Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2011; 2:478-483. doi:10.7150/jca.2.478 This volume Cite
Short Research Communication
The First Type III Domain of Fibronectin is Associated with the Expression of Cytokines within the Lung Tumor Microenvironment
Mingzhe Zheng1, David M. Jones2, Carol Horzempa1, Aparna Prasad1, Paula J. McKeown-Longo1 ![]()
1. Center for Cell Biology and Cancer Research, Albany Medical College, Albany, New York, USA
2. Department of Pathology, Albany Medical College, Albany, New York, USA
Received 2011-8-4; Accepted 2011-9-23; Published 2011-9-27
Citation:
Zheng M, Jones DM, Horzempa C, Prasad A, McKeown-Longo PJ. The First Type III Domain of Fibronectin is Associated with the Expression of Cytokines within the Lung Tumor Microenvironment. J Cancer 2011; 2:478-483. doi:10.7150/jca.2.478. https://www.jcancer.org/v02p0478.htm
Other stylesAbstract
Recent studies have pointed to changes in tissue mechanics as a contributory element to the development of malignancies. Increased tissue rigidity is associated with the unfolding of the Type III domains of fibronectin within the extracellular matrix. The consequences of this unfolding on cellular functions within the lung are not well understood. In the present study, we evaluated the effect of a peptide representing a partially unfolded intermediate of the first Type III repeat of fibronectin (FnIII-1c) on inflammatory gene expression in adult human lung fibroblast cells. FnIII-1c induced expression of cytokines, CXCL1-3, IL-8 and TNF-α, by lung fibroblast cells. The increase in IL-8 expression was dependent on Toll-like receptor 2 and NFκB. Immunohistochemistry of tissue arrays representing squamous cell carcinoma of the lung revealed extensive stromal staining for IL-8 and fibronectin fibrils which were co-aligned with myofibroblasts. These data suggest a model in which unfolding of FnIII domains secondary to myofibroblast-generated tension may induce the release of cytokines by stromal fibroblasts present within the lung tumor.
Keywords: lung cancer, inflammation, fibronectin, cytokine
INTRODUCTION
Lung cancer is the leading cause of cancer-related death with a world-wide annual death rate of 1.3 million people (1). In spite of developments in surgical techniques, chemotherapy and radiation oncology, the overall 5 year survival rate (15%) has remained unchanged for decades. Inflammation mediated destruction of the extracellular matrix, such as that which occurs in chronic obstructive pulmonary disease (COPD), asthma, tuberculosis and emphysema, is associated with increased risk of lung cancer (2-4). The association between lung inflammatory disease and lung cancer has been recognized for several decades, however, the mechanisms underlying their linkage are not understood. Recent studies have suggested that chronic inflammation and extracellular matrix remodeling may provide a mechanistic link between COPD and the development of lung cancer (2).
Increased deposition of plasma fibronectin into the lung extracellular matrix is seen in lung cancer and has been implicated in promoting tumor cell proliferation, invasion and resistance to chemotherapy (5-10). Changes in the mechanical properties of lung tissue are seen in a number of disease states including cancer, COPD, asthma and emphysema, where changes in the remodeling and stiffening of the extracellular matrix accompany disease progression. A recent study of lung fibrosis in bleomycin treated mice, has shown that early fibrotic lesions in the lung are associated with large (as much as 30 fold) changes in the lung tissue stiffness. These large increases in tissue rigidity occur in focal areas in association with extracellular matrix remodeling and infiltration of myofibroblasts (11). The role of tissue stiffening in the initiation and progression of lung cancer has not been studied, but studies on breast cancer show that increased tissue stiffening is positively correlated with disease progression (12).
The secondary structure of the fibronectin molecule is organized into individually folded domains termed Types I, II and III. The Type III domains are folded into structurally stable beta-pleated sheets which unfold and refold in response to changes in intracellular tension. The biological role of many of these domains is not well understood, but accumulating evidence has indicated that the Type III domains of fibronectin present within the matrix become unfolded as the matrix becomes more rigid (13).
Steered molecular dynamics has been used to predict stable intermediate structures of partially unfolded fibronectin Type III domains. One such intermediate is recapitulated in a peptide comprised of the C-terminal two-thirds of the first FnIII domain, FnIII-1c (14). In the present study, we show that addition of FnIII-1c to adult lung fibroblasts causes a large increase in the expression of inflammatory cytokines including CXCL1-3 and IL-8. Staining of tissue microarrays of squamous cell carcinoma shows extensive myofibroblast infiltration into a fibronectin rich stroma coupled with increased expression of IL-8. Our studies are consistent with a model in which myofibroblast driven unfolding of the secondary structure of matrix fibronectin plays a role in the initiation and/or maintenance of chronic inflammation within the tumor microenvironment.
MATERIALS AND METHODS
Analysis of gene expression
Primary human adult lung fibroblasts were grown in lung/cardiac fibroblast growth medium (Cell Applications, Inc. San Diego, CA) and used from passage 3-6. In most experiments, cell monolayers were serum-starved overnight with serum-free medium (lung/cardiac fibroblast basal medium containing 0.1% BSA) before treatment. Recombinant His-tagged fibronectin modules, III-1c and III-13, were prepared as described previously (15). Levels of contaminating endotoxin (<0.25 units/nmole of protein) in recombinant proteins were determined using the limulus amebocyte lysate assay, QCL-1000 (Lonza, Walkersville, MD). Total RNA was isolated using RNAeasy Plus Mini Kit (Qiagen, Valencia, CA). The RT2 First Strand Kit was used to convert RNA into first strand cDNA. Mixtures of cDNA and SYBR® Green qPCR Mastermix were applied to the Human Inflammatory Response and Autoimmunity PCR Array Plates (SABiosciences, Frederick, MD). A MyiQ cycler system (Bio-Rad Laboratories) was used for real-time PCR detection. The data were analyzed using the Excel-based PCR array data analysis templates provided by the manufacturer. The inhibitors of NFκB signaling PS-1145 (Sigma-Aldrich) and BAY 11-7082 (Calbiochem) were dissolved in DMSO. IL-8 protein concentration was measured by ELISA (ELISA Set from BD Biosciences, San Diego, CA). Blocking antibodies to human TLR2 and TLR4 were from R&D Systems (Minneapolis, MN).
Tissue section staining
Non-small cell human lung carcinoma tissue with self-matching normal adjacent tissue microarray panels (US Biomax Inc, Rockville, MD), were processed for immuno-staining using peroxidase-based ABC system (Vector Laboratories, Burlingame, CA). The primary antibodies and their dilution were: 10 μg/ml IgG of anti-human FnIII-1 mouse monoclonal antibody 9D2, 25 μg/ml IgG of anti-human IL-8 mouse monoclonal antibody (R&D System, Minneapolis, MN ) or 1:1600 dilution ascites of anti-α smooth muscle actin mouse monoclonal antibody (A2547, Sigma, St. Louis, MO). Color was developed by reaction with 3,3'-Diaminobenzidine. Tissue sections were counterstained with hematoxylin.
RESULTS
FnIII-1c induces the expression of inflammatory cytokines in adult lung fibroblasts
To determine the effect of FnIII domain unfolding on gene expression, we incubated human adult lung fibroblast cells with the FnIII-1c peptide. This peptide recapitulates a stable intermediate structure which is formed when fibronectin undergoes unfolding in response to cellular force (14). After 2 hours of incubation with FnIII-1c, RNA was extracted from cells and gene expression was analyzed by microarray. As shown in Fig. 1A, several genes were upregulated in response to FnIII-1c and the five most highly upregulated genes (CXCL-1,2,3, IL-8 and TNF-α) are shown in Fig. 1B. IL-8, which exhibited over a 100 fold increase in expression in response to FnIII-1c was chosen for further characterization. The microarray results for IL-8 were confirmed by ELISA, which demonstrated a dose-dependent increase in the amount of IL-8 protein present in the conditioned medium of lung fibroblasts treated with FnIII-1c. The FnIII-13 domain served as control and had no effect on IL-8 expression (Fig. 1C).
FnIII-1 induction of IL-8 requires TLR-2 and NFκB
Expression of inflammatory cytokines is often under the control of the transcription factor, NFκB, which is translocated to the nucleus following activation. To determine whether FnIII-1c activated NFκB, nuclear lysates were prepared from lung fibroblasts treated with increasing doses of FnIII-1c. Western blot analysis of lysates indicated that within an hour of FnIII-1c treatment, there was a dose dependent increase in the accumulation of the p65/relA subunit of NFκB in the nucleus (Fig. 2A). To verify a role for NFkB in the induction of IL-8, cells were pretreated with two different inhibitors of NFκB activation, PS-1178 and BAY11-7072. Incubation of cells with FnIII-1c resulted in a large increase in the amount of IL-8 present in the conditioned medium when compared with the control, FnIII-13 (Fig. 2B). In the presence of either inhibitor, FnIII-1c induction of IL-8 was decreased by 85%, confirming a role for NFkB in the induction of IL-8 by FnIII-1c. Activation of NFκB can be regulated by the Toll-like family of receptors (TLRs), transmembrane receptors which regulate the innate immune system. To evaluate a role for TLRs in the induction of IL-8, cells were incubated with FnIII-1c in the presence of blocking antibody to either TLR2 or TLR4. Blocking antibody to TLR2 inhibited IL-8 expression in response to FnIII-1c (Fig. 2C). Neither control antibodies nor blocking antibody to TLR4 inhibited IL-8 secretion in response to FnIII-1c. Taken together, these experiments indicate that the induction of IL-8 expression in lung fibroblasts by FnIII-1c occurs through the TLR2 dependent activation of NFκB.
Fibronectin is preferentially expressed in the stroma of human squamous cell carcinoma
Tissue microarrays containing samples of squamous cell carcinoma of the lung were stained for fibronectin using the 9D2 monoclonal antibody to fibronectin which recognizes an epitope within the FnIII-1 domain (16). Fig. 3A shows heavy staining for fibronectin in two samples of squamous cell carcinoma (panels a and b) in the stroma (stroma) adjacent to the infiltrating border of carcinoma cells (tumor). The stroma show extensive fibrillar staining for fibronectin with aligned fibroblasts (F) and infiltrating inflammatory cells(I). There was also light to moderate diffuse staining for fibronectin within the tumor. Staining for fibronectin in normal lung tissue was less pronounced (panel c).
Figure 1
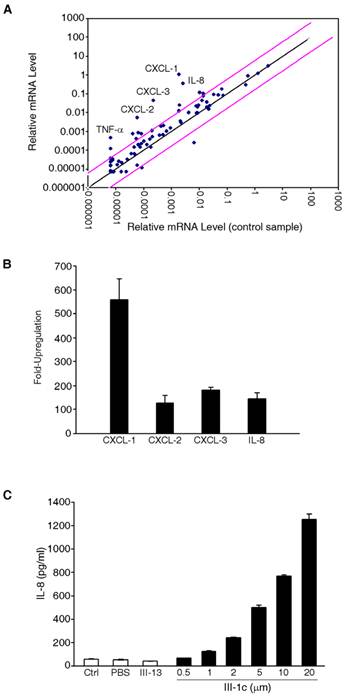
FnIII-1c induces expression of inflammatory genes in lung fibroblasts. (A) Monolayers of adult lung human fibroblasts were treated with 10 μM FnIII-1c or PBS in serum-free culture medium supplemented with 0.1% BSA for 2 hrs. Expression profiling of genes was performed using the Human Inflammatory Response PCR Array. Pink lines indicate a 5-fold change in baseline. Five inflammatory genes (labeled) were highly upregulated in response to FnIII-1c. (B) The fold induction of inflammatory genes by FnIII-1c is shown. Values represent the average fold increase over housekeeping genes from 3 separate experiments. (C) Human adult lung fibroblasts were treated with increasing concentrations of FnIII-1c or FnIII-13 for 4 hrs. IL-8 concentration in the culture medium was determined by ELISA. Error bars represent mean ± SD.
Figure 2
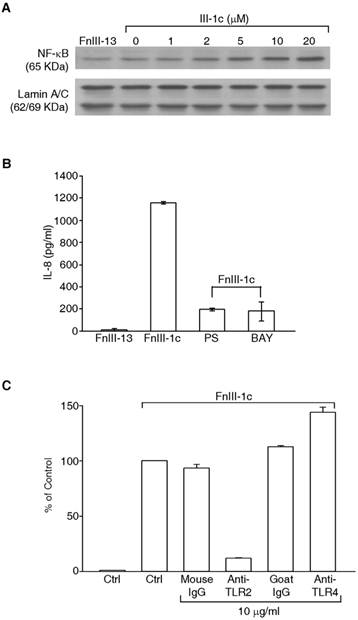
FnIII-1c induces TLR2-dependent expression of IL-8 in human adult lung fibroblasts. (A) Human adult lung fibroblasts were incubated with the indicated concentrations of FnIII-1c with PBS for 1 hr. The nuclear fraction was isolated and analyzed by immunoblotting for the presence of the NFκB protein, p65/rel A. Nuclear lamin A/C served as a loading control. (B) Cell were pretreated for 2 hrs with inhibitors of NFκB signaling, 10 μM of BAY11-7082 or 10 μM of PS-1145. Cell then received 10 μM FnIII-1c for FnIII-13 for an additional 4 hrs. Conditioned medium from cells was collected and IL-8 was measured by ELISA. (C) Cells were pretreated with blocking antibodies to human TLR2, TLR4, or control IgG for 1 hr prior to the addition of 10 μM FnIII-1c for 4 hrs. IL-8 present in the conditioned medium was analyzed by ELISA. Positive control was set as 100%. Error bars represent mean ± SD.
The stroma of human lung tumors is enriched for myofibroblasts and IL-8
Figure 3B shows sequential sections of a squamous cell carcinoma of the lung which have been stained for either smooth muscle cell actin (panel a) or IL-8 (panel b). The tumor stroma (stroma) stained heavily for smooth muscle actin and the staining pattern appeared fibrillar, similar to that seen with fibronectin (Fig. 3A), indicating that the fibroblasts aligned along the fibronectin fibrils are myofibroblasts. IL-8 staining was present in both the stroma and the carcinoma regions of the tumor (panel b). Control staining was negative (panel c). These data indicate that in squamous cell carcinoma both fibronectin and IL-8 are heavily expressed in the tumor stroma and that the increased fibronectin within the tumor stroma is associated with an influx of myofibroblasts which are aligned along fibronectin fibers. These data are consistent with a model in which myofibroblast contractility dependent remodeling of fibronectin matrix initiates the release of inflammatory cytokines within the lung tumor microenvironment.
DISCUSSION
Chronic inflammation and fibrotic disease are believed to be contributing factors in the progression of lung cancer in high-risk patients (2,3). Our data are consistent with a model in which changes in the secondary structure of matrix fibronectin driven by increased rigidity of lung tissue is part of a feed forward mechanism controlling the release of inflammatory mediators into the lung microenvironment. The cytokines released in response to FnIII-1c, IL-8, CXCL1, 2, and 3, are CXCR2 ligands which promote both inflammation and angiogenesis through the recruitment of inflammatory cells and the mobilization of bone marrow endothelial cells (17,18). Our data suggest that unfolding of the FnIII-1 domain within fibronectin present in the tumor stroma promotes tumor progression through the release of cytokines from resident lung fibroblasts.
The FnIII-1c peptide used in this study was originally called anastellin and shown to have anti-angiogenic and anti-tumor properties (19). We subsequently showed that FnIII-1c had inhibitory effects on signaling pathways regulating endothelial microvessel cell proliferation (20,21). In contrast, when incubated with fibroblast cells, FnIII-1c had little effect on growth and instead activated signaling pathways leading to the release of cytokines, which are known to be proangiogenic (20,22). These data suggest that the FnIII-1 domain of fibronectin may have both pro- and anti-angiogenic roles within the tumor microenvironment.
Figure 3
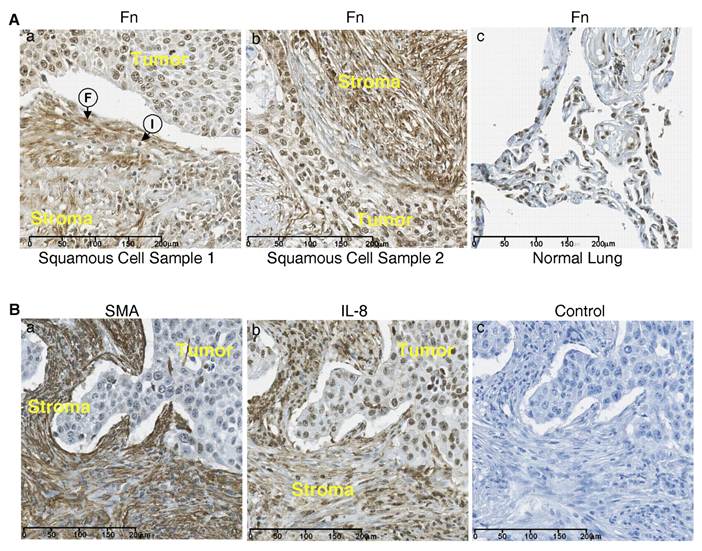
The stroma of squamous cell carcinoma is enriched in fibronectin. (A) Tissue sections from two lung squamous cell carcinomas (panels a and b) and a section of normal lung tissue (panel c) were stained for fibronectin . Fibroblasts (F) and Immune cells (I) are present. (B) Sequential sections of squamous cell carcinoma were stained for IL-8 (panel a) and smooth muscle actin (SMA) (panel b). Control tissue stained with only secondary antibody (panel c).
Although the function of TLRs in the regulation of inflammation in response to bacterial pathogens has been recognized for many years, more recent studies have pointed to a role of TLRs in mediating non-septic inflammatory responses. It is now becoming clear that TLRs may play a role in the initiation and/or maintenance of inflammatory processes during organ dysfunction and tissue injury, thereby contributing to chronic illness. Both TLR2 and TLR4 are expressed in resident lung cells and both TLR2 and TLR4 have been reported to be activated through endogenous molecules including fragments of matrix proteins, such as hyaluronic acid (HA), the EDA domain of fibronectin (FnEDA) as well as heat shock proteins and β-amyloid (23,24). Accumulating evidence indicates that TLR signaling is exceedingly complex and the elicited immune response is both ligand- and cell-type specific (25).
We have shown in a previous study, that addition of Fn III-1c to human dermal fibroblasts results in the NFkB dependent release of inflammatory mediators, including IL-8 (22). In dermal fibroblasts, induction of cytokine expression was dependent on TLR4 rather than TLR2. The basis for the involvement of distinct TLR receptors in the two cell types is not known. The ability of FnIII-1c to signal through either TLR2 or TLR4 suggests that FNnIII-1c does not bind directly to TLRs to initiate signaling, but instead interacts with TLR signaling complexes by means of a co-receptor or ancillary binding protein.
The assembly of TLR containing signaling complexes which differentially function to regulate unique patterns of cytokine expression suggests that it may be possible to selectively target arms of TLR signaling pathways to treat disease without sacrificing pathways controlling host-defense. Our data suggest that force-dependent remodeling of the fibronectin matrix promotes inflammation with the lung. The identification of mechanically sensitive response elements within the fibronectin matrix, and their associated signaling pathways will provide novel targets for the treatment of chronic lung inflammation and may also represent a unique opportunity for developing therapies directed at early stage intervention in that population (i.e., former smokers, patients with COPD) who are at high risk for the development of lung cancer.
Acknowledgements
This study was supported by grant W81XWH-10-1-0755 from the DoD Lung Cancer Research Program of the Office of the Congressionally-Directed Medical Research Program and by grant CA-069612 from the National Institutes of Health.
CONFLICT OF INTEREST
The authors have declared that no conflict of interest exists.
References
1. Jemal A, Siegl R, Ward E, Hao Y, Xu J, Murray T. et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58(2):71-96
2. Houghton AM, Mouded M, Shapiro SD. Common origins of lung cancer and COPD. Nature Med. 2008;14:1023-4
3. Mackinnon AC, Kopatz J, Sethi T. The molecular and cellular biology of lung cancer: identifying novel therapeutic strategies. Brit Med Bulletin. 2010;95(47):61
4. Lee G, Walser TC, Dubinett SM. Chronic inflammation, chronic obstructive pulmonary disease, and lung cancer. Current Opin Pulmon Med. 2009;15:303-7
5. Liu G, Meng X, Jih Y, Bai J, Zhao Y, Cui X. et al. Inhibitory role of focal adhesion kinase on anoikis in the lung cancer cell A549. Cell Biol Int. 2008;32:663-70
6. Jia D, Yan M, Wang X, Liang L, Liu L, Kong H. et al. Development of a highly metastatic model that reveals a crucial role of fibronectin in lung cancer cell migration and invasion. BMC Cancer. 2010;10:364
7. Malik G, Knowles LM, Dhir R, Xu S, Yang S, Ruoslahti E. et al. Plasma fibronectin promotes lung metastasis by contributions to fibrin clots and tumor cell invasion. Cancer Res. 2010;70:4327-34
8. Roman J, Rizenthaler JD, Roser-Page S, Sun X, Han S. α5β1-integrin expression is essential for tumor progression in experimental lung cancer. Am J Respir Cell Mol Biol. 2010;43:684-91
9. Zheng Y, Ritzenthaler JD, Roman J, Han S. Nicotine stimulates human lung cancer cell growth by inducing fibronectin expression. Am J Respir Cell Mol Biol. 2007;376:681-90
10. Hartmann TN, Burger JA, Glodek A, Fujii N, Burger M. CXCR4 chemokine receptor and integrin signaling co-operate in mediating adhesion and chemoresistance in small cell lung cancer (SCLC) cells. Oncogene. 2005;24:4462-71
11. Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM. et al. Feedback amplifications of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol. 2010;190:693-708
12. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT. et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891-906
13. Antia M, Baneys G, Kubow KE, Vogel V. Fibronectin in aging extracellular matrix fibrils is progressively unfolded by cells and elicits an enhanced rigidity response. Farady Discuss. 2008;139:229-49
14. Gao M, Craig D, Lequin O, Campbell ID, Vogel V, Schulten K. Structure and functional significance of mechanically unfolded fibronectin type III1 intermediates. Proc Natl Acad Sci. 2003;100:14784-9
15. Klein RM, Zheng M, Ambesi A, van de Water L, McKeown-Longo PJ. Stimulation of extracellular matrix remodeling by the first type III repeat in fibronectin. J Cell Sci. 2003;116:4663-74
16. Gui L, Wojciechowski K, Gildner CD, Nedelkovska H, Hocking DC. Identification of the heparin-binding determinants within fibronectin repeat III1: role in cell spreading and growth. J Biol Chem. 2006;281:34816-25
17. Raman D, Baugher PJ, Thu YM, Richmond A. Role of chemokines in tumor growth. Cancer Lett. 2007;256:137-65
18. Jones CP, Pitchford SC, Lloyd CM, Rankin SM. CXCR2 mediates the recruitment of endothelial progenitor cells during allergic airways remodeling. Stem Cells. 2009;27:3074-81
19. Yi M, Ruoslahti E. A fibronectin fragment inhibits tumor growth, angiogenesis, and metastasis. Proc Natl Acad Sci. 2001;98:620-4
20. Ambesi A, Klein RM, Pumiglia KM, McKeown-Longo PJ. Anastellin, a fragment of the first type III repeat of fibronectin, inhibits extracellular signal-regulated kinase and causes G1 arrest in human microvessel endothelial cells. Cancer Res. 2005;65:148-56
21. Ambesi A, McKeown-Longo PJ. Anastellin, the angiostatic fibronectin peptide, is a selective inhibitor of lysophospholipid signaling. Mol Cancer Res. 2009;7(2):255-65
22. You R, Zheng M, McKeown-Longo PJ. The first type III repeat in fibronectin activates an inflammatory pathway in dermal fibroblasts. J Biol Chem. 2010;285:36255-9
23. Lorne E, Dupont H, Abraham E. Toll-like receptors 2 and 4: initiators of non-septic inflammation in critical care medicine? Inten Care Med. 2010;36:1826-35
24. Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A. et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155-61
25. Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators of Inflammation. 2010 Epub
Author contact
![]() Corresponding author: Paula J. McKeown-Longo, Ph.D., Center for Cell Biology and Cancer Research, Mail Code 165, Albany Medical College, 47 New Scotland Avenue, Albany, NY 12208. Telephone: (518) 265-5651; Fax: (518) 262-5669; E-mail: mckeowpamc.edu
Corresponding author: Paula J. McKeown-Longo, Ph.D., Center for Cell Biology and Cancer Research, Mail Code 165, Albany Medical College, 47 New Scotland Avenue, Albany, NY 12208. Telephone: (518) 265-5651; Fax: (518) 262-5669; E-mail: mckeowpamc.edu
Citation styles
APA
Zheng, M., Jones, D.M., Horzempa, C., Prasad, A., McKeown-Longo, P.J. (2011). The First Type III Domain of Fibronectin is Associated with the Expression of Cytokines within the Lung Tumor Microenvironment. Journal of Cancer, 2, 478-483. https://doi.org/10.7150/jca.2.478.
ACS
Zheng, M.; Jones, D.M.; Horzempa, C.; Prasad, A.; McKeown-Longo, P.J. The First Type III Domain of Fibronectin is Associated with the Expression of Cytokines within the Lung Tumor Microenvironment. J. Cancer 2011, 2, 478-483. DOI: 10.7150/jca.2.478.
NLM
Zheng M, Jones DM, Horzempa C, Prasad A, McKeown-Longo PJ. The First Type III Domain of Fibronectin is Associated with the Expression of Cytokines within the Lung Tumor Microenvironment. J Cancer 2011; 2:478-483. doi:10.7150/jca.2.478. https://www.jcancer.org/v02p0478.htm
CSE
Zheng M, Jones DM, Horzempa C, Prasad A, McKeown-Longo PJ. 2011. The First Type III Domain of Fibronectin is Associated with the Expression of Cytokines within the Lung Tumor Microenvironment. J Cancer. 2:478-483.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.