Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2011; 2:435-442. doi:10.7150/jca.2.435 This volume Cite
Research Paper
Gemcitabine Overcomes Erlotinib Resistance in EGFR-Overexpressing Cancer Cells through Downregulation of Akt
Chandra Bartholomeusz1,3* ![]() , Fumiyuki Yamasaki1,2,4*, Hitomi Saso1,3, Kaoru Kurisu4, Gabriel N. Hortobagyi3, Naoto T. Ueno1,2,3
, Fumiyuki Yamasaki1,2,4*, Hitomi Saso1,3, Kaoru Kurisu4, Gabriel N. Hortobagyi3, Naoto T. Ueno1,2,3 ![]()
1. Breast Cancer Translational Research Laboratory, The University of Texas MD Anderson Cancer Center, Houston, Texas;
2. Department of Stem Cell Transplantation and Cellular Therapy, The University of Texas MD Anderson Cancer Center, Houston, Texas;
3. Department of Breast Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, Texas;
4. Graduate School of Biomedical Sciences, Hiroshima University, Hiroshima, Japan
*These authors contributed equally to this work.
Received 2011-7-20; Accepted 2011-8-7; Published 2011-8-7
Citation:
Bartholomeusz C, Yamasaki F, Saso H, Kurisu K, Hortobagyi GN, Ueno NT. Gemcitabine Overcomes Erlotinib Resistance in EGFR-Overexpressing Cancer Cells through Downregulation of Akt. J Cancer 2011; 2:435-442. doi:10.7150/jca.2.435. https://www.jcancer.org/v02p0435.htm
Other stylesAbstract
A phase III clinical trial showed gemcitabine chemotherapy combined with epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor erlotinib significantly improved overall survival in patients with advanced pancreatic cancer. Therefore, we studied whether addition of gemcitabine to erlotinib in cancer cells having intrinsic or acquired erlotinib resistance could restore chemosensitization in these cells. We studied the synergistic effect of erlotinib and gemcitabine in EGFR-overexpressing A-431 cells with acquired erlotinib resistance and in intrinsic erlotinib-resistant triple negative breast cancer (TNBC) BT-549, MDA-MB-231 and MDA-MB-468 cell lines. Erlotinib and gemcitabine were synergistic in both parental intrinsically erlotinib-sensitive A-431 cells (combination index = 0.69 at the effective dose [ED50]) and in two A-431 cell pools that had acquired erlotinib resistance (combination indices = 0.63 and 0.49 at ED50). The synergistic effect of erlotinib and gemcitabine on cancer cells did not require sensitivity to erlotinib provided that erlotinib can inhibit EGFR. The restoration of sensitivity by gemcitabine occurred through downregulation of phosphorylated Akt (p-Akt), which suggests that PI3K-PTEN-Akt activity is important to the synergism between the two agents. In A-431 parental cells, treatment with gemcitabine followed by erlotinib - but not the reverse sequence - was superior to erlotinib alone. The importance of the order of administration maybe due to the downregulation of p-Akt by gemcitabine in a dose- and time-dependent manner in cells with intrinsic or acquired erlotinib resistance. Our data show that gemcitabine increased the cytotoxic effect of erlotinib by downregulating p-Akt in EGFR-overexpressing cells with either intrinsic or acquired erlotinib resistance.
Keywords: gemcitabine, erlotinib, resistance, A-431, breast cancer
Introduction
Epidermal growth factor receptor (EGFR), a member of the ErbB family of receptor tyrosine kinases, is overexpressed in many solid malignancies and is associated with poor clinical prognosis. As a result, EGFR is considered to be an attractive target for cancer treatment1,2. Two pharmacologic approaches one with monoclonal antibodies and the other with EGFR tyrosine kinase inhibitors targeting EGFR have been successfully developed3.
Erlotinib is a small molecule EGFR inhibitor that targets EGFR's adenosine triphosphate (ATP)-binding sites, resulting in the inhibition of EGFR tyrosine kinase activity. Erlotinib was the first anti-EGFR agent to be used to extend the survival of patients with advanced non-small cell lung cancer4 and currently is being investigated as a therapy for several types of tumors in multiple clinical trials3,5. Erlotinib was recently approved by the United States Food and Drug Administration for combination use with gemcitabine in the treatment of advanced pancreatic cancer3,5. Erlotinib offers clinical benefit to a subset of patients with non-small cell lung cancer. However, most of these patients demonstrate disease progression within 1 year, and whether erlotinib should be included in a combination chemotherapeutic regimen at that point has been unclear.
However, resistance to EGFR tyrosine kinase inhibitors presents a therapeutic challenge; some tumors seem to resist these drugs intrinsically, and cancers that initially respond frequently acquire resistance6,7,8. The phosphatidylinositol 3-kinase (PI3K)-PTEN-Akt pathway is thought to be involved in intrinsic mechanisms of resistance to EGFR inhibitors, including erlotinib9,10,11,12, and we have previously shown that it is involved in acquired resistance to erlotinib8. Therefore, it is possible that targeting the PI3K-PTEN-Akt pathway in combination with erlotinib treatment would enhance cytotoxicity and increase the efficacy of erlotinib and other EGFR inhibitors. Intrinsic and acquired erlotinib resistances are associated with upregulation of Akt activity. Targeting PI3K-PTEN-Akt specifically offers a theoretical advantage because it is the convergence point for a number of upstream signaling pathways; thus, multiple pathways could be blocked by inhibition of PI3K-PTEN-Akt alone. However, no therapeutically useful direct PI3K-PTEN-Akt inhibitors have been identified to date. Because gemcitabine is the only drug that is recognized to have a combination effect with erlotinib, we decided to investigate whether gemcitabine targets the PI3K-PTEN-Akt pathway when combined with erlotinib.
With this background in mind, the purpose of this study was to assess the role of PI3K-PEN-Akt inhibition in gemcitabine' s ability to reverse erlotinib resistance. We designed a study to determine whether the addition of gemcitabine to erlotinib in cancer cells having intrinsic or acquired erlotinib resistance could restore chemosensitization in these cells, and if so, whether the PI3K-PTEN-Akt pathway was involved. Herein, we report that when gemcitabine was followed by erlotinib treatment, it synergistically decreased the viability of cancer cells by restoring sensitivity to erlotinib. The restoration of sensitivity by gemcitabine occurred through downregulation of p-Akt, which suggests that PI3K-PTEN-Akt activity is involved in the synergistically increased apoptosis and is an important mechanism for understanding and overcoming erlotinib resistance.
Materials and Methods
Chemotherapeutic agents and cell lines. Erlotinib was dissolved in dimethyl sulfoxide at 5 mmol/L as described previously13. Clinical-grade gemcitabine (Eli Lilly and Co, Indianapolis, IN) was purchased and dissolved in sterile water at 100 mM.
For studies of cells with acquired resistance to erlotinib, we previously established A-431 epidermoid cancer cells8 that overexpress EGFR and are sensitive to erlotinib. We developed two pools of erlotinib-resistant A-431 cells by continuously exposing them to erlotinib (10 μmol/L for pool 1 and 5 μmol/L for pool 2) for 6 months as described previously8. The cells regained morphologic characteristics similar to those of the parental line after 6 months of erlotinib exposure. Erlotinib resistance was confirmed as described previously8. The resistant cells were maintained in medium without erlotinib for at least 1 week before each experiment was performed.
For studies of cells with intrinsic resistance to erlotinib, we also obtained triple-negative BT-549, MDA-MB-231, and MDA-MB-468 cells from the American Type Culture Collection (Manassas, VA). These are EGFR-overexpressing breast cancer cell lines that are intrinsically resistant to erlotinib13.
Dose-response studies. A-431 parental cells and samples from both pools of A-431 cells with acquired erlotinib resistance were seeded at 500 cells/well in 96-well dishes. After 24 h, the cells were treated in quadruplicate for 72 h with 2-fold serial dilutions of erlotinib alone, gemcitabine alone, or both drugs simultaneously at a fixed ratio of 2 μM erlotinib:7 nM gemcitabine. After 72 h, a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed as described previously13. All experiments were performed at least three times. Growth inhibition was expressed as the percentage of remaining viable cells relative to the number of viable cells in untreated cultures, which was set at 100%. Combination indices (CIs) were obtained using the commercial software package CalcuSyn (Biosoft, Cambridge, UK)14. Drug synergism, addition, and antagonism were defined by CI values of <1.0, 1.0, and >1.0, respectively. For each drug, at least three doses above and three doses below the individual effective dose (ED50) were tested to determine the median effect as calculated by the computer software.
The dose-response study was also performed with BT-549, MDA-MB-231, and MDA-MB-468 breast cancer cells to assess the combination effect of erlotinib and gemcitabine on intrinsic erlotinib resistance. The procedure was exactly the same except that the ratio was 2 μM erlotinib: 10 nM gemcitabine.
Time course studies. We tried three time courses to investigate the combination effect of erlotinib and gemcitabine. In the first schedule, erlotinib and gemcitabine were given simultaneously. In the second schedule, the cells were treated with gemcitabine for 72 h and then with erlotinib for another 72 h. In the third schedule, the cells were treated with erlotinib for 72 h and then with gemcitabine for another 72 h.
Trypan blue exclusion assay. In order to assess cell viability and thereby gauge the efficacy of combination therapy on each of the types of cells, we performed a trypan blue exclusion assay as described previously8. Briefly, A-431 parental cells and samples from both pools of erlotinib-resistant cells were plated and treated with 2 μM erlotinib, 7 nM gemcitabine, or both drugs simultaneously. After 72 h of exposure, floating and adhering cells were collected by trypsinization and counted under a microscope.
Cell cycle analysis. As a measure of apoptosis, cell-cycle analysis was performed to determine the percentage of cells in the sub-G1 phase of the cell cycle, as described in our previous report13. Briefly, A-431 parental cells and samples from both pools of erlotinib-resistant cells were treated with 2 μM erlotinib, 7 nM gemcitabine, or both drugs simultaneously. After 72 h of drug treatment, the cells were fixed overnight in 70% ethanol and then resuspended in propidium iodide. DNA content was measured using a FACScan cytometer (Becton Dickinson, Franklin Lakes, NJ).
Western blotting. Western blotting was performed as described previously13. Immunoblotting and immunoprecipitation were performed with the following antibodies: mouse anti-EGFR (Ab-12; Lab Vision, Fremont, CA); mouse anti-p-Akt (Ser 473, #4051) and rabbit anti-Akt (#9272; both from Cell Signaling Technology, Beverly, MA); and mouse anti-β-actin (A-5441; Sigma Chemical, St. Louis, MO). Signals were detected using an Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE).
Results
Synergistic effect of erlotinib and gemcitabine in cell lines with acquired erlotinib resistance
The EGFR-overexpressing A-431 cells (parental erlotinib-sensitive cells and two pools of erlotinib-resistant cells) were treated with serial dilutions of erlotinib, gemcitabine, or both drugs simultaneously at a fixed ratio spanning the ED50 of each drug. Table 1 shows the affected fraction of cells versus the CI (synergistic) values for the drug mixture for all three cell lines.
In the A-431 parental cells, the drug combination resulted in a CI value of 0.69 at ED50, indicating synergism between erlotinib and gemcitabine. In both erlotinib-resistant pools of cells, the drug combination resulted in CI values very similar to that of the parental cells (with CI values of 0.63 for pool 1 and 0.49 for pool 2). The sensitivities of both pools of erlotinib-resistant cells to gemcitabine were very similar (ED50 pool 1, 11.8 nM; ED50 pool 2, 10.3 nM). Therefore, erlotinib was shown to enhance the cytotoxic effects of gemcitabine in EGFR-overexpressing cancer cells even after acquired erlotinib-resistance.
Table 1
Synergistic Effect of Erlotinib and Gemcitabine in Cell Lines with Acquired Erlotinib Resistance
| A-431 | Combination Indices | ||||
|---|---|---|---|---|---|
| Cell Line | ED50 | ED75 | ED90 | Dm | |
| Mixture | Parental | 0.69257 | 0.70776 | 0.72472 | 0.83689 |
| Pool 1 | 0.62572 | 0.68561 | 0.76407 | 1.89010 | |
| Pool 2 | 0.49079 | 0.49590 | 0.51086 | 1.20910 | |
| Erlotinib | Parental | 1.76766 | |||
| (µM) | Pool 1 | 28.8355 | |||
| Pool 2 | 15.4557 | ||||
| Gemcitabine | Parental | 13.3674 | |||
| (nM) | Pool 1 | 11.8092 | |||
| Pool 2 | 10.2576 | ||||
Dm means the median effect dose, it is analogous to the IC50)
Increase in apoptosis after erlotinib and gemcitabine treatment
We performed flow-cytometric cell-cycle analysis to determine whether the results of the dose-response assays were a reflection of cell-cycle arrest or apoptosis. Similar to other DNA-damaging agents, gemcitabine promotes apoptosis15,16, and we expected gemcitabine treatment to induce apoptosis in our A-431 cells. Indeed, the percentage of parental cells treated with 7 nM gemcitabine that were in the sub-diploid (sub-G1) phase was 5 times higher than the percentage of untreated parental cells in the sub-diploid phase (Fig. 1). This percentage increased to a further 3 to 4 times higher in parental cells treated with both 2 μM erlotinib and 7 nM gemcitabine. Both erlotinib- resistant cell pools demonstrated similar synergistic increases in apoptosis after treatment with gemcitabine and erlotinib (Fig. 1).
Fig 1
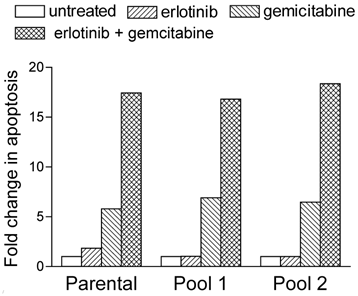
Erlotinib and gemcitabine synergistically increased their rate of apoptosis in EGFR-overexpressing A-431 epidermoid cancer cells with intrinsic (parental) or acquired (pools 1 and 2) erlotinib resistance. Cells were treated with 2 µM erlotinib, 7 nM gemcitabine, or both simultaneously. After 72 h of drug treatment, the cells were fixed, resuspended in propidium iodide, and analyzed for DNA content using a FACScan cytometer. The extent of changes in sub-diploid content (sub-G1 cell-cycle phase) relative to untreated cells was assessed to determine the levels of apoptosis.
Optimal timing of gemcitabine and erlotinib treatment
We next examined whether the order in which gemcitabine and erlotinib were administered influenced cell survival in erlotinib-resistant cells. We first examined the level of cytotoxicity in response to three different schedules of gemcitabine and erlotinib. In the first schedule, erlotinib and gemcitabine were given simultaneously, which resulted in the enhancement of erlotinib-mediated cytotoxicity by gemcitabine (see Fig. 1). In the second schedule, A-431 parental cells were treated with gemcitabine for 72 h and then with erlotinib for another 72 h, and a similar synergistic effect was observed (Fig. 2A). In contrast to the first and second schedules, when the cells were treated with erlotinib first and then gemcitabine, there was no additive effect (Fig. 2B).
Downregulation of p-Akt expression by gemcitabine
Since resistance to erlotinib is known to be associated with increased levels of p-Akt, a prosurvival molecule downstream of EGFR, we examined whether treatment of cells with gemcitabine would decrease the expression level of p-Akt and whether gemcitabine could overcome erlotinib resistance by inhibiting Akt activity. In previous studies, we have shown that by treating cells with an Akt inhibitor IV, erlotinib sensitivity was restored in erlotinib-resistant pools of A-431 cells8. Accordingly, we investigated the p-Akt expression level in A-431 cells after treatment with gemcitabine. As expected, the level of p-Akt was decreased after treatment with gemcitabine at doses of 3.5-14 nM in both erlotinib-sensitive parental cells and erlotinib-resistant cells (Fig. 3) and in the TNBC cell lines BT-549 and MDA-MB-468 after treatment with gemcitabine at a dose of 100nM (Fig. 4).
Fig 2
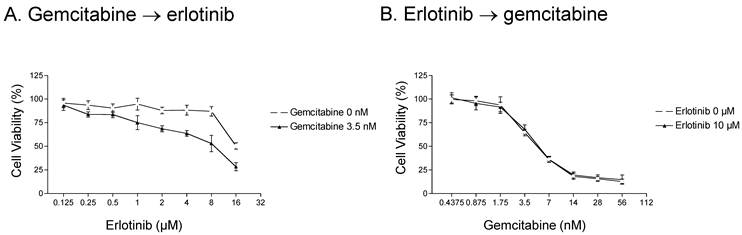
Effect of erlotinib and gemcitabine treatment schedule on parental A-431 cells. A. Cells were treated with 0 or 3.5 nM gemcitabine for 72 h and then with 0.125-16 μM erlotinib for 72 h. A synergistic effect was observed. B. In contrast, a synergistic effect was not observed when cells were treated with 0 or 10 μM erlotinib for 72 h and then with 0.4375-56 nM gemcitabine for 72 h. Note that the concentration along the x-axis scale in each graph doubles at each tick mark.
Fig 3
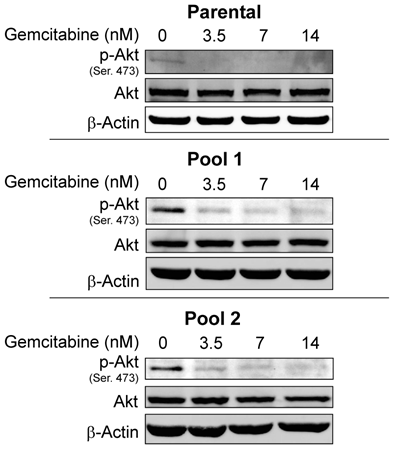
Protein expression levels of Akt and p-Akt were analyzed by Western blotting after treatment with gemcitabine in both erlotinib-sensitive A-431 cells (parental) and erlotinib-resistant A-431 cells (two pools). p-Akt but not Akt was downregulated in both erlotinib-sensitive and erlotinib-resistant cells. β-actin was used as a loading control.
Fig 4
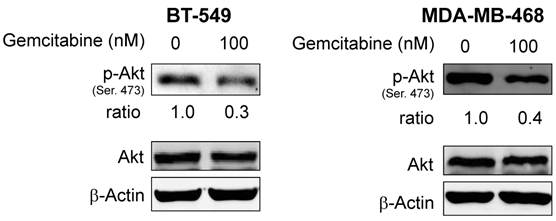
Protein expression levels of Akt and p-Akt were analyzed by Western blotting after treatment with gemcitabine in erlotinib-resistant BT-549 and MDA-MB-468 breast cancer cells. p-Akt but not Akt was downregulated in both cell lines. β-actin was used as a loading control.
Synergistic effect of erlotinib and gemcitabine in breast cancer cell lines with intrinsic erlotinib resistance
Finally, we examined the combined effect of erlotinib and gemcitabine in breast cancer cell lines intrinsically resistant to erlotinib. We used TNBC cell lines BT-549, MDA-MB-468, and MDA-MB-231 because these cell lines express EGFR and high levels of unphosphorylated Akt and are intrinsically resistant to erlotinib. The cells were treated with serial dilutions of erlotinib, gemcitabine, or both drugs simultaneously at a fixed ratio spanning the ED50 of each drug. The measurements of cell viability at the various drug concentrations are shown in Table 2. In BT-549, MDA-MB-468, and MDA-MB-231 cells at the ED50, the drug combination resulted in CI values of 0.39, 0.82, and 0.71, respectively, indicating synergism between these two agents. The sensitivity levels (in terms of ED50) of these cells to gemcitabine alone were 342 nM in BT-549 cells, 29.2 nM in MDA-MB-468 cells, and 10.3 nM in MDA-MB-231 cells. These results suggest that a synergistic effect of erlotinib and gemcitabine was also seen in breast cancer lines with intrinsic erlotinib resistance EGFR-overexpressing TNBC.
Similar to as was seen in A-431 cells, MDA-MB-468 breast cancer cells treated with 8 nM gemcitabine had similar percentages of cells that were in the sub-G1phase as did untreated cells, whereas this percentage increased to 1.9 times that of untreated cells and 1.7 times that of just erlotinib in MDA-MB-468 cells administered both 2 μM erlotinib and 8 nM gemcitabine (data not shown).
Table 2
Synergistic Effect of Erlotinib and Gemcitabine in Cell Lines with Intrinsic Erlotinib Resistance
| Combination Indices | |||||
|---|---|---|---|---|---|
| Cell Line | ED50 | ED75 | ED90 | Dm | |
| Mixture | BT-549 | 0.39275 | 0.26629 | 0.18403 | 6.89283 |
| MDA-MB-468 | 0.81612 | 0.70875 | 0.65044 | 2.88089 | |
| MDA-MB-231 | 0.70510 | 0.55634 | 0.45635 | 2.42506 | |
| Erlotinib | BT-549 | 23.59856 | |||
| (µM) | MDA-MB-468 | 8.90787 | |||
| MDA-MB-231 | 25.7763 | ||||
| Gemcitabine | BT-549 | 342.3832 | |||
| (nM) | MDA-MB-468 | 29.2352 | |||
| MDA-MB-231 | 10.3228 | ||||
Discussion
In this study, we showed that gemcitabine increases the cytotoxic effect of erlotinib via downregulation of p-Akt. This synergistic effect, in the form of enhanced apoptosis, was observed both in cells with intrinsic erlotinib resistance and cells with acquired erlotinib resistance8.
Through these findings, we have shown that the restoration of sensitivity by gemcitabine occurred through downregulation of p-Akt, which suggests that PI3K-PTEN-Akt activity is critical to the synergistic effect of the two drugs on cell death
Our findings are consistent with those of other groups suggesting that erlotinib given first, followed by gemcitabine, had no effect, whereas gemcitabine followed by erlotinib produced a synergistic effect. Our previous studies revealed that erlotinib can target EGFR in cells with either intrinsic erlotinib resistance13 or acquired erlotinib resistance8 by inhibiting the phosphorylation of EGFR. Another study demonstrated that erlotinib significantly inhibited the phosphorylation of EGFR and other downstream kinases and significantly promoted gemcitabine-induced apoptosis in vivo17. Our in vitro model of EGFR-driving cancer cells now indicates that the growth-inhibitory effects in such cells of erlotinib combined with gemcitabine could apply even after the cells have acquired erlotinib resistance. The molecular mechanisms behind the synergism between erlotinib and gemcitabine most likely involved the decreased p-Akt expression caused by gemcitabine. In addition, other investigators have also shown that the administration of gemcitabine first followed by gefitinib increased the therapeutic index of such therapy over that of gemcitabine alone18, and cytotoxic synergism was also found to result when cells were exposed to concurrent pemetrexed and erlotinib or sequential pemetrexed followed by erlotinib in both erlotinib-sensitive and erlotinib-resistant human non-small cell lung cancer cell lines. These studies support the importance of drug scheduling in the treatment of cancer7. The latter regimen may be more effective because treatment with erlotinib causes G1 cell-cycle arrest8, thereby reducing the cytotoxic effect of gemcitabine, as gemcitabine mainly affects cells in the S phase of the cell cycle.
To clarify the mechanisms behind the synergism between gemcitabine and erlotinib, other investigators have shown that gemcitabine increases EGFR -phosphorylation and subsequently induces the EGFR degradation that may be a mechanism for gemcitabine-mediated cell death19. This is an interesting observation because the degradation of EGFR may result in downregulation of p-Akt in some cell lines, leading to apoptosis, which is consistent with our findings. Also, genistein (a naturally occurring isoflavone in soybeans) has been reported to increase the cytotoxic effect induced by erlotinib in certain pancreatic cancer cells through the inhibition of Akt and nuclear factor-κB20. That study and our results suggest that downstream molecules of the Akt pathway are involved in the synergistic effect of erlotinib and gemcitabine.
We have shown that one of the molecular mechanisms involved in the development of acquired erlotinib resistance is p-Akt upregulation subsequent to PTEN downregulation8. Multiple molecular mechanisms of resistance to EGFR tyrosine kinase inhibitors exist; they include mutations in the EGFR tyrosine kinase domain, modification of other pathways, such as those of HER3, IGF-R, and E-cadherin8, hepatocyte growth factor, and amplification of MET proto-oncogenes21. These mechanisms may vary from one tumor or patient to the next. Therefore, even though we observed an enhanced response to erlotinib after treating our erlotinib-resistant cell pools with gemcitabine, other erlotinib-resistant cell pools derived from other EGFR-overexpressing lines might not yield the same results. Hence, further elucidating the mechanisms by which these cancers develop erlotinib resistance is imperative. Another possibility is that temporarily interrupting erlotinib therapy and administering non-erlotinib chemotherapy to patients with erlotinib-resistant cancers could eventually restore cell sensitivity to erlotinib. In a previous in vitro study, a 6-month interruption of a continuous maintenance regimen of gefitinib did restore cell sensitivity to gefitinib21.
Our results showed that gemcitabine increased the cytotoxic effect of erlotinib by downregulating p-Akt in EGFR-overexpressing cells with either intrinsic or acquired erlotinib resistance. These findings indicate that further studies are needed to understand the molecular mechanisms by which gemcitabine acts synergistically with erlotinib. We can speculate that the continued treatment of erlotinib-resistant cancers with a combination of erlotinib and gemcitabine may be effective. The enhanced understanding of the molecular mechanism behind these agents elucidated by our findings may help to develop more effective therapeutic targets for enhancing the synergism of other agents with erlotinib.
Acknowledgements
This work was supported by the Nellie B. Connally Breast Cancer Research Fund; National Institutes of Health R01 grant CA123318-01A1 (NU); Japan Society for the Promotion of Science Grant-in-Aid for Scientific Research and grants-in-aid from the Japan Society for the Promotion of Science grant No. 22591612 (FY); and National Cancer Institute Cancer Center Support Grant CA-16672 (support of the Flow Cytometry and Cellular Imaging Core Facility and the Media Preparation Core Facility at The University of Texas MD Anderson Cancer Center). We thank Sunita Patterson, Maude Veech, and Elizabeth L. Hess, ELS(D), Department of Scientific Publications at MD Anderson Cancer Center, for their expert editorial review.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Ciardiello F, Tortora G. A novel approach in the treatment of cancer: targeting the epidermal growth factor receptor. Clin Cancer Res. 2001;7:2958-70
2. Ono M, Kuwano M. Molecular mechanisms of epidermal growth factor receptor (EGFR) activation and response to gefitinib and other EGFR-targeting drugs. Clin Cancer Res. 2006;12:7242-51
3. Bareschino MA, Schettino C, Troiani T, Martinelli E, Morgillo F, Ciardiello F. Erlotinib in cancer treatment. Ann Oncol. 2007;18(Suppl 6):vi35-41
4. Shepherd FA, Rodrigues Pereira J, Ciuleanu T. et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123-32
5. Rocha-Lima CM, Soares HP, Raez LE, Singal R. EGFR targeting of solid tumors. Cancer Control. 2007;14:295-304
6. Bunn PAJr. Can acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors be overcome by different small-molecule tyrosine kinase inhibitors? J Clin Oncol. 2007;25:2504-5
7. Tabernero J. The role of VEGF and EGFR inhibition: implications for combining anti-VEGF and anti-EGFR agents. Mol Cancer Res. 2007;5:203-20
8. Yamasaki F, Johansen MJ, Zhang D. et al. Acquired resistance to erlotinib in A-431 epidermoid cancer cells requires down-regulation of MMAC1/PTEN and up-regulation of phosphorylated Akt. Cancer Res. 2007;67:5779-88
9. Engelman JA, Janne PA, Mermel C. et al. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci U S A. 2005;102:3788-93
10. Maiello MR, D'Alessio A, De Luca A. et al. AZD3409 inhibits the growth of breast cancer cells with intrinsic resistance to the EGFR tyrosine kinase inhibitor gefitinib. Breast Cancer Res Treat. 2007;102:275-82
11. Ono M, Hirata A, Kometani T. et al. Sensitivity to gefitinib (Iressa, ZD1839) in non-small cell lung cancer cell lines correlates with dependence on the epidermal growth factor (EGF) receptor/extracellular signal-regulated kinase 1/2 and EGF receptor/Akt pathway for proliferation. Mol Cancer Ther. 2004;3:465-72
12. Haas-Kogan DA, Prados MD, Tihan T. et al. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst. 2005;97:880-7
13. Yamasaki F, Zhang D, Bartholomeusz C. et al. Sensitivity of breast cancer cells to erlotinib depends on cyclin-dependent kinase 2 activity. Mol Cancer Ther. 2007;6:2168-77
14. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27-55
15. Bold RJ, Chandra J, McConkey DJ. Gemcitabine-induced programmed cell death (apoptosis) of human pancreatic carcinoma is determined by Bcl-2 content. Ann Surg Oncol. 1999;6:279-85
16. Cartee L, Kucera GL, Willingham MC. Induction of apoptosis by gemcitabine in BG-1 human ovarian cancer cells compared with induction by staurosporine, paclitaxel and cisplatin. Apoptosis. 1998;3:439-49
17. Ng SS, Tsao MS, Nicklee T, Hedley DW. Effects of the epidermal growth factor receptor inhibitor OSI-774, Tarceva, on downstream signaling pathways and apoptosis in human pancreatic adenocarcinoma. Mol Cancer Ther. 2002;1:777-83
18. Chun PY, Feng FY, Scheurer AM, Davis MA, Lawrence TS, Nyati MK. Synergistic effects of gemcitabine and gefitinib in the treatment of head and neck carcinoma. Cancer Res. 2006;66:981-8
19. Feng FY, Varambally S, Tomlins SA. et al. Role of epidermal growth factor receptor degradation in gemcitabine-mediated cytotoxicity. Oncogene. 2007;26:3431-9
20. El-Rayes BF, Ali S, Ali IF, Philip PA, Abbruzzese J, Sarkar FH. Potentiation of the effect of erlotinib by genistein in pancreatic cancer: the role of Akt and nuclear factor-kappaB. Cancer Res. 2006;66:10553-9
21. Ando K, Ohmori T, Inoue F. et al. Enhancement of sensitivity to tumor necrosis factor alpha in non-small cell lung cancer cells with acquired resistance to gefitinib. Clin Cancer Res. 2005;11:8872-9
Author contact
![]() Corresponding author: Naoto T. Ueno, MD, PhD and Chandra Bartholomeusz, MD, PhD, Department of Breast Medical Oncology, Unit 1354, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Boulevard, Houston, TX 77030. Phone: 713-792-8754 or 713-745-1086 Fax: 713-794-4747. E-mail: nuenoorg and chbarthoorg
Corresponding author: Naoto T. Ueno, MD, PhD and Chandra Bartholomeusz, MD, PhD, Department of Breast Medical Oncology, Unit 1354, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Boulevard, Houston, TX 77030. Phone: 713-792-8754 or 713-745-1086 Fax: 713-794-4747. E-mail: nuenoorg and chbarthoorg
Citation styles
APA
Bartholomeusz, C., Yamasaki, F., Saso, H., Kurisu, K., Hortobagyi, G.N., Ueno, N.T. (2011). Gemcitabine Overcomes Erlotinib Resistance in EGFR-Overexpressing Cancer Cells through Downregulation of Akt. Journal of Cancer, 2, 435-442. https://doi.org/10.7150/jca.2.435.
ACS
Bartholomeusz, C.; Yamasaki, F.; Saso, H.; Kurisu, K.; Hortobagyi, G.N.; Ueno, N.T. Gemcitabine Overcomes Erlotinib Resistance in EGFR-Overexpressing Cancer Cells through Downregulation of Akt. J. Cancer 2011, 2, 435-442. DOI: 10.7150/jca.2.435.
NLM
Bartholomeusz C, Yamasaki F, Saso H, Kurisu K, Hortobagyi GN, Ueno NT. Gemcitabine Overcomes Erlotinib Resistance in EGFR-Overexpressing Cancer Cells through Downregulation of Akt. J Cancer 2011; 2:435-442. doi:10.7150/jca.2.435. https://www.jcancer.org/v02p0435.htm
CSE
Bartholomeusz C, Yamasaki F, Saso H, Kurisu K, Hortobagyi GN, Ueno NT. 2011. Gemcitabine Overcomes Erlotinib Resistance in EGFR-Overexpressing Cancer Cells through Downregulation of Akt. J Cancer. 2:435-442.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.