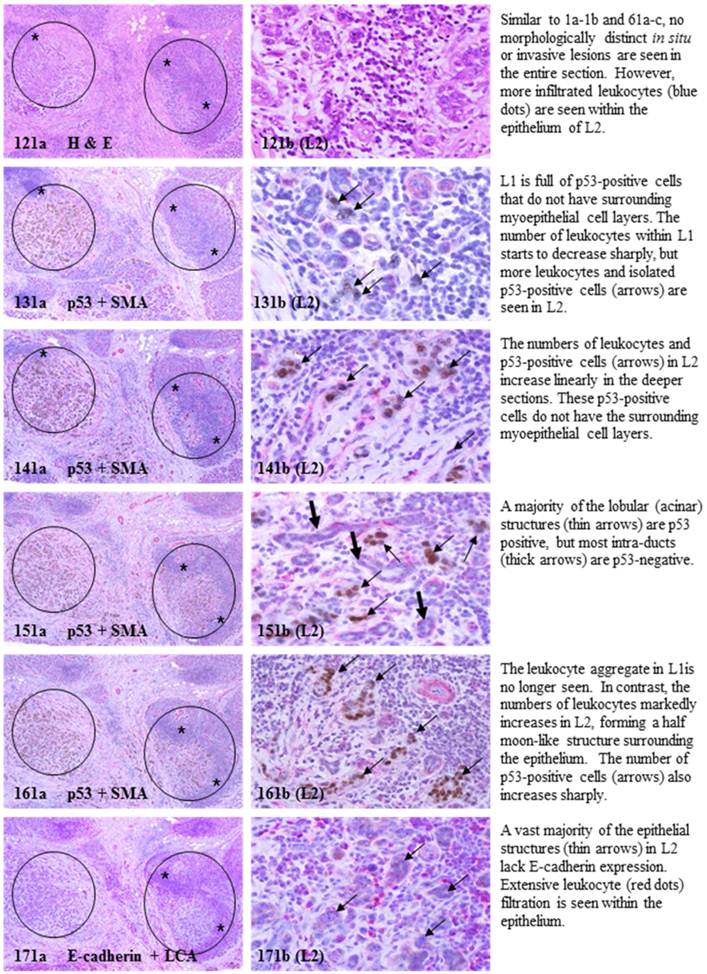
MMA in lobule distant from invasive lesion.
Figure 2
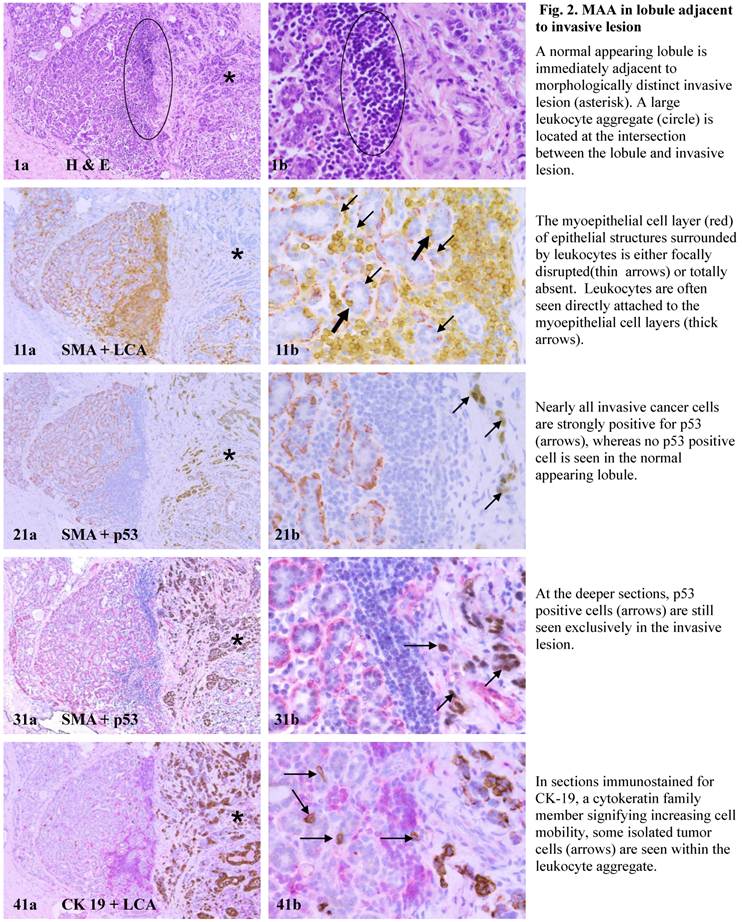
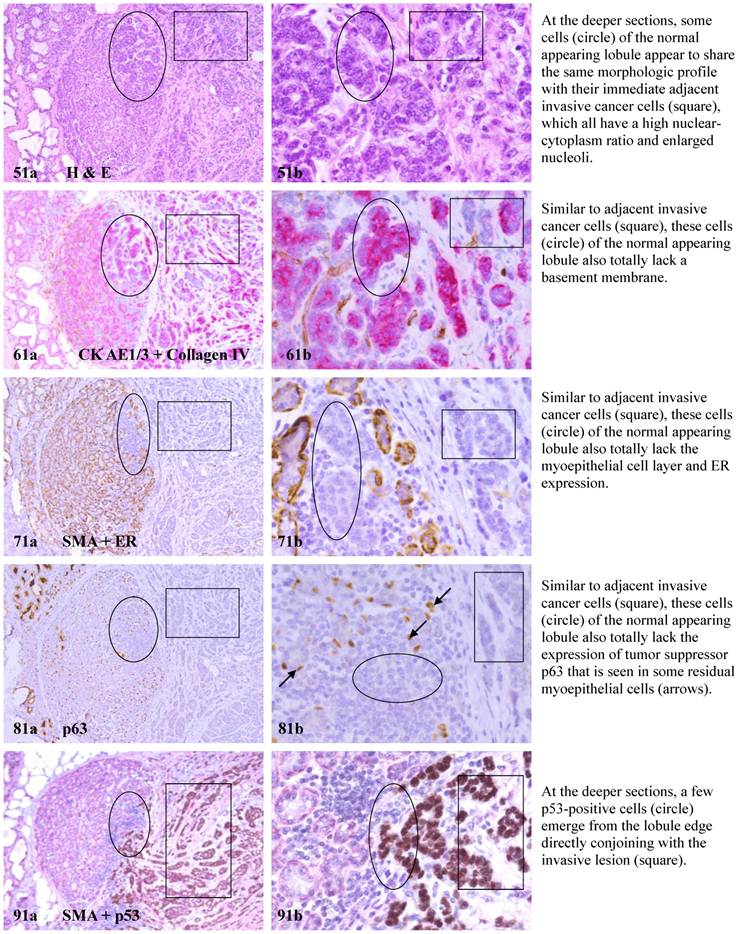
MAA in lobule adjacent to invasive lesion.
The mechanism(s) for progression of these normal- or hyperplastic-appearing tissues is also unknown, but it appears to be triggered by leukocyte infiltration for the following reasons: (1). Pregnancy associated and inflammatory breast cancers, which have extensive leukocyte infiltration, have the most aggressive clinical course and worst prognosis among breast malignancies [43,44], (2).Increased leukocyte infiltration correlated with substantially elevated tumor cell proliferation in prostate tumors [45], (3). Increased leukocyte infiltration correlated with progression of oral epithelium from hyperkeratosis to dysplasia, and to carcinoma [46], and (4). Pre-invasive prostate tumors with chronic inflammation had a significantly higher rate of subsequent invasive tumors than morphologically similar lesions without chronic inflammation [47].
Our recent in vitro study has revealed that protease-degraded collagen I fragments could function as a specific mediator to attract macrophage infiltration [48]. Thus, the formation of leukocyte aggregates within these clusters or lobules is likely to result from increased degradation of the myoepithelial cells and the basement membrane. Our previous studies in multiple types of human tumors, including those in breast, prostate, cervix, and lung, have revealed that leukocytes could facilitate tumor cell invasion or metastasis through 3-correlated pathways [32,49]: (1). The physical movement of leukocytes into the epithelial cells disrupts the inter-cellular junctions and cell surface adhesion molecules, causing the disassociation of tumor cells from the tumor core, (2). Leukocytes are conjoined with some of these tumor cells through plasma membrane fusion, creating tumor cell-leukocyte chimeras (TLCs), and (3). The leukocytes of TLCs impart migratory capacity to the associated tumor cell partners, physically dragging them to different tissue sites.
Thus, the entire luminal cell population within these clusters or lobules could directly invade the stroma when its entire surrounding myoepithelial cell layer become degenerated and disrupted. Although these cells might not possess all the properties of invasive cancer cells, the changed microenvironment may act as a second “hit” to trigger a cascade reaction of malignant transformation that rapidly alters the genetic and biochemical profiles of these cells. This speculation is supported by the fact that major single-gene defects, such as a mutation in the BRCA1 or BRCA2, could cause significantly higher cancer incidence and early cancer onset in their inheritance patterns [50]. This speculation is further supported by a recent study, which has shown that interstitial flow emanated from tumors into their microenvironment could promotes tumor cell invasion by influencing cell behavior and modulating cell-cell interactions [51,52].
Due to these unique presentations, these “sick” lobules or cell cluster may represent the “seeds” or precursors for cancer of unknown primary site, which is one of the 10 most frequent cancers worldwide and ranks as the 4th most common cause of cancer-related death [53-55]. The development of early, uncommon, systemic metastasis, and resistance to therapy are hallmarks of this clinical entity or condition/outcome. In addition, these “sick” lobules or tissues could also be potentially associated with childhood cancer [36,37], in which diagnostic or therapeutic radiation exposure function as a cancer “initiator” or “promoter”.
However, a conclusive classification of these normal appearing structures could not be made at present for the following reasons: (1) aberrant expression of p53 or e-cebB2 is not a conclusive sign of tumor malignancy, (2) a genome-wide comparison with distinct malignant lesions has not been made, (3) the large lobule structures are seen mainly in pregnancy-associated cancer, which may not be highly representative for the general population, (4) no clinical follow-up data are available for these cases, and (5) we need to test more samples to demonstrate statistical significance. On the other hand, as it is estimated that an average of 16 years (30-doubling times) are needed for a cancer-initiating cell to develop into a 10-mm, clinically detectable tumor [7-9], it is also possible that these malignancy-associated alterations in these normal or hyperplastic appearing structures may be focal and transient with no significant consequences.
Supplementary Material
Comments from well recognized experts in the field.
Acknowledgements
This study was supported in part by research grants DAMD17-01-1-0129, DAMD17-01-1-0130, PC051308 from Congressionally Medical Research Programs, BCTR0706983 from The Susan G. Komen Breast Cancer Foundation, 2006CB910505 from the Ministry of Chinese Science and Technology Department, and 2008-02 from the US Military Cancer Institute and Henry M. Jackson Foundation to Dr. Yan-gao Man.
Some of the experimental procedures of this study were conducted in the Armed Forces Institute of Pathology (AFIP) and the American Registry of Pathology (ARP), in which Dr. Man served as the director of Gynecologic and Breast Research Laboratory from January 1,1995 to March 31, 2011. Dr. Man is very grateful to Dr. Renata B. Greenspan, the former director of the AFIP, to Dr. Florabel G. Mullick, the current director of the AFIP, and to Dr. William A. Gardner, the executive director of the ARP for their guidance and unreserved support to his research projects.
As the entire Research Section of the AFIP had been permanently disestablished and the Diagnostic Section of the AFIP has been replaced by the Joint Pathology Center by the US Congress, Dr. Man has accepted a senior scientist position kindly offered by the Henry Jackson Foundation.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Armitage P, Doll R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer. 1954;8(1):1-12
2. Moolgavkar SH, Knudson AGJr. Mutation and cancer: a model for human carcinogenesis. J Natl Cancer Inst. 1981;66(6):1037-1052
3. Frank SA. Age-specific incidence of inherited versus sporadic cancers: a test of the multistage theory of carcinogenesis. Proc Natl Acad Sci U S A. 2005;102(4):1071-1075
4. Goldfarb RH, Liotta LA. Proteolytic enzymes in cancer invasion and metastasis. Semin Thromb Hemost. 1986;12:294-307
5. Clarke R, Brunner N, Katzenellenbogen BS. Progression of human breast cancer cells from hormone-dependent to hormone-independent growth both in vitro and in vivo. Proc Natl Acad Sci USA. 1989;86:3649-3653
6. Hu M, Yao J, Carroll DK, Weremowicz S, Chen H, Carrasco D. et al. Regulation of in situ to invasive breast carcinoma transition. Cancer Cell. 2008;13(5):394-406
7. von FD, Weber E, Hoeffken W. et al. Growth rate of 147 mammary carcinomas. Cancer. 1980;45:2198-2207
8. Arnerlov C, Emdin SO, Lundgren B. et al. Breast carcinoma growth rate described by mammographic doubling time and S-phase fraction. Correlations to clinical and histopathologic factors in a screened population. Cancer. 1992;70:1928-1934
9. Talmadge JE. Clonal selection of metastasis within the life history of a tumor. Cancer Res. 2007;67(24):11471- 11475
10. King TA, Li W, Brogi E, Yee CJ, Gemignani ML, Olvera N. et al. Heterogenic loss of the wild-type BRCA allele in human breast tumorigenesis. Ann Surg Oncol. 2007;14(9):2510-2518
11. Ko SS, Na YS, Yoon CS, Park JY, Kim HS, Hur MH. et al. The significance of c-erbB-2 overexpression and p53 expression in patients with axillary lymph node--negative breast cancer: a tissue microarray study. Int J Surg Pathol. 2007;15(2):98-109
12. Logullo AF, Lopes AB, Nonogaki S, Soares FA, Netto MM, Nishimoto IN, Brentani MM. C-erbB-2 expression is a better predictor for survival than galectin-3 or p53 in early-stage breast cancer. Oncol Rep. 2007;18(1):121-126
13. Tsuda H. Gene and chromosomal alterations in sporadic breast cancer: correlation with histopathological features and implications for genesis and progression. Breast Cancer. 2009;16(3):186-201
14. Tramm T, Kim JY, Tavassoli FA. Diminished number or complete loss of myoepithelial cells associated with metaplastic and neoplastic apocrine lesions of the breast. Am J Surg Pathol. 2011;35(2):202-211
15. Deng G, Lu Y, Zlotnikov G, Thor AD, Smith HS. Loss of heterozygosity in normal tissue adjacent to breast carcinomas. Science. 1996;274(5295):2057-2059
16. Moinfar F, Man YG, Bratthauer GL, Ratschek M, Tavassoli FA. Genetic abnormalities in mammary ductal intraepithelial neoplasia-flat type (Clinging ductal carcinoma in situ)-A simulator of normal mammary epithelium. Cancer. 2000;88:2072-2081
17. Gardner WA, Culberson DE. Atrophy and proliferation in the young adult Prostate. J Urol. 1987;137:53-56
18. Gardner WA. Hypothesis: Pediatric Origins of Prostate Cancer. Hum Path. 1995;26:1291-1292
19. Bennett BD, Gardner WA. Embryonal hyperplasia of the prostate. The Prostate. 1985;3:411-417
20. Malins DC, Johnson PM, Barker EA. Cancer-related changes in prostate DNA as men age and early identification of metastasis in prostate tumors. Proc Natl Acad Sci USA. 2003;100:5401-5406
21. Malins DC, Anderson KM, Gilman NK. Development of a cancer DNA phenotype prior to tumor Formation. Proc Natl Acad Sci Sci USA. 2004;101:10721-10725
22. Malins DC, Gilman NK, Green VM. A DNA phenotype in healthy prostates, conserved in tumors and adjacent normal cells, implies a relationship to carcinogenesis. Proc Natl Acad Sci USA. 2005;102:19093- 19096
23. Man YG, Nieburgs HE. A subset of cell clusters with malignant features in morphologically normal and hyperplastic breast tissues. Cancer Detect Prev. 2006;30(3):239-247
24. Man YG. Focal degeneration of aged or injured myoepithelial cells and the resultant auto- immunoreactions are trigger factors for breast tumor invasion. Medical Hypotheses. 2007;69(6):1340-1357
25. Man YG, Gardner WA. Focal degeneration of basal cells and the resultant auto-immunoreactions: a novel mechanism for prostate tumor progression and invasion. Medical Hypotheses. 2008;70:387-408
26. Man YG, Gardner WA. Bad seeds produce bad crops: a single step-process of prostate carcinogenesis and progression. Int J Biol Sci. 2008;4:246-258
27. Man YG. Bad seeds produce bad crops: a single step-process of breast carcinogenesis and progression. Bioscience Hypotheses. 2008;1:147-155
28. Hsiao YH, Chou MC, Fowler C, Mason JT, Man YG. Breast cancer heterogeneity: mechanisms, proofs, and implications. Journal of Cancer. 2010;1:6-13
29. Man YG. Tumor cell budding from focally disrupted tumor capsules: a common pathway for all breast cancer subtype derived invasion? J Cancer. 2010;1:27-31
30. Man YG. A seemingly most effective target for early detection and intervention of prostate tumor invasion. J Cancer. 2010;1:63-69
31. Hsiao YH, Deng CX, Mason JT, Chou MC, Man YG. Hidden malignant cells within leukocyte aggregates in normal or hyperplastic tissues of pregnancy-associated breast cancer. Cancer Epidemiol. 2011 [Epub ahead of print]
32. Man YG, Mason J, Harley R, Kim YH, Zhu KM, Gardner WA. Leukocyte-facilitated tumor dissemination: a novel model for tumor cell dissociation and metastasis. J Cell Biochem. 2011 doi: 10.1002/jcb.23035
33. Hsiao YH, Tsai HD, Chou MC, Man YG. The myoepithelial cell layer may serve as a potential trigger factor for different outcomes of stage-matched invasive lobular and ductal breast cancers. Int J Biol Sci. 2011;7:147-153
34. Tot T. The theory of the sick breast lobe and the possible consequences. Int J Surg Pathol. 2007;15(4):369-375
35. Tot T. The origins of early breast carcinoma. Semin Diagn Pathol. 2010;27(1):62-68
36. Kaatsch P. Epidemiology of childhood cancer. Cancer Treat Rev. 2010;36(4):277-285
37. Kleinerman RA. Cancer risks following diagnostic and therapeutic radiation exposure in children. Pediatr Radiol. 2006;36(Suppl 2):121-125
38. Tobacman JK. Filament disassembly and loss of mammary myoepithelial cells after exposure to lambdacarrageenan. Cancer Res. 1997;57:2823-2826
39. Sapino A, Macri L, Tonda L, Bussolati G. Oxytocin enhances myoepithelial cell differentiation and proliferation in the mouse mammary gland. Endocrinology. 1993;133:838-842
40. Zou Z, Anisowicz A, Hendrix MJ, Thor A, Neveu M, Sheng S. et al. Maspin, a serpin with tumor-suppressing activity in human mammary epithelial cells. Science. 1994;263:526-529
41. Barbareschi M, Pecciarini L, Cangi MG. et al. p63, a p53 homologue, is a selective nuclear marker of myoepithelial cells of the human breast. Am J Surg Pathol. 2001;25:1954-1960
42. Sternlight MD, Barsky SH. The myoepithelial defense: a host defense against cancer. Med Hypotheses. 1997;48:37-46
43. Rodriguez AO, Chew H, Cress R, Cress R, Xing G, McElvy S, Danielsen B, Smith L. Evidence of poorer survival in pregnancy-associated breast cancer. Obstet Gynecol. 2008;112(1):71-78
44. Mathelin C, Annane K, Treisser A, Chenard MP, Tomasetto C, Bellocq JP, Rio MC. Pregnancy and post-partum breast cancer: a prospective study. Anticancer Res. 2008;28:2447-2452
45. Smith CJ, Gardner WAJr. Inflammation-proliferation: possible relationships in the prostate. Prog Clin Biol Res. 1987;239:317-325
46. Gannot G, Gannot I, Vered H, Buchner A, Keisaris Y. Increase in immune cell infiltration with progression of oral epithelium from hyperkeratiosis to dysplasia and carcinoma. Br J Cancer. 2002;86:1444- 1448
47. MacLennan GT, Eisenberg R, Fleshman RL, Taylor JM, Fu P, Resnick MI, Gupta S. The influence of chronic inflammation in prostatic carcinogenesis: a 5-year follow-up study. J Urol. 2006;176:1012-1016
48. OBrien JO, Lyons T, Bell KP, Monks J, Lucia S, Wilson RS, Hines L, Man YG, Borges V, Schedin P. Alternatively activated macrophages and collagen I remodeling characterize the post partum involuting mammary gland across species. Am J Pathol. 2010;176(3):1241-1255
49. Man YG, Harley R, Mason J, Gardner WA. Contributions of leukocytes to tumor invasion and metastasis: the “piggy-back” hypothesis. Cancer Epidem. 2010;34(1):3-6
50. Frank SA. Genetic predisposition to cancer - insights from population genetics. Nat Rev Genet. 2004;5(10):764- 772
51. Shieh AC, Rozansky HA, Hinz B, Swartz MA. Tumor cell invasion is promoted by interstitial flow-induced matrix priming by stromal fibroblasts. Cancer Res. 2011;71(3):790-800
52. Shieh AC, Swartz MA. Regulation of tumor invasion by interstitial fluid flow. Phys Biol. 2011Feb7;8(1):015012
53. Pentheroudakis G, Briasoulis E, Pavlidis N. Cancer of unknown primary site: missing primary or missing biology? Oncologist. 2007;12(4):418-425
54. Carlson HR. Carcinoma of unknown primary: searching for the origin of metastasis. JAAPA. 2009;22(8):18-21
55. Pentheroudakis G, Pavlidis N. Probing the unknown in cancer of unknown primary: which way is the right way? Ann Oncol. 2010 [Epub ahead of print]
Author contact
![]() Corresponding author: Yan-gao Man, MD., PhD., Diagnostic and Translational Research Center, Henry Jackson Foundation, 401 Professional Drive, Gaithersburg, 20879. Phone: 240-833-4969; Fax: 240-833-4940; E-mail: ymannorg
Corresponding author: Yan-gao Man, MD., PhD., Diagnostic and Translational Research Center, Henry Jackson Foundation, 401 Professional Drive, Gaithersburg, 20879. Phone: 240-833-4969; Fax: 240-833-4940; E-mail: ymannorg
Citation styles
APA
Man, Y.g., Grinkemeyer, M., Izadjoo, M., Stojadinovic, A. (2011). Malignant Transformation and Stromal Invasion from Normal or Hyperplastic Tissues: True or False?. Journal of Cancer, 2, 413-424. https://doi.org/10.7150/jca.2.413.
ACS
Man, Y.g.; Grinkemeyer, M.; Izadjoo, M.; Stojadinovic, A. Malignant Transformation and Stromal Invasion from Normal or Hyperplastic Tissues: True or False?. J. Cancer 2011, 2, 413-424. DOI: 10.7150/jca.2.413.
NLM
Man Yg, Grinkemeyer M, Izadjoo M, Stojadinovic A. Malignant Transformation and Stromal Invasion from Normal or Hyperplastic Tissues: True or False?. J Cancer 2011; 2:413-424. doi:10.7150/jca.2.413. https://www.jcancer.org/v02p0413.htm
CSE
Man Yg, Grinkemeyer M, Izadjoo M, Stojadinovic A. 2011. Malignant Transformation and Stromal Invasion from Normal or Hyperplastic Tissues: True or False?. J Cancer. 2:413-424.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.