Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2014; 5(5):336-343. doi:10.7150/jca.7202 This issue Cite
Research Paper
Identification of a Novel Compound That Suppresses Breast Cancer Invasiveness by Inhibiting Transforming Growth Factor-β Signaling via Estrogen Receptor α
Natsuka Goto1*, Hiromi Hiyoshi1,2* ![]() , Ichiaki Ito1, Keisuke Iida 3, Yuka Nakajima1,2, Kazuo Nagasawa3, Junn Yanagisawa1,2
, Ichiaki Ito1, Keisuke Iida 3, Yuka Nakajima1,2, Kazuo Nagasawa3, Junn Yanagisawa1,2 ![]()
1. Graduate School of Life and Environmental Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba 305-8577, Japan.
2. Center for Tsukuba Advanced Research Alliance, University of Tsukuba, 1-1-1 Tennodai, Tsukuba 305-8577, Japan.
3. Faculty of Technology, Tokyo University of Agriculture and Technology-TUAT, 2-24-16 Naka-cho, Koganei-shi, Tokyo 185-0031, Japan.
* equal contribution.
Received 2013-7-19; Accepted 2014-2-16; Published 2014-3-23
Citation:
Goto N, Hiyoshi H, Ito I, Iida K, Nakajima Y, Nagasawa K, Yanagisawa J. Identification of a Novel Compound That Suppresses Breast Cancer Invasiveness by Inhibiting Transforming Growth Factor-β Signaling via Estrogen Receptor α. J Cancer 2014; 5(5):336-343. doi:10.7150/jca.7202. https://www.jcancer.org/v05p0336.htm
Other stylesAbstract
Breast cancer is the most frequently diagnosed cancer and the leading cause of death by cancer among females worldwide. An overwhelming majority of these deaths is because of metastasis. Estrogen stimulates and promotes growth of breast tumors, whereas transforming growth factor-beta (TGF-β) signaling promotes invasion and metastasis. We previously reported that estrogen and estrogen receptor alpha (ERα) suppressed breast cancer metastasis by inhibiting TGF-β signaling, whereas antiestrogens that suppress breast cancer growth, such as the selective ER modulator tamoxifen (TAM) or the pure antiestrogen fulvestrant (ICI 182,780), cannot suppress TGF-β signaling or breast cancer invasiveness. Therefore, we predicted that a compound that inhibits TGF-β signaling but does not facilitate ERα signaling would be ideal for suppressing breast cancer invasiveness and growth. In the present study, we identified an ideal candidate compound, N-23. Like estrogen, N-23 strongly decreased expression of TGF-β/Smad target gene plasminogen activator inhibitor-1 (PAI-1), but it did not increase the expression of ERα target gene pS2. While estrogen decreased the levels of phosphorylated Smad2 and Smad3, N-23 had no effect. In addition, TGF-β-dependent recruitment of Smad3 to the PAI-1 gene promoter was inhibited in the presence of estrogen or N-23. We also investigated the effects of N-23 on proliferation, migration, and invasion of breast cancer cells. In contrast to estrogen, N-23 inhibited the cellular proliferation of breast cancer cells. Moreover, we showed that N-23 suppressed the migration and invasion of breast cancer cells to the same extent as by estrogen. Taken together, our findings indicate that N-23 may be a candidate compound that is effective in inhibiting breast cancer progression.
Keywords: TGF-β, ERα, breast cancer, compounds, invasion and metastasis.
Introduction
Breast cancer is the most frequently diagnosed cancer and the leading cause of death by cancer among females worldwide. Breast cancer accounts for 23% (1.38 million) of all new cancer cases and 14% (458,400) of all deaths by cancer [1]. Breast cancer is typically hormone-dependent; exposure to estrogen promotes breast cancer cell growth and proliferation. The effects of estrogen are mediated by the binding of estrogen receptors (ER), ERα and ERβ, which are members of the nuclear receptor superfamily that function as ligand-induced transcription factors [2, 3]. ERα is the major ER subtype in mammary epithelium, and it plays an important role in breast cancer progression [4, 5].
Upon estrogen binding, ligand-activated ERα binds to the estrogen responsive element (ERE) in the target gene promoter and stimulates gene expression [6-8]. Because estrogen plays a significant role in the stimulation and growth of breast cancer cells, antiestrogens such as the selective ER modulator (SERM) including tamoxifen (TAM), or fulvestrant [ICI 182,780 (ICI)] are used in endocrine therapy for ER-positive breast cancer [9]. TAM has both estrogen agonist and antagonist effects that are specific for its target tissue. In breast tissue, TAM acts primarily as an estrogen antagonist. Unlike TAM, ICI is a pure ER antagonist that does not exhibit agonist effects and downregulates ER protein [10].
The transforming growth factor-beta (TGF-β) superfamily is a large, evolutionarily conserved family of secreted multifunctional peptides involved in almost every aspect of cellular behavior [11]. TGF-β binding to TGF-β receptors triggers phosphorylation of Smad family members [12, 13]. Smad2 and Smad3 are both receptor-activated Smads (R-Smad), whereas Smad4 serves as a common partner (Co-Smad) for all R-Smads [14]. The overwhelming majority of deaths due to cancer are because of metastasis [15], and several studies have demonstrated that TGF-β/Smad pathway plays a crucial role in cancer metastasis [16, 17]. These studies suggest that regulation of TGF-β signaling is important for breast cancer therapy.
In our previous study, we found that ERα inhibited TGF-β/Smad signaling by promoting degradation of phosphorylated Smads (pSmad) in an estrogen-dependent manner [18]. In addition, we provided evidence that estrogen and ERα suppress breast cancer metastasis by inhibiting TGF-β signaling [19]. Thus, it is suggested that estrogen has an inhibitory effect on breast cancer invasiveness. However, estrogen enhances breast cancer growth by promoting ERα signaling [20]. Thus, estrogen also has promotive effects on breast cancer cell proliferation. In contrast, antiestrogens have negative effects on breast cancer growth [20, 21], but do not inhibit TGF-β signaling or breast cancer invasiveness [19]. Based on these findings, we consider that a compound that inhibits TGF-β signaling without enhancing ERα signaling is ideal for inhibiting breast cancer progression.
In the present study, we screened for compounds that demonstrated such activity and then identified N-23 as an ideal compound. We demonstrated that the inhibitory effect of N-23 on TGF-β signaling is mediated by ERα. In addition, we showed that N-23 suppresses TGF-β signaling by inhibiting TGF-β-dependent recruitment of Smad3 to the TGF-β target gene promoter. Moreover, N-23 suppresses the proliferation and invasiveness of breast cancer cells. Our observations suggest that N-23 is a candidate compound that is effective for the inhibition of breast cancer progression.
Materials and Methods
Cell culture. Highly migratory MCF-7 cell line termed MCF-7-M5 was generated previously [19]. MCF-7 and MCF-7-M5 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS).
Real-time RT-PCR. Real-time reverse transcription-PCR was performed essentially as described previously [22]. Cells were homogenized in 1 ml of Sepazol and total RNA was extracted according to the manufacturer's instructions (Nacalai tesque). cDNA was synthesized from total RNA using RevatraAce reverse transcriptase (TOYOBO) and oligo dT primer. Real-time PCRs were performed to amplify fragments representing for the indicated mRNA expression using the Thermal Cycle DiceTM TP800 (TaKaRa) and SYBR Premix Ex TaqTM (TaKaRa). The primer sequences are as follows: pS2 forward primer: 5'-TGCTGTTTCGACGACACCGTT-3'; reverse primer: 5'-AGGCAGATCCCTGCAGAAGT-3', PAI-1 forward primer: 5'-GCAGGACATCCGGGAGAGA-3'; reverse primer: 5'-CCAATAGCCTTGGCCTGAGA-3'.
RNA interference. For transfection of siRNAs, cells at 30 to 50% confluency were transfected with 20 nM of siRNA using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocol. The siRNA duplexes human ERα, 5'-GCUACUGUGCAGUGUGCAAUGACUA-3'. StealthTM RNAi negative control Med GC (Invitrogen) was used as a negative control.
Western blotting. Cells were lysed in TNE buffer [10 mM Tris-HCl (pH 7.8), 1% Nonidet P-40 (NP-40), 0.15 M NaCl, and 1 mM ethylenediaminetetraacetic acid (EDTA)], and then immunoblotted with the appopriate antibodies. The antibodies used in this study were: anti-Smad2/3 (BD), Smad3 (Abcam), phospho-Smad2 (Cell Signaling), phospho-Smad3 (BIOSOURCE) and β-actin (Sigma) antibodies. Specific proteins were visualized using an enhanced chemiluminescence (ECL) Western blot detection system (Millipore).
Chromatin immunoprecipitation (ChIP) assay. ChIP assay was performed as previously described [23]. Nuclear proteins were cross-linked to genomic DNA by adding formaldehyde and then cross-linking was stopped by adding glycin. Cells were washed with PBS and harvested by scraping. After centrifugation, the cell pellets were resuspended in SDS lysis buffer [1% SDS, 10 mM EDTA, protease inhibitors (NacalaiTesque) and 50 mM Tris-HCl (pH 8.1)] and the lysates were sonicated to result in DNA fragments of 300 to 1,000 bp in length. After centrifugation and removal of aliquot (whole-cell extract) as input sample, supernatants were diluted with ChIP dilution buffer [0.01% SDS, 1.1% TritonX-100, 1.2 mM EDTA, 16.7 mM NaCl, protease inhibitors and 16.7 mM Tris-HCl (pH 8.1)]. Samples were incubated with normal rabbit IgG or anti-Smad3 antibody (Abcam) overnight at 4°C with rotation. The immuno-complexes were collected with protein G Sepharose beads (GE healthcare). The beads were washed with the following buffers: low salt wash buffer [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl and 20 mM Tris-HCl (pH 8.1)], high salt wash buffer [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 500 mM NaCl and 20 mM Tris-HCl (pH 8.1)] and LiCl wash buffer [0.25 mM LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA and 10 mM Tris-HCl (pH 8.1)]. Finally, the beads were washed with TE buffer [1 mM EDTA and 10 mM Tris-HCl (pH 8.0)]. The immuno-complexes were then eluted with elution buffer (1% SDS, 10 mM DTT and 100 mM NaHCO3). After centrifugation, the supernatant was collected and the cross-linking was reversed by adding NaCl. The remaining proteins were digested by proteinase K. The DNA was phenol precipitated, ethanol precipitated, and measured by real-time PCR. The primers for real-time PCR are as follows: forward primer: 5'-GCAGGACATCCGGGAGAGA-3'; reverse primer: 5'-CCAATAGCCTTGGCCTGAGA-3' for PAI-1 gene upstream region.
Proliferation assay. Cells (5 × 104) were seeded in phenol red-free DMEM containing 4% charcoal-stripped FBS. After 24 h (day 0), the medium was exchanged for phenol red-free DMEM containing 1% charcoal-stripped FBS with dimethyl sulfoxide (DMSO) (Vehicle) or estrogen (10-8 M) or N-23 (10-6 M). At day 1, 3 and 5, cells were trypsinized and treated with 0.2% trypan blue. Viable cells were counted using Countess Automated Cell Counter (Invitrogen). At day2 and 4, the medium was exchanged for phenol red-free DMEM containing 1% charcoal-stripped FBS with ligands.
Tumor xenograft models. Female BALB/c nu/nu mice at 5 weeks of age were purchased from CLEA Japan. The mice were kept in a pathogen-free environment under controlled conditions of light and humidity. MCF-7 cells were cultured as monolayers, trypsinized and resuspended in Matrigel at 1.0 × 108 cells per ml. Each mouse was injected subcutaneously with 100 μl of cell suspension (1 × 107 cells) in both flanks. N-23 was subcutaneously injected in the scruff of the neck (45 mg/kg) for every 2 days. Tumor growth was monitored twice each week by measuring the tumor size using calipers; tumor volume was determined using the formula V = 1/2 × larger diameter × (smaller diameter)2. At day 63, tumors were excised, weighed and fixed with 20% formalin. All animal experiments were performed in accordance with institutional guidelines.
Invasion and migration assay. The invasive potential of MCF-7-M5 cells was tested with Matrigel invasion chambers (24-well format, 8 μm pore size; BD Biosciences). After incubation in DMEM containing 4% charcoal-stripped FBS for 24 h, cells were incubated with vehicle alone (DMSO) or estrogen (10-8 M) or N-23 (10-6 M), and transferred into insert chambers for 36 h. After incubation, the cells on the upper surface of the filter were removed, and invading cells were fixed in methanol. Fixed cells were stained with crystal violet and counted under a microscope. Migration assays were performed using the same procedure, except that the insert chambers were not coated with Matrigel and cells in chamber were incubated for 24 h.
Statistical analysis. Significance of differences was determined by Student t-test analyses.
Results
Screening for compounds that do not enhance ERα signaling but inhibit TGF-β signaling
We previously reported that ERα and estrogen suppress breast cancer metastasis by inhibiting TGF-β/Smad signaling via a non-genomic pathway [18]. According to these findings, it was shown that estrogen suppresses breast cancer invasiveness. However, estrogen promotes breast cancer growth by facilitating the ERα pathway [20]. In contrast, antiestrogens, such as TAM or ICI, have suppressive effects on breast cancer tumorigenesis [20, 21], but do not inhibit either TGF-β signaling or breast cancer invasiveness [19]. Based on these reports, we predicted that a compound that does not enhance ERα signaling but inhibits TGF-β signaling would be effective in inhibiting breast cancer progression. Therefore, we screened for compounds that do not enhance ERα signaling but inhibit TGF-β signaling.
Screening was performed using luciferase reporter assays with reporter plasmids containing 3 × EREs or TGF-β responsive elements (9 × CAGA). Through this screening, we identified N-1 as a candidate compound (Fig. 1A). To confirm the effects of N-1 on ERα transcriptional activity and TGF-β signaling, we examined the mRNA levels of the ERα target gene pS2 and the TGF-β/Smad target gene plasminogen activator inhibitor-1 (PAI-1) (Fig. 1B and C). Estrogen increased pS2 expression, whereas N-1 did not affect it with or without estrogen (Fig. 1B and Supplementary Figure 1A). PAI-1 expression was increased with TGF-β treatment, but this was attenuated by estrogen or N-1 treatment (Fig. 1C). These results indicate that N-1 does not enhance the ERα pathway but inhibits TGF-β signaling.
Identification of the compound N-23, which does not enhance ERα signaling and inhibits TGF-β signaling through ERα
Although N-1 can suppress TGF-β signaling, its inhibition of PAI-1 expression was weak compared with that of estrogen (Fig. 1C). Therefore, we synthesized new N-1 derivatives (N-20, N-23, N-26, and N-27; Fig. 2A) and investigated the effects of these N-1 derivatives on pS2 and PAI-1 expression (Fig. 2B and C). We observed that N-1 derivatives showed various effects on pS2 and PAI-1 expression (Fig. 2B and C). Only N-20 increased pS2 expression (Fig. 2B), whereas TGF-β-induced expression of PAI-1 was strongly attenuated by N-20 and N-23, as compared with N-1, N-26, and N-27 (Fig. 2C). These results suggest that only N-23 strongly inhibits TGF-β signaling without enhancing ERα signaling (Fig. 2B, C and Supplementary Figure 1B). Based on these results, we identified N-23 as an ideal compound.
The inhibition of TGF-β signaling by estrogen is mediated through ERα via a non-genomic function [18]. Therefore, we examined whether the inhibitory effect of N-23 on TGF-β signaling was mediated via ERα by knocking down ERα with siRNA (Fig. 2D). We observed that knockdown of ERα expression in MCF-7 human breast cancer cells abolished the inhibitory effect of N-23 on PAI-1 expression (Fig. 2E). These data suggest that N-23 does not enhance the ERα pathway but strongly suppresses TGF-β signaling via ERα.
Figure 1
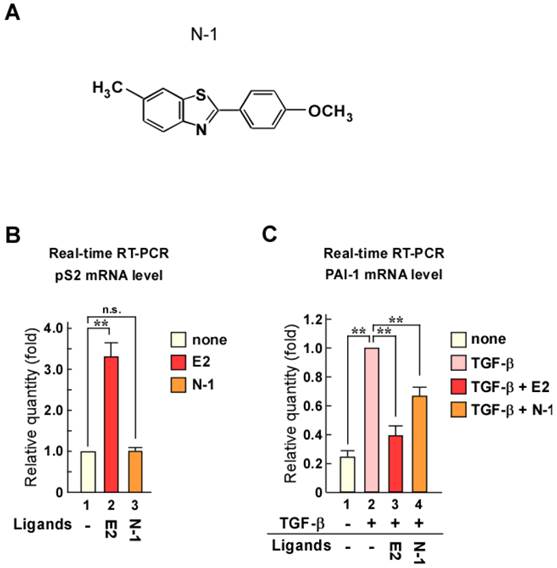
Identification of a compound that does not enhance ERα signaling but inhibits TGF-β signaling. (A) Structure of the candidate compound N-1. (B, C) N-1 does not enhance ERα pathway but suppresses TGF-β signaling. MCF-7 cells were cultured in the absence or presence of TGF-β (1 ng/mL), estrogen (E2, 10-8 M), or N-1 (10-6 M). The mRNA levels of pS2 (B) and PAI-1 (C) were quantified by real-time RT-PCR. ** indicates p < 0.01 and n.s. indicates p > 0.05.
Figure 2
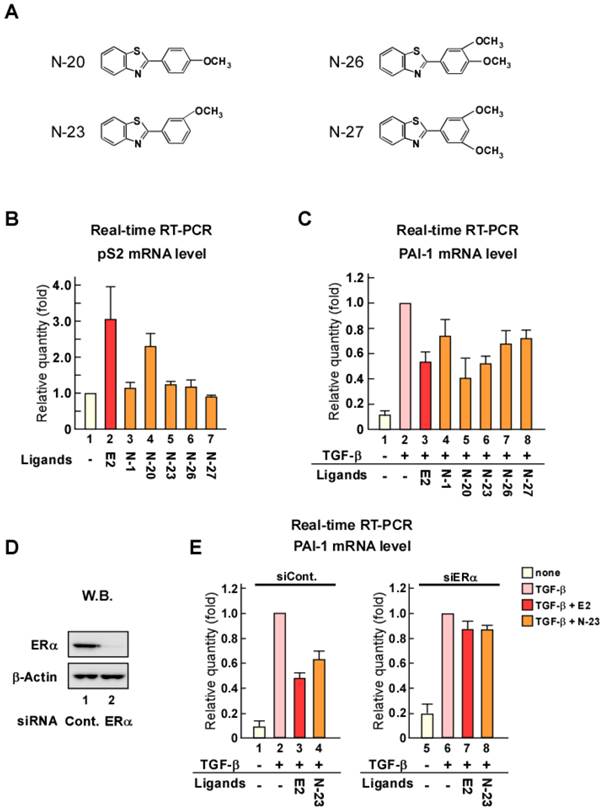
N-23 does not facilitate ERα signaling but strongly suppresses TGF-β signaling as compared with N-1. (A) Structures of N-1 derivatives. (B, C) Effects of N-1 derivatives on ERα and TGF-β signaling. MCF-7 cells were cultured in the absence or presence of TGF-β (1 ng/mL), E2 (10-8 M), N-1 (10-6 M), N-20 (10-6 M), N-23 (10-6 M), N-26 (10-6 M), or N-27 (10-6 M). The mRNA levels of pS2 (B) and PAI-1 (C) were quantified using real-time RT-PCR. (D) ERα is knocked down by treatment with siRNA targeting ERα. MCF-7 cells were transfected with siRNA targeting ERα. Western blotting was used to examine the levels of ERα and β-Actin. (E) Knocking down ERα expression eliminates the effects of N-23 on TGF-β-induced PAI-1 expression. ERα expression was knocked down in MCF-7 cells cultured in the presence or absence of TGF-β (1 ng/mL), E2 (10-8 M), or N-23 (10-6 M). The mRNA level of PAI-1 was quantified using real-time RT-PCR.
N-23 suppresses TGF-β signaling by inhibiting TGF-β-dependent recruitment of Smad3 to the PAI-1 promoter
When TGF-β binds to its cell surface receptors, the activated receptors phosphorylate Smad2 and Smad3, which are key transducers of TGF-β signaling. We previously reported that ERα forms a protein complex with Smad and Smurf, a ubiquitin ligase for Smad [18]. In addition, we demonstrated that Smad ubiquitination and subsequent degradation by ERα are enhanced in an estrogen-dependent manner that results in inhibition of TGF-β signaling. Therefore, to explore the mechanism underlying the inhibition of TGF-β signaling by N-23, we evaluated the effect of N-23 on Smad and pSmad protein levels using Western blotting (Fig. 3A). Total Smad and pSmad protein levels were reduced by estrogen as in our previous study, whereas N-23 had no effect (Fig. 3A, lanes 3 and 4). These results indicate that the inhibitory effect of N-23 on TGF-β signaling is not mediated by Smad or pSmad degradation, unlike estrogen. Next, we examined Smad3 recruitment to the promoter region in the PAI-1 gene since Smad3 has DNA-binding ability [24]. Chromatin immunoprecipitation (ChIP) assay demonstrated that the interaction between Smad3 and the PAI-1 promoter is potentiated by TGF-β, and Smad3 recruitment was suppressed in the presence of estrogen or N-23 (Fig. 3B). These results indicate that N-23 suppresses TGF-β signaling by inhibiting TGF-β-dependent recruitment of Smad3 to the PAI-1 promoter.
N-23 suppresses the proliferation and invasiveness of breast cancer cells
As previously mentioned, estrogen/ERα enhances cellular growth and breast cancer tumorigenesis [20], whereas TGF-β/Smad signaling facilitates metastasis [16, 17]. We showed that N-23 did not enhance the ERα pathway, but strongly inhibited TGF-β signaling. Therefore, we investigated the effects of N-23 on breast cancer cell proliferation. Consistent with previous reports, estrogen promoted proliferation of MCF-7 breast cancer cells (Fig. 4A). In contrast, N-23 inhibited cellular growth (Fig. 4A). We also showed that N-23 suppressed breast cancer tumor growth using mouse xenograft models (Fig. 4B and C). We then examined whether N-23 suppressed breast cancer migration and invasion using the highly migratory MCF-7 cell line MCF-7-M5, which we previously generated [19]. In a transwell migration assay, N-23 suppressed migration of MCF-7-M5 cells to the same extent as estrogen (Fig. 4D). Consistent with this result, the invasive potential of breast cancer cells was suppressed by both estrogen and N-23 (Fig. 4E). These results indicate that N-23 suppresses breast cancer cell proliferation and invasiveness.
Discussion
Approximately two-thirds of all breast cancers express ER. Therefore, ER expression is predictive of response to endocrine therapy to prevent estrogen stimulation for proliferation. To antagonize ER signaling, ICI and SERMs including TAM are used in breast cancer therapy. SERMs have been used for several years as the first-line hormonal therapy for postmenopausal patients. Furthermore, TAM is an effective treatment for all stages of hormone-responsive breast cancer and can prevent breast cancer in high-risk women [25]. Nevertheless, breast cancer is still the leading cause of death by cancer in females all over the world. Majority of deaths are because of metastasis in breast cancer patients as well as in other cancer patients.
Figure 3
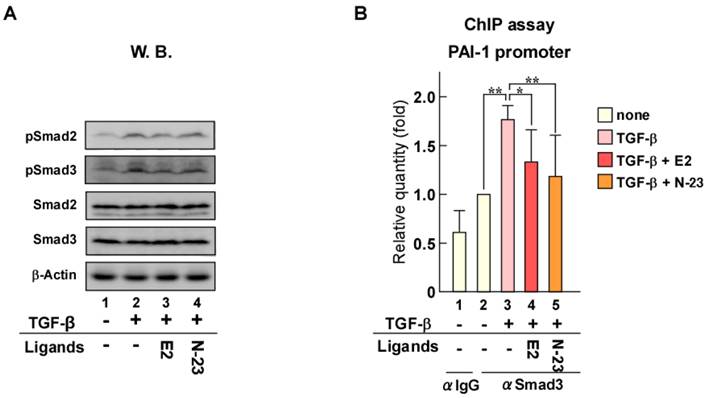
N-23 suppresses TGF-β signaling by inhibiting TGF-β-dependent recruitment of Smad3 to the PAI-1 promoter. (A) N-23 does not affect pSmad protein levels. MCF-7 cells were treated with or without TGF-β (1 ng/mL), E2 (10-8 M), or N-23 (10-6 M). Western blotting was used to examine the levels of pSmad2, pSmad3, Smad2, Smad3, and β-Actin. (B) N-23 inhibits TGF-β-dependent recruitment of Smad3 to the PAI-1 promoter. MCF-7 cells were cultured in the presence or absence of TGF-β (1 ng/mL), E2 (10-8 M), or N-23 (10-6 M). A ChIP assay was performed with control IgG or anti-Smad3 antibodies. Immunoprecipitated DNA was examined using real-time RT-PCR and primers specific for the PAI-1 promoter. Samples were normalized to the amount of input DNA. ** indicates p < 0.01 and * indicates p < 0.05.
Figure 4
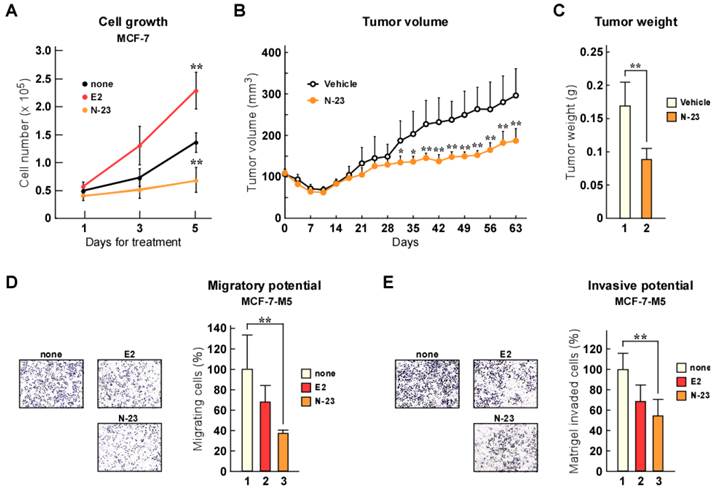
N-23 suppresses both proliferation and invasiveness of breast cancer cells. (A) Proliferation of MCF-7 cells is inhibited by N-23 treatment. MCF-7 cells were cultured in the presence or absence of E2 (10-8 M) or N-23 (10-6 M). At the indicated time points, the number of cells were counted using Countess Automated Cell Counter (Invitrogen). (B, C) N-23 suppresses breast cancer tumor growth in vivo. Nude mice were injected in both flanks with MCF-7 cells and injected with DMSO (Vehicle) or N-23 (45 mg/kg) for every 2 days. Tumor growth was monitored twice each week (B) and xenografts were removed and weighed after 63 days (C). (D, E) MCF-7-M5 cells were seeded onto filters in uncoated (D) or Matrigel matrix-coated (E) top chambers in the absence or presence of E2 (10-8 M) or N-23 (10-6 M). After incubation, migrated or invaded cells were stained using crystal violet and counted under a microscope. ** indicates p < 0.01 and * indicates p < 0.05.
TGF-β signaling is known to participate in prometastatic functions. Genetic manipulation of the TGF-β pathway in tumor cell lines and experimental animal models has validated the facilitative role of TGF-β in metastasis during late cancer progression. In addition, exogenous TGF-β facilitates the invasiveness and metastatic behavior of breast cancer cells in vivo [17]. TGF-β normally stimulates production of extracellular matrix proteins and induces the proteolytic activity of cancer cells by increasing matrix-degrading enzyme expression [26]. Increased TGF-β levels contribute to generation of more invasive cancer cells by reducing cellular adhesion and increasing cancer cell motility and proteolytic activity.
In a previous study, we showed that estrogen suppressed breast cancer invasion by inhibiting TGF-β signaling via a non-genomic pathway. ERs are known to bind with their specific response element ERE, and regulate its downstream target genes [6, 7]; this process is known as the genomic pathway. However, it has been recently established that ERs regulate several transcriptional factors through protein-protein interactions, and this process is defined as the non-genomic pathway. From these observations, it was revealed that estrogen does not use the genomic functions of ERα to inhibit TGF-β signaling. Thus, we speculate that some ERα ligands could separately regulate the genomic or non-genomic functions of ERα.
In addition, we showed that in contrast to estrogen, TAM and ICI could not inhibit TGF-β signaling or breast cancer invasiveness. Several studies have reported the effects of antiestrogens on breast cancer invasiveness. TAM inhibits MCF-7 cell proliferation, but increases their invasiveness by upregulating collagenase IV expression [27] and promoting an invasive phenotype through association with the Src kinase pathway and pro-invasive gene expression [28]. It was also reported that ICI increases P-cadherin expression and facilitates breast cancer cell invasion [29]. Therefore, clinical treatment with TAM or ICI suppresses breast cancer tumorigenesis but cannot suppress breast cancer metastasis.
In the present study, we screened for compounds that inhibit TGF-β signaling but do not enhance ERα signaling. Our identified compound, N-23, inhibited TGF-β signaling via ERα, but it did not enhance ERα pathway. This result confirms that the inhibition of TGF-β signaling by ERα does not require ERα genomic functions. We previously demonstrated that estrogen inhibited TGF-β signaling by promoting pSmad2 and pSmad3 degradation. Unlike estrogen, N-23 did not decrease pSmad2 or pSmad3 protein levels. However, estrogen and N-23 suppressed TGF-β-dependent recruitment of Smad3 to the PAI-1 gene promoter. Our data indicate that the mechanism of TGF-β signaling suppression by N-23 is different from that of estrogen. Further experiments are necessary to determine the mechanisms for the inhibition of TGF-β signaling by N-23.
Our previous study showed that antiestrogens could not suppress breast cancer invasiveness, whereas they inhibited cell proliferation of breast cancer cells. On the other hand, we demonstrated that N-23 has suppressive effects on both cell growth and invasiveness of breast cancer cells. Our findings suggest that N-23 is a candidate compound that effectively inhibits breast cancer progression. These observations provide new insight into breast cancer therapy and indicate that compounds that separately regulate ERα and TGF-β signaling may be useful for developing effective endocrine therapies.
Supplementary Material
Supplementary Figure 1Acknowledgements
This work was supported by grants from Japan Science and technology Agency (JST), and from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69-90 doi:10.3322/caac.20107
2. Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K. et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835-9
3. McKenna NJ, O'Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 2002;108:465-74
4. Curtis Hewitt S, Couse JF, Korach KS. Estrogen receptor transcription and transactivation: Estrogen receptor knockout mice: what their phenotypes reveal about mechanisms of estrogen action. Breast Cancer Res. 2000;2:345-52
5. Warner M, Nilsson S, Gustafsson JA. The estrogen receptor family. Curr Opin Obstet Gynecol. 1999;11:249-54
6. McDonnell DP, Norris JD. Connections and regulation of the human estrogen receptor. Science. 2002;296:1642-4 doi:10.1126/science.1071884
7. McKenna NJ, Lanz RB, O'Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev. 1999;20:321-44
8. Elliston JF, Fawell SE, Klein-Hitpass L, Tsai SY, Tsai MJ, Parker MG. et al. Mechanism of estrogen receptor-dependent transcription in a cell-free system. Mol Cell Biol. 1990;10:6607-12
9. Buzdar AU. Endocrine therapy in the treatment of metastatic breast cancer. Sem in Oncol. 2001;28:291-304
10. Morris C, Wakeling A. Fulvestrant ('Faslodex')--a new treatment option for patients progressing on prior endocrine therapy. Endocr Relat Cancer. 2002;9:267-76
11. Sporn MB, Roberts AB. TGF-beta: problems and prospects. Cell Regul. 1990;1:875-82
12. Abdollah S, Macias-Silva M, Tsukazaki T, Hayashi H, Attisano L, Wrana JL. TbetaRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2-Smad4 complex formation and signaling. J Biol Chem. 1997;272:27678-85
13. Liu X, Sun Y, Constantinescu SN, Karam E, Weinberg RA, Lodish HF. Transforming growth factor beta-induced phosphorylation of Smad3 is required for growth inhibition and transcriptional induction in epithelial cells. Proc Natl Acad Sci USA. 1997;94:10669-74
14. Wharton K, Derynck R. TGFbeta family signaling: novel insights in development and disease. Development. 2009;136:3691-7 doi:10.1242/dev.040584
15. Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 2010;70:5649-69 doi:10.1158/0008-5472.CAN-10-1040
16. Deckers M, van Dinther M, Buijs J, Que I, Lowik C, van der Pluijm G. et al. The tumor suppressor Smad4 is required for transforming growth factor beta-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res. 2006;66:2202-9 doi:10.1158/0008-5472.CAN-05-3560
17. Kang Y, He W, Tulley S, Gupta GP, Serganova I, Chen CR. et al. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci USA. 2005;102:13909-14 doi:10.1073/pnas.0506517102
18. Ito I, Hanyu A, Wayama M, Goto N, Katsuno Y, Kawasaki S. et al. Estrogen inhibits transforming growth factor beta signaling by promoting Smad2/3 degradation. J Biol Chem. 2010;285:14747-55 doi:10.1074/jbc.M109.093039
19. Goto N, Hiyoshi H, Ito I, Tsuchiya M, Nakajima Y, Yanagisawa J. Estrogen and antiestrogens alter breast cancer invasiveness by modulating the transforming growth factor-beta signaling pathway. Cancer Sci. 2011;102:1501-8 doi:10.1111/j.1349-7006.2011.01977.x
20. Osborne CK, Hobbs K, Clark GM. Effect of estrogens and antiestrogens on growth of human breast cancer cells in athymic nude mice. Cancer Res. 1985;45:584-90
21. Howell A, Osborne CK, Morris C, Wakeling AE. ICI 182,780 (Faslodex): development of a novel, "pure" antiestrogen. Cancer. 2000;89:817-25
22. Murayama A, Ohmori K, Fujimura A, Minami H, Yasuzawa-Tanaka K, Kuroda T. et al. Epigenetic control of rDNA loci in response to intracellular energy status. Cell. 2008;133:627-39 doi:10.1016/j.cell.2008.03.030
23. Kajiro M, Hirota R, Nakajima Y, Kawanowa K, So-ma K, Ito I. et al. The ubiquitin ligase CHIP acts as an upstream regulator of oncogenic pathways. Nat Cell Biol. 2009;11:312-9 doi:10.1038/ncb1839
24. Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091-100 doi:10.1093/emboj/17.11.3091
25. Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM. et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst. 1998;90:1371-88
26. Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807-21 doi:10.1038/nrc1208
27. Thompson EW, Reich R, Shima TB, Albini A, Graf J, Martin GR. et al. Differential regulation of growth and invasiveness of MCF-7 breast cancer cells by antiestrogens. Cancer Res. 1988;48:6764-8
28. Borley AC, Hiscox S, Gee J, Smith C, Shaw V, Barrett-Lee P. et al. Anti-oestrogens but not oestrogen deprivation promote cellular invasion in intercellular adhesion-deficient breast cancer cells. Breast Cancer Res. 2008;10:R103. doi:10.1186/bcr2206
29. Paredes J, Stove C, Stove V, Milanezi F, Van Marck V, Derycke L. et al. P-cadherin is up-regulated by the antiestrogen ICI 182,780 and promotes invasion of human breast cancer cells. Cancer Res. 2004;64:8309-17 doi:10.1158/0008-5472.CAN-04-0795
Author contact
![]() Corresponding author: Junn Yanagisawa, Hiromi Hiyoshi, Graduate School of Life and Environmental Sciences, University of Tsukuba, Tsukuba Science City, Ibaraki 305-8577, Japan. Tel: 81-29-853-7323 Fax: 81-29-853-7322 E-mail: junnytsukuba.ac.jp, hiromi_hiyoshitsukuba.ac.jp.
Corresponding author: Junn Yanagisawa, Hiromi Hiyoshi, Graduate School of Life and Environmental Sciences, University of Tsukuba, Tsukuba Science City, Ibaraki 305-8577, Japan. Tel: 81-29-853-7323 Fax: 81-29-853-7322 E-mail: junnytsukuba.ac.jp, hiromi_hiyoshitsukuba.ac.jp.
Citation styles
APA
Goto, N., Hiyoshi, H., Ito, I., Iida, K., Nakajima, Y., Nagasawa, K., Yanagisawa, J. (2014). Identification of a Novel Compound That Suppresses Breast Cancer Invasiveness by Inhibiting Transforming Growth Factor-β Signaling via Estrogen Receptor α. Journal of Cancer, 5(5), 336-343. https://doi.org/10.7150/jca.7202.
ACS
Goto, N.; Hiyoshi, H.; Ito, I.; Iida, K.; Nakajima, Y.; Nagasawa, K.; Yanagisawa, J. Identification of a Novel Compound That Suppresses Breast Cancer Invasiveness by Inhibiting Transforming Growth Factor-β Signaling via Estrogen Receptor α. J. Cancer 2014, 5 (5), 336-343. DOI: 10.7150/jca.7202.
NLM
Goto N, Hiyoshi H, Ito I, Iida K, Nakajima Y, Nagasawa K, Yanagisawa J. Identification of a Novel Compound That Suppresses Breast Cancer Invasiveness by Inhibiting Transforming Growth Factor-β Signaling via Estrogen Receptor α. J Cancer 2014; 5(5):336-343. doi:10.7150/jca.7202. https://www.jcancer.org/v05p0336.htm
CSE
Goto N, Hiyoshi H, Ito I, Iida K, Nakajima Y, Nagasawa K, Yanagisawa J. 2014. Identification of a Novel Compound That Suppresses Breast Cancer Invasiveness by Inhibiting Transforming Growth Factor-β Signaling via Estrogen Receptor α. J Cancer. 5(5):336-343.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.