Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(7):593-603. doi:10.7150/jca.11238 This issue Cite
Research Paper
Differential Expression of ADAM23, CDKN2A (P16), MMP14 and VIM Associated with Giant Cell Tumor of Bone
André Luis Giacometti Conceição1,*, Erica Babeto1,*, Natalia Maria Candido1, Fernanda Craveiro Franco1, Débora Aparecida Pires de Campos Zuccari2, Jane Lopes Bonilha3, José Antônio Cordeiro4, Marilia Freitas Calmon1, Paula Rahal1 ![]()
1. Laboratory of Genomics Studies, UNESP, São José do Rio Preto, Brazil.
2. Center for the Study of Cancer Prognosis, FAMERP, São José do Rio Preto, Brazil.
3. Department of Pathology, FAMERP, São José do Rio Preto, Brazil.
4. Department of Epidemiology and Collective Health, FAMERP, São José do Rio Preto, Brazil.
* These authors contributed equally to this work
Received 2014-12-4; Accepted 2015-2-10; Published 2015-5-23
Citation:
Conceição ALG, Babeto E, Candido NM, Franco FC, de Campos Zuccari DAP, Bonilha JL, Cordeiro JA, Calmon MF, Rahal P. Differential Expression of ADAM23, CDKN2A (P16), MMP14 and VIM Associated with Giant Cell Tumor of Bone. J Cancer 2015; 6(7):593-603. doi:10.7150/jca.11238. https://www.jcancer.org/v06p0593.htm
Other stylesAbstract
Though benign, giant cell tumor of bone (GCTB) can become aggressive and can exhibit a high mitotic rate, necrosis and rarely vascular invasion and metastasis. GCTB has unique histologic characteristics, a high rate of multinucleated cells, a variable and unpredictable growth potential and uncertain biological behavior. In this study, we sought to identify genes differentially expressed in GCTB, thus building a molecular profile of this tumor. We performed quantitative real-time polymerase chain reaction (qPCR), immunohistochemistry and analyses of methylation to identify genes that are putatively associated with GCTB. The expression of the ADAM23 and CDKN2A genes was decreased in GCTB samples compared to normal bone tissue, measured by qPCR. Additionally, a high hypermethylation frequency of the promoter regions of ADAM23 and CDKN2A in GCTB was observed. The expression of the MAP2K3, MMP14, TIMP2 and VIM genes was significantly higher in GCTB than in normal bone tissue, a fact that was confirmed by qPCR and immunohistochemistry. The set of genes identified here furthers our understanding of the molecular basis of GCTB.
Keywords: Giant cell tumor of bone, gene expression, hypermethylation, immunohistochemistry
Introduction
Giant cell tumor of bone (GCTB), also known as osteoclastoma [1], accounts for approximately 5% of all primary bone tumors. GCTB is an expansible osteolytic tumor that most often arises at the end of a long bone in a skeletally mature patient [2]. This neoplasm usually affects young adults - approximately two-thirds of the patients are between 20 and 40 years of age [3]. GCTB most commonly involves the distal femur, proximal tibia, distal radius, proximal humerus and the sacral bone, with a slight predominance in females [4, 5].
Histologically, GCTB comprises 3 distinct cell types: multinucleated osteoclast-like giant cells, monocytic round-shaped macrophage-like cells, spindle-shaped and fibroblast-like stromal cells. The stromal cells of GCTB are the primary neoplastic cells; they are the only proliferating cell component in long-term culture [6]. Although their exact origin has yet to be determined, the stromal cells may descend from either an osteoblastic lineage or bone marrow mesenchymal cells, which might both regulate the formation of multinucleated osteoclast-like giant cells in the neoplasm [7]. The stromal cells of GCTB may drive the macrophage-like cells to undergo fusion to form multinucleated osteoclast-like giant cells, and the latter eventually cause aggressive bone resorption and skeletal destruction [8].
The World Health Organization has classified GCTB as an aggressive, potentially malignant lesion [4]. Clinically, GCTB is considered benign, but it often becomes an aggressive lesion with a tendency for local recurrence. Depending on the type of treatment and the local presentation of the tumor, recurrence rates range from 0 to 65% [9]. GCTB is one of the rare benign tumors that can grow [10] intravascularly and give rise to distant metastases; slow growth and lung metastasis occur in 2 - 10% of cases [11-13].
The Epithelial Mesenchymal Transition (EMT) is a phenomenon which cancer cell acquires plasticity that confers features such as metastasis and recurrences. The EMT process is regulated by a number of genes distributed in compartments of cancer cell [14, 15]. To elucidate the molecular mechanisms involved in the tumorigenesis of GCTB, selected genes have been identified by analyzing gene expression [16-18]. Few studies, however, have investigated the genetic profile of GCTB, we have searched for to identify differentially expressed genes involved in tumorigenesis. Elucidation of these genes and the molecular mechanisms that may regulate their expression remain to be identified to achieve a better understanding of the biology of GCTB [19, 20].
The MAP2K3 gene is related to cell proliferation. In several types of advanced cancers that lead to cell proliferation, this gene has been shown to promote cancer cell invasion and metastasis [21]. The ADAM23, CDH2, CDKN2A (p16), CDK4, CXCL14, MMP14, NFκB, SNAI1, TIMP2, TIMP3, VIM and ZEB1 genes encode proteins involved in different stages of cell transformation, including cell motility, adjacent tissue invasion, tumor progression, dissemination through the vasculature and finally proliferation from a micro-metastasis to macroscopic secondary tumor [22-31].
In this study, we selected genes involved in cellular differentiation and proliferation in giant cell tumors of bone and analyzed their expression by qPCR. The genes ADAM23 and CDKN2A were underexpressed in giant cell tumor of bone, and we report that the hypermethylation of the promoter regions of these genes controls their expression in GCTB. Furthermore, we showed, by immunohistochemistry, that overexpression of the MAP2K3, MMP14, TIMP2 and VIM genes was significantly higher in CGTB than in non-neoplastic samples. Because of their role in the cellular mechanisms involved in the alteration of cellular homeostasis, these genes may be involved in the development of GCTB.
Material and methods
GCTB Sample Collection
The samples were collected from 42 patients with primary GCTB, including 23 fresh samples of GCTB, 24 paraffin-embedded samples of GCTB, 9 fresh, histologically normal bone tissue samples of femur, and 3 paraffin-embedded samples of histologically normal medullar bone tissues of femur, all of which were confirmed by pathologists. Histologically normal samples were extracted from the margin of tumors of the femur. Tissues were obtained from the Tumor Bank at The Pio XII Foundation/IBILCE-UNESP, São Paulo, Brazil. The use of all patient-derived material was approved by the institution's Research Ethics Board at The Pio XII Foundation of the Cancer Hospital of Barretos, and informed consent was obtained individually from the patients. The diagnosis of GCTB was established by a biopsy prior to surgical excision, and the patients had no other malignancies at the time of the surgery. The tissues were obtained during surgery from patients undergoing tumor resection, and the diagnosis of GCTB was verified post-operatively by a histopathologist. Microdissection was performed on the collected samples. The fresh samples were used to perform the quantitative real-time polymerase chain reaction, and paraffin-embedded samples were used in the methylation and immunohistochemistry analyses.
RNA Extraction
Total RNA was isolated from GCTB tissue and normal tissue using TRIzol (Life Technologies, Grand Island, NY, USA) and following the protocol instructions. For qPCR, approximately 5 μg of total RNA from each sample were used to synthesize cDNA with a High-Capacity cDNA Archive Kit (Life Technologies, Grand Island, NY, USA), according to the manufacturer's instructions. β-ACTIN (ACTB) amplification served as a control for cDNA quality.
Quantitative Real-time Polymerase Chain Reaction (qPCR)
Twenty genes related to tumorigenesis, progression, cell migration and tumor malignancy were selected. We used 23 fresh CGTB samples and a pool of total RNA from a subset of 9 fresh tissue samples of normal bone, defined as the normal reference (control group). Gene-specific primers for qPCR were designed for optimal hybridization kinetics using the Primer 3.0 program (provided by the Whitehead/MIT Center for Genome Research, Cambridge, MA, USA).
Quantitative real-time PCR involved an ABI prism 7300 sequencer detector system and SYBR Green PCR Core Reagent (Life Technologies, Grand Island, NY, USA). The reaction mixture (20 μL total volume) contained 25 ng of cDNA, gene-specific forward and reverse primers for each gene, and 10 μL of 2× Quantitative SYBR Green PCR Master Mix. Relative quantification was performed using the CT values and triplicate reactions for GCTB samples and a reference sample from each gene and from the endogenous control (glyceraldehyde-3-phosphate dehydrogenase; GAPDH). The primers were designed in different exons, and the sequences are available in Table 1. Therefore, the relative expression of each specific gene was calculated by using the formula: R =
= (E target)ΔCt target (control - sample)/(E endogenous)ΔCt endogenous (control - sample), as previously described [32]. The cut-off for the analysis of gene expression was ≥ 2 for increases and decreases in expression. A value below this cut-off was considered to indicate that the increase or decrease in expression was not significant.
(E target)ΔCt target (control - sample)/(E endogenous)ΔCt endogenous (control - sample), as previously described [32]. The cut-off for the analysis of gene expression was ≥ 2 for increases and decreases in expression. A value below this cut-off was considered to indicate that the increase or decrease in expression was not significant.
DNA Extraction
DNA samples of fresh tissue were isolated using TRIzol (Life Technologies, Grand Island, NY, USA). For the extraction of DNA from paraffin, the samples were deparaffinized with xylene, and the tissue samples were digested in a buffer (100 mmol/L NaCl, 10 mmol/L Tris-HCl pH 8.0, 25 mmol/L ethylenediamine tetraacetic acid (EDTA) and 1% sodium dodecyl sulfate) containing 20 mg/mL proteinase K at 50°C for 3 days. Total DNA was isolated using phenol-chloroform extraction and ethanol precipitation. The DNA pellets were resuspended with 20 mL of TE buffer (10 mmol/L Tris-HCl, 1 mmol/L EDTA, pH 8.0) and stored at -20°C until PCR amplification. β-Globin gene (HBB) amplification served as a control for DNA quality.
Bisulfite Modification of DNA and Methylation-Specific PCR
The genomic DNA extracted from tissues was modified using bisulfite treatment according to Calmon et al. [33]. The DNA methylation status in the CpG island promoter was determined using the previously described MSP procedure, which used primers specific for the methylated (M) or unmethylated (U) sequences of the bisulfite-modified DNA. The primers used for each gene in the PCR reaction were specific to methylated and unmethylated DNA (Table 2). Bisulfite-modified DNA from peripheral blood lymphocytes from a healthy individual was previously treated and untreated with CpG methyltransferase (M.SssI) (New England Biolabs, Ipswich, MA, USA) and served as a positive control for hypermethylated and unmethylated DNA. A blank control containing all the PCR components (except template DNA) was also included in all of the experiments. Reaction products were separated using electrophoresis on an 8% polyacrylamide gel and stained with silver nitrate.
Table 1
Primer sequences used in quantitative real-time polymerase chain reaction
| Gene | Primer Sequence (5' - 3') | Size of product |
|---|---|---|
| ADAM23 | F:CCACTCGATTCCAAGGGTAAAGT R:ATGCAGGTGGCTTCATTACTACAC | 64bp |
| CDH2 | F:ATATGGCCTTTCAAACACAGC R:CGTCATGGCAGTAAACTCTGG | 82bp |
| CDKN2A (p16) | F:ACCAGAGGCAGTAACCATGC R:AAGTTTCCCGAGGTTTCTCAG | 99bp |
| CDK4 | F:CCCGAAGTTCTTCTGCAGTC R:CTGGTCGGCTTCAGAGTTTC | 119bp |
| CXCL4 | F:GAAATGAAGCCAAAGTACCCG R:TACAACGCCTGGAACG | 152bp |
| GAPDH | F:ACCCACTCCTCCACCTTTGA R:CTGTTGCTGTAGCCAAATTCGT | 79bp |
| MAP2K3 | F:GCCTATGGGGTGGTAGAGAAG R:TTGATGTCCAGGTCCATGAG | 102bp |
| MMP14 | F:CACTGCCTACGAGAGGAAGG R:GAGCAGCATCAATCTTGTCG | 149bp |
| NFκB | F:CCAGCTGGCAGGTATTTGAC R:TCAGCCAGCTGTTTCATGTC | 100bp |
| SNAI1 | F:CTCTTTCCTCGTCAGGAAGC R:AGGGCTGCTGGAAGGTAAAC | 95bp |
| TIMP2 | F:GTAGTGATCAGGGCCAAAGC R:TCTCAGGCCCTTTGAACATC | 124bp |
| TIMP3 | F:GGGGAAGAAGCTGGTAAAGG R:CCACAGAGACTCTCGGAAGC | 135bp |
| VIM | F:GAAATTGCAGGAGGAGATGC R:ATTCCACTTTGCGTTCAAGG | 116bp |
| ZEB1 | F:TGCACTGAGTGTGGAAAAGC R:AAGCGTTTCTTGCAGTTTGG | 107bp |
Table 2
Primer sequences used in methylation-specific polymerase chain reaction
| Gene | External Primer | T°C | Primer sequence | T°C | Size of product M | |
|---|---|---|---|---|---|---|
| Methylated sequence (5'-3') | Unmethylated sequence (5'-3') | |||||
| ADAM23 | F:ATTGTTTTTTGGTTAGAATGTCG | F:ATTGTTTTTTTGTTAGAATGTTG | 59 | 100 | ||
| R:TAAAAAAAACACAAAAAACCGAACG | R:TAAAAAAAACACAAAAAACCAAACA | |||||
| CDKN2A (p16) | F:GGAGAGGGGGAGAGTAGGT | 60 | F:CGGGGAGTAGTATGGAGTCGGCGGC | F:TGGGGAGTAGTATGGAGTTGGTGGT | 64 | 81 |
| R:CTACAAACCCTCTACCCACCT | R: GACCCCGAACCGCGACCGTAA | R: CAACCCCAAACCACAACCATAA | ||||
T= annealing temperature (°C), U= Unmethylated sequence, M= Methylated sequence.
Immunohistochemistry
Unstained 4.5 µm sections were cut from each paraffin block, deparaffinized and rehydrated using routine techniques. Endogenous peroxidase activity was blocked with 0.3% H2O2 for 30 min in a citrate buffer (10 mM, pH-6) at 95°C. Monoclonal antibodies used were anti-human MMP14 (Abcam Inc., Cambridge, MA, USA) (1:50 dilution), anti-human TIMP2 (Chemicon/Millipore, Billerica, MA, USA) (1:150 dilution), anti-human VIM (Dako, Carpinteria, CA, USA) (1:100 dilution) and polyclonal rabbit anti-human MAP2K3 (Abcam Inc., Cambridge, MA, USA) (1:50 dilution). They were applied and incubated at 4°C overnight. Afterward, sections were incubated with a biotinylated secondary antibody and exposed to a streptavidin complex (HRP Ready-to Use, DakoCytomation, Carpinteria, CA, USA). Positive reactions were visualized with 3,3' diaminobenzidine tetrahydrochloride (DAB, Signet® Laboratories, Dedham, MA, USA), followed by counterstaining with hematoxylin.
Normal pancreas tissue was used as a positive control for TIMP2, and breast carcinoma tissue was used as the positive control for MAP2K3, MMP14 and VIM. Sections treated without primary antibodies were used as negative controls. TIMP2, MAP2K3, MMP14 and VIM densitometric analyses were conducted with an Axioskop II microscope (Zeiss, Germany) using the Software AxiovisionTM (Zeiss). For these analyses, 3 different fields from each tumor fragment were used, and 20 different points were analyzed to calculate an average intensity of immunoreactivity. The values were obtained as arbitrary units (a.u.).
Statistical Analysis
Minitab Student 14 software was employed to perform the statistical analyses, with significance set at p < 0.05. Relative expression levels detected by qPCR in CGTBs samples were transformed into natural logarithms. The Wilcoxon signed-rank test was applied to compare the gene expression levels in tumor tissue and normal bone tissue. The Kruskal-Wallis test was used to determine whether there was an association between gene expression and recurrence or metastasis. The data from protein expression were obtained using immunohistochemistry and were statistically examined with the Mann-Whitney test.
Results
Patients
The study population involved 42 patients. Twenty-one (50%) were male and 21 (50%) were female. Their minimum and maximum ages were 13 and 74 years, respectively, with a mean of 35.9 years ± 14.9. The most common locations of the GCTB were femur (26%), tibia (21%) and radius (14%). Eight patients (17%) experienced a recurrence between 11 and 42 months after surgery, and 6 patients (12.7%) experienced metastases between 1 and 65 months after surgery (Table 3).
Table 3
Epidemiological, clinical and pathological characteristics of patients diagnosed with GCTB
| Sample | Age(y) | Sex | Anatomic location | Outcome | |
|---|---|---|---|---|---|
| Recurrence (date) | Metastasis (date) | ||||
| 1F | 37 | M | Proximal tibia L | ned | 19 months |
| 2F | 41 | F | Distal radius R | ned | ned |
| 3F | 24 | M | Proximal femur L | ned | ned |
| 4F/P | 52 | F | Distal femur L | ned | ned |
| 5F | 17 | M | Scapula L | ned | ned |
| 6F | 32 | F | Olecranon R | 11 months | ned |
| 7F/P | 52 | M | Distal femur L | 11 months | ned |
| 8F/P | 74 | F | Distal femur L | 19 months | ned |
| 9F | 35 | M | Distal femur L | ned | ned |
| 10F | 33 | M | Proximal tibia R | 20 months | ned |
| 11F | 21 | F | Proximal tibia R | ned | ned |
| 12F | 37 | F | Proximal tibia L | ned | ned |
| 13F | 13 | F | * | ned | 41 months |
| 14F/P | 28 | F | Distal femur R | 19 months | ned |
| 15F | 19 | F | Sacrum | ned | ned |
| 16F | 22 | F | Distal fibula R | ned | ned |
| 17F | 16 | F | Ischium R | ned | ned |
| 18F | 22 | M | Distal fibula L | ned | 20 months |
| 19F | 27 | M | Proximal humerus L | ned | No |
| 20F | 69 | F | Occipital | ned | ned |
| 21F | 24 | M | Proximal tibia R | 22 months | ned |
| 22F | 46 | F | Distal radius L | ned | ned |
| 23F | 58 | F | Hemipelvis R | ned | ned |
| 24P | 55 | M | Proximal tíbia L | ned | ned |
| 25P | 22 | F | Ischium R | ned | 65 months |
| 26P | 57 | M | Proximal tibia L | 42 months | ned |
| 27P | 34 | M | Proximal tibia R | ned | ned |
| 28P | 24 | M | Distal radius L | ned | ned |
| 29P | 27 | M | Distal femur L | ned | ned |
| 30P | 51 | F | Scapula L | Yes* | 07 months |
| 31P | 31 | M | Wrist R | ned | ned |
| 32P | 22 | F | Distal radius R | ned | ned |
| 33P | 41 | M | Distal femur L | ned | 01 months |
| 34P | 24 | F | Wrist L | ned | ned |
| 35P | 41 | M | Distal femur R | ned | ned |
| 36P | 55 | M | Distal radius L | ned | ned |
| 37P | 41 | F | Distal radius L | ned | ned |
| 38P | 35 | M | Thumb L | ned | ned |
| 39P | 39 | M | Proximal fibula L | ned | ned |
| 40P | 53 | M | Proximal tibia R | ned | ned |
| 41P | 38 | F | Distal femur L | ned | ned |
| 42P | 19 | F | Distal femur L | ned | ned |
Abbreviations: f: fresh tumor; p: paraffin-embedded tumor; f: female; m: male; r: right; l: left; ned: no evidence of disease, * = no information was obtained.
Expression of the Selected Genes
The CXCL14, CDH2, CDK4, MAP2K3, MMP14, SNAI1, TIMP2, TIMP3, VIM and ZEB1 genes were overexpressed in the GCTB samples compared to the reference samples (p < 0.001), and the fold change of gene expression ranged from 2.03 to 6.58. CDKN2A (p16) and ADAM23 were significantly downregulated in tumor tissues compared to normal bone tissue (p < 0.001), and the fold change of gene expressions was found to be -6.46 and -4.70, respectively. The NFκB gene was not differentially expressed (Figure 1).
Methylation Analysis of the ADAM23 and CDKN2A (p16) genes
After gene expression analysis, the methylation pattern in the promoter region of the ADAM23 and CDKN2A (p16) genes was analyzed using MSP-PCR in 26 GCTB samples.
Hypermethylation in the promoter regions of the ADAM23 and CDKN2A (p16) genes was detected in 92% (24/26) and in 83.3% (20/24) of the tumors analyzed, respectively (Figure 2 and Table 4). The hypermethylation of ADAM23 and CDKN2A (p16) was statistically associated with the presence of GCTB (p < 0.001 and p < 0.05, respectively). In addition, the sets of primers for the genes used in this study gave no indication of hypermethylation in the normal lymphocytes analyzed. The hypermethylation of the ADAM23 gene was detected in 90.5% of the non-recurrent tumors samples and in 90.5% of the non-metastatic tumor samples. The promoter region of the ADAM23 gene was hypermethylated in all the samples that exhibited metastasis or recurrence. Because of the small number of samples, however, no significant association could be found between the presence of hypermethylation in the promoter regions of ADAM23 and CDKN2A (p16) genes and the clinicopathologic parameters of these genes in GCTB.
Table 4
Methylation pattern of ADAM23 and CDKN2A (p16) in 26 GCTB samples
| Samples | ADAM23 | p16 |
|---|---|---|
| 3F | ˜ | ˜ |
| 4F | ˜ | ˜ |
| 7F | ˜ | ˜ |
| 9F | ˜ | ˜ |
| 10F | ˜ | ˜ |
| 11F | ˜ | ˜ |
| 12F | ˜ | ˜ |
| 13F | ˜ | ˜ |
| 14F | ˜ | ˜ |
| 15F | ˜ | ˜ |
| 18F | ˜ | X |
| 19F | ˜ | ˜ |
| 23F | ˜ | ˜ |
| 24P | ˜ | ˜ |
| 25P | ˜ | X |
| 28P | ˜ | ˜ |
| 29P | ™ | ˜ |
| 30P | ˜ | ™ |
| 35P | ˜ | ™ |
| 37P | ˜ | ˜ |
| 38P | ˜ | ˜ |
| 39P | ˜ | ˜ |
| 40P | ˜ | ™ |
| 41P | ˜ | ˜ |
| 42P | ™ | ˜ |
legend: ˜: methylated genes; ™: unmethylated genes and x: not amplified.
Figure 1
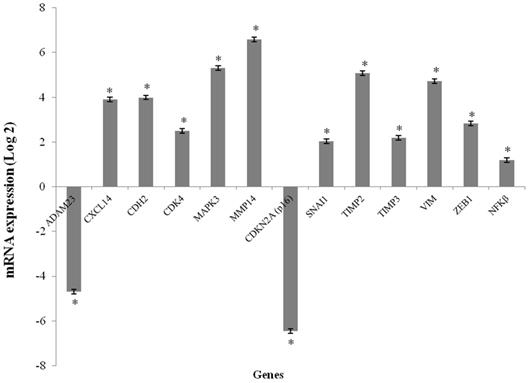
mRNA expression levels media in GCTB samples. mRNA expression of the selected genes using qPCR. The results are shown as the fold change in expression relative to normal bone of femur. (*p < 0.001, Wilcoxon Signed Rank Test)
Figure 2
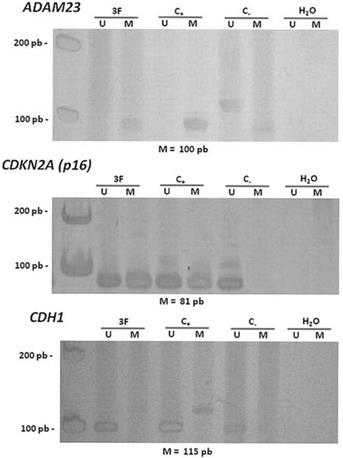
Representative data showing the methylation status of the promoter regions of the genes ADAM23 and CDKN2A (p16). Lanes U and M correspond to unmethylated and methylated reactions, respectively. In each case, 3F indicates GCTB patient sample, C- indicates DNA from lymphocyte, C+ indicates in vitro methylated DNA (IVD), H2O indicates negative PCR control. On the left: molecular weight marker; and below: size of methylated PCR product.
Immunohistochemistry of the Overexpressed Genes (MAP2K3, MMP14, TIMP2 and VIM)
Immunohistochemistry was performed on 24 samples of GCTB and on the normal tissue margins of the tumors. Immunohistochemical staining of GCTB samples confirmed the presence of MMP14, MAP2K3, TIMP2 and VIM in the GCTB microenvironment. Staining of the samples for MMP14, MAP2K3, TIMP2 and VIM was positive in the cytoplasm of multinucleated giant cells, stromal cells and monocytic cells (Figure 3A-D). In normal bone tissue there was very weak staining in different cell types of these 4 gene products (Figure 3 E-H) and breast carcinoma and normal pancreas showed immunostaining, as representative positive controls for the selected gene products (Figure 3I-L). GCTB samples were found to have an increased expression of MMP14, MAP2K3, TIMP2 and VIM compared to the normal tissue margins of the tumor (p < 0.05) (Figure 3M).
Figure 3
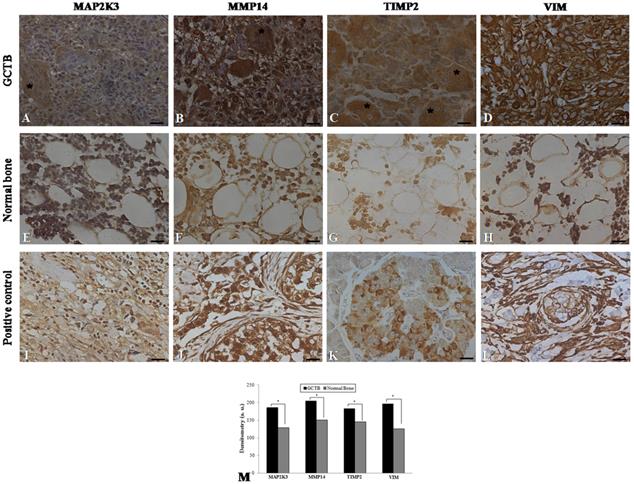
Immunohistochemistry in GCTB and control samples. Protein detection of (A) MAP2K3, (B) MMP14, (C) TIMP2 and (D) VIM in giant cell tumor of bone samples by immunohistochemistry. (E-H) Normal bone samples were used as negative controls and (I, J, L) breast carcinoma and (K) normal pancreas were used as positive controls. The presence of multinucleated giant cells is marked with asterisk. (M) Graphic of densitometry of the immunostaining of MAP2K3, MMP14, TIMP2 and VIM in the samples analyzed. Bars = 20 μm. (*p < 0.05, Mann-Whitney test).
Discussion
Giant cell tumor of bone is a rare type of bone tumor that is generally benign and characterized histologically by multinucleated giant cells with a background of mononuclear stromal cells [34]. GCTB is a tumor that, while benign, often becomes aggressive and exhibits local recurrence and small rates of metastasis [5, 35]. This type of tumor belongs to a group of benign tumors of bone, including osteoblastoma, chondroblastoma and chondromyxoid fibroma that share characteristics that are both benign and aggressive [36, 37]. GCTB has unique histological characteristics consisting of mononuclear cells and multinucleated giant cells with a variable and unpredictable growth potential [10]. Thus, it has several characteristics of aggressive, including a high mitotic rate, necrosis and recurrence after resection [38]. However, the identification of aggressive GCTB is often difficult because these tumors are rare and diagnostic criteria remain poorly defined [39]. The molecular mechanisms responsible for the post-surgical recurrence and rarely metastasis of GCTB are not well understood. To better understand this rare type of tumor and to avoid recurrences, it is important to elucidate the molecular biology of the tumor and the genes involved in tumorigenesis of giant cell tumor of bone.
Tumor cells are characterized by changes in gene expression and transcriptional inactivation caused by epigenetic events that can initiate the expansion of altered cells during the early stages of tumorigenesis [40]. Methylation is a major epigenetic modification process in human cells; changes in methylation patterns play an important role in the genesis of tumors via transcriptional inactivation. The methylation of the C-5 methyl group in DNA's 5'-cytosine results in the formation of 5-methylcytosine. Methyl groups decrease the binding affinity between the promoter regions of DNA and transcription factors, which silences genes that play important roles in maintaining cellular homeostasis [41]. Methylation events play an important role in tumor progression and are frequent in transformed cells. The methylation of CpG islands is found in gene promoter regions, which may be associated with the repair of damaged DNA, metastases and invasiveness of the cancer genesis process [42]. The process of hypermethylation in the promoter region was reported in giant cell tumor of bone, for example, in the genes UCHL1, IGFBP4 and RUNX3, which are involved in the regulation of cell proliferation, apoptosis and cell transformation [43] [44]. Therefore, we investigated if hypermethylation in the promoter region regulates the expression of the genes ADAM23 and CDKN2A (p16) in giant cell tumor of bone.
The hypermethylation of the promoters of the ADAM23 and CDKN2A (p16) genes is a mechanism of silencing these genes, similar to hypermethylation in other tumor types [22, 45]. This is the first time that hypermethylation in the promoter region of the ADAM23 and CDKN2A (p16) genes has been described in this type of tumor.
The ADAM23 gene is located on chromosome 2q33 and encodes ADAM domain 23; this constitutes an Adam family of type I transmembrane glycoproteins that has a common structural organization, including a metalloprotease and disintegrin domain. This protein possesses a potent adhesion domain and is involved in cell-cell and cell-matrix interactions [33], which allow for contact between cells. This contact can prevent growth through contact inhibition, particularly in normal cells.
An analysis of the ADAM23 gene promoter suggests that methylation is an active epigenetic event in the silencing of gene activity. Hypermethylation of the promoter region of the ADAM23 gene has been associated with advanced breast, brain, gastric, and head and neck cancers [33, 46-48]. Furthermore, one study shows that the promoter region of this gene tends to be more frequently methylated in metastatic gastric carcinomas [49]. Hypermethylation of the ADAM23 gene in primary breast tumors is significantly associated with an incidence of metastases. The loss of ADAM23 expression may promote αvβ3 integrin activation leading to enhance tumor cell migration [50, 51]. In our study, we observed that the most GCTB samples exhibited hypermethylation in the promoter region of the ADAM23 gene. Thus, we suggest that the silencing of ADAM23 could lead to GCTB progression because of the loss of contact inhibition and abnormal cell-matrix interactions that lead to uncontrolled cell proliferation.
The CDKN2A gene, also known as p16INK4, is considered a tumor suppressor gene because it is frequently observed in human cancers to be silenced through deletion or via an inactivating mutation [52-56]. Typically, p16 induces cell cycle arrest and prevents cell division by inhibiting the cyclin-dependent kinases CDK4 and CDK6, as well as CDK-mediated phosphorylation of the retinoblastoma gene [57]. The CDKN2A protein plays an important role in suppressing cell cycle progress, and downregulation of p16 could increase cell proliferation and contribute to the development of a variety of cancers [58, 59]. Moreover, CDKN2A downregulation has been confirmed using immunohistochemistry analysis in chondrosarcoma cell lines [60] and the presence of CDKN2A gene in enchondromas leads to low proliferative activity and the inhibition of cell-cycle progression [61]. The CDKN2A gene protects cells from undergoing transformation and protects the genome from mutagenic events. It also induces apoptosis in cells that escape the control of the cell [62].
Methylation of cytosine at CpG sites in the CDKN2A gene promoter, which results in the silencing of p16 expression, occurs in many cell lines, including those of colorectal cancer and various primary carcinomas of colon, breast, bladder, ovary, lung and bone marrow [63-68]. Aberrant inactivation of the CDKN2A gene by methylation is a frequent early event in multiple human cancers [69, 70], which means it is a factor of interest as a biomarker of pre-malignant alterations [71]. In this study, CDKN2A gene expression is suppressed possibly because of the high frequency of DNA methylation in its promoter region. The loss of CDKN2A activity as a consequence of mutations or promoter hypermethylation is a common step in the genesis and progression of tumors [62] because the inactivation of CDKN2A allows tumor cells to pass through the G1 cell cycle checkpoint [72]. Thus, we suggest that the inactivation of CDKN2A in GCTB contributes to inappropriate mitotic divisions in the giant cells leading to increased cell proliferation.
Epithelial mesenchymal transition (EMT) is essential for driving plasticity during cancer progression. Furthermore, the recurrence after therapy, common in GCTB, is one cancer-related function of EMT, cancer cells to trans-differentiate to mesenchymal cancer cells with an increased expression of many genes [14]. Through gene expression and immunohistochemical analyses, 4 overexpressed genes in GCTB have been identified. The expression of MMP14, MAP2K3, TIMP2 and VIM genes were significantly higher in GCTB than in non-neoplastic samples. Changes in these genes might be critical to the development of GCTB.
The MMP14 gene, which belongs to the family of zinc metalloendopeptidases, is thought to be responsible for the accelerated breakdown of the extracellular matrix (ECM), an essential event in tumorigenesis. Generally, MMPs at the metastastic sites are expressed at a higher level compared to corresponding primary tumors [73]. MMP-9 is expressed in giant cell tumor of bone and because normal cells such as fibroblasts do not synthesize MMP-9, the production of MMP-9 may be important for the migration of cells into the blood stream, lymphatic vessels or adjacent normal tissues [74]. MMP-13 has been reported to accelerate bone remodeling and promotes proliferation while inhibiting apoptosis in human osteoblast-like cells, and its expression is upregulated in giant cell tumor of bone [75-77]. Our study observed, for the first time, the overexpression of the MMP14 gene in multinucleated giant cells, stromal cells and monocytic cells of GCTB. MMP14 has been detected in bone lesions from metastatic prostate cancer, and MMP14 immunostaining was detected in 80% of brain metastases from lung adenocarcinomas [78]. The data support the hypothesis that high levels of MMP14 might play a role in the aggressiveness of giant cell tumor of bone, which has also been suggested for breast cancer [79].
The VIM has shown an association in different types of cancer, including prostate cancer, breast cancer, gastric cancer and gallbladder cancer [80-83]. Studies of the ectopic overexpression of vimentin have shown the invasive behavior of epithelial carcinoma cells, which reveal the association of vimentin with tumor invasiveness and motility [84, 85]. One previous study found expression of VIM in mononuclear cells of GCTB [86]. Our results show that the increased expression of vimentin in multinucleated giant cells, stromal cells and monocytic cells of GCTB is significantly associated with GCTB, and therefore, the increased expression of vimentin may act as a new indicator of this tumor. Zhao et al. [87] demonstrated that vimentin affects prostate cancer cell motility and invasion, and vimentin could be a predictive marker of tumors that might progress to metastatic disease. The MAP2K3 gene is translated into a mitogen-activated protein kinase 3 and is involved in the signal transduction that controls proliferation and programmed cell death [88]. MAP2K3 protein is involved in the Ras-MKK3-p38-signaling cascade, the components of which may confer an invasive phenotype to the cell [89]. Our study shows that the MAP2K3 gene is overexpressed in GCTB compared to normal tissue. The data support the hypothesis the MAP2K3 protein is implicated in tumor invasion and growth [90, 91].
The metallopeptidase inhibitor 2 (TIMP2) gene belongs to the TIMP family. Its function is to inhibit the activity of matrix metallopeptidases [92]. The binding of TIMP2 with MT1-MMP stimulates invasion in the most aggressive cancers, and TIMP2 expression is associated with both tumor recurrence and a poor prognosis. Our study shows that the TIMP2 gene is overexpressed in GCTB compared to normal tissue. Giannopoulos et al. [93] found increased TIMP2 concentration in pancreatic cancer cells and this concentration correlates with the degree of lymphatic vessel infiltration, which ultimately leads to lymphatic node metastases.
In summary, hypermethylation of the promoter region of ADAM23 and CDKN2A occurs in GCTB. The silencing of these genes may contribute to tumor progression because of a possible relationship between these genes and the adhesion domains involved in cell-cell and cell-matrix interactions, which can negatively regulate cell growth. The study also shows that overexpression of the MAP2K3, MMP14, TIMP2 and VIM genes was significantly higher in GCTB than in non-neoplastic samples. The expression of these genes may be involved in any step of the development of GCTB because of their role in the cellular mechanisms involved in the alteration of cellular homeostasis. For the first time, we were able to identify altered genes in GCTB and provide insight into tumor biology. Once these genes have been characterized in GCTB, molecular genetic tools may be used to explore the biological processes involved in this disease. In addition, our approach may provide relevant information and motivation in the development of new therapies for GCTB. Further studies with larger cohorts are required to evaluate whether the identified genes are associated with any clinicopathologic parameters of GCTB. These findings contribute to a better understanding of the tumorigenesis of GCTB and may therefore help to improve diagnosis and patient outcome.
Acknowledgements
We are grateful to Sandra Morini MD, Valter Penna MD and André Stellet MD for collaborating with us to develop this study.
Funding
The study was supported by São Paulo State Research Foundation - FAPESP (2007/52554-9; 2009/08829-9).
Competing interests
The authors disclose that they have no financial interests in the subject of this paper.
References
1. Wulling M, Delling G, Kaiser E. The origin of the neoplastic stromal cell in giant cell tumor of bone. Human pathology. 2003;34:983-93
2. Balke M, Campanacci L, Gebert C, Picci P, Gibbons M, Taylor R. et al. Bisphosphonate treatment of aggressive primary, recurrent and metastatic Giant Cell Tumour of Bone. BMC cancer. 2010;10:462. doi:10.1186/1471-2407-10-462
3. Campanacci M, Baldini N, Boriani S, Sudanese A. Giant-cell tumor of bone. The Journal of bone and joint surgery American volume. 1987;69:106-14
4. Bassiony AA, Abdelrahman M, Abdelhady A, Assal MK. Resection arthrodesis for the management of aggressive giant cell tumor of the distal femur. Indian journal of orthopaedics. 2009;43:67-71 doi:10.4103/0019-5413.44432
5. Karpik M. Giant Cell Tumor (tumor gigantocellularis, osteoclastoma) - epidemiology, diagnosis, treatment. Ortopedia, traumatologia, rehabilitacja. 2010;12:207-15
6. Matsumoto Y, Okada Y, Fukushi J, Kamura S, Fujiwara T, Iida K. et al. Role of the VEGF-Flt-1-FAK pathway in the pathogenesis of osteoclastic bone destruction of giant cell tumors of bone. Journal of orthopaedic surgery and research. 2010;5:85. doi:10.1186/1749-799X-5-85
7. Matsubayashi S, Nakashima M, Kumagai K, Egashira M, Naruke Y, Kondo H. et al. Immunohistochemical analyses of beta-catenin and cyclin D1 expression in giant cell tumor of bone (GCTB): a possible role of Wnt pathway in GCTB tumorigenesis. Pathology, research and practice. 2009;205:626-33 doi:10.1016/j.prp.2009.02.011
8. Lau CP, Huang L, Tsui SK, Ng PK, Leung PY, Kumta SM. Pamidronate, farnesyl transferase, and geranylgeranyl transferase-I inhibitors affects cell proliferation, apoptosis, and OPG/RANKL mRNA expression in stromal cells of giant cell tumor of bone. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 2011;29:403-13 doi:10.1002/jor.21249
9. Klenke FM, Wenger DE, Inwards CY, Rose PS, Sim FH. Recurrent giant cell tumor of long bones: analysis of surgical management. Clinical orthopaedics and related research. 2011;469:1181-7 doi:10.1007/s11999-010-1560-9
10. Alberghini M, Kliskey K, Krenacs T, Picci P, Kindblom L, Forsyth R. et al. Morphological and immunophenotypic features of primary and metastatic giant cell tumour of bone. Virchows Archiv: an international journal of pathology. 2010;456:97-103 doi:10.1007/s00428-009-0863-2
11. Viswanathan S, Jambhekar NA. Metastatic giant cell tumor of bone: are there associated factors and best treatment modalities? Clinical orthopaedics and related research. 2010;468:827-33 doi:10.1007/s11999-009-0966-8
12. Gong L, Liu W, Sun X, Sajdik C, Tian X, Niu X. et al. Histological and clinical characteristics of malignant giant cell tumor of bone. Virchows Archiv: an international journal of pathology. 2012;460:327-34 doi:10.1007/s00428-012-1198-y
13. Wan R, Zhang W, Xu J, Hao P, Yang Y, Shen Y. et al. The outcome of surgical treatment for recurrent giant cell tumor in the appendicular skeleton. Journal of orthopaedic science: official journal of the Japanese Orthopaedic Association. 2012;17:464-9 doi:10.1007/s00776-012-0228-6
14. Kiesslich T, Pichler M, Neureiter D. Epigenetic control of epithelial-mesenchymal-transition in human cancer. Molecular and clinical oncology. 2013;1:3-11 doi:10.3892/mco.2012.28
15. Liu H, Zhang X, Li J, Sun B, Qian H, Yin Z. The biological and clinical importance of epithelial-mesenchymal transition in circulating tumor cells. Journal of cancer research and clinical oncology. 2014 doi:10.1007/s00432-014-1752-x
16. Smith LT, Mayerson J, Nowak NJ, Suster D, Mohammed N, Long S. et al. 20q11.1 amplification in giant-cell tumor of bone: Array CGH, FISH, and association with outcome. Genes, chromosomes & cancer. 2006;45:957-66 doi:10.1002/gcc.20354
17. Gamberi G, Benassi MS, Bohling T, Ragazzini P, Molendini L, Sollazzo MR. et al. Prognostic relevance of C-myc gene expression in giant-cell tumor of bone. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 1998;16:1-7 doi:10.1002/jor.1100160102
18. Pazzaglia L, Conti A, Chiechi A, Novello C, Magagnoli G, Astolfi A. et al. Differential gene expression in classic giant cell tumours of bone: Tenascin C as biological risk factor for local relapses and metastases. Histopathology. 2010;57:59-72 doi:10.1111/j.1365-2559.2010.03597.x
19. Ueda Y, Imai K, Tsuchiya H, Fujimoto N, Nakanishi I, Katsuda S. et al. Matrix metalloproteinase 9 (gelatinase B) is expressed in multinucleated giant cells of human giant cell tumor of bone and is associated with vascular invasion. The American journal of pathology. 1996;148:611-22
20. Guenther R, Krenn V, Morawietz L, Dankof A, Melcher I, Schaser KD. et al. Giant cell tumors of the bone: molecular profiling and expression analysis of Ephrin A1 receptor, Claudin 7, CD52, FGFR3 and AMFR. Pathology, research and practice. 2005;201:649-63 doi:10.1016/j.prp.2005.07.005
21. Guldal CG, Ahmad A, Korshunov A, Squatrito M, Awan A, Mainwaring LA. et al. An essential role for p38 MAPK in cerebellar granule neuron precursor proliferation. Acta neuropathologica. 2012 doi:10.1007/s00401-012-0946-z
22. Choi JS, Kim KH, Jeon YK, Kim SH, Jang SG, Ku JL. et al. Promoter hypermethylation of the ADAM23 gene in colorectal cancer cell lines and cancer tissues. International journal of cancer Journal international du cancer. 2009;124:1258-62 doi:10.1002/ijc.24023
23. Gao P, Xing AY, Zhou GY, Zhang TG, Zhang JP, Gao C. et al. The molecular mechanism of microRNA-145 to suppress invasion-metastasis cascade in gastric cancer. Oncogene. 2013;32:491-501 doi:10.1038/onc.2012.61
24. Sabir M, Baig RM, Mahjabeen I, Kayani MA. Novel germline CDK4 mutations in patients with head and neck cancer. Hereditary cancer in clinical practice. 2012;10:11. doi:10.1186/1897-4287-10-11
25. Gu XL, Ou ZL, Lin FJ, Yang XL, Luo JM, Shen ZZ. et al. Expression of CXCL14 and its anticancer role in breast cancer. Breast cancer research and treatment. 2012;135:725-35 doi:10.1007/s10549-012-2206-2
26. Sepiashvili L, Hui A, Ignatchenko V, Shi W, Su S, Xu W. et al. Potentially novel candidate biomarkers for head and neck squamous cell carcinoma identified using an integrated cell line-based discovery strategy. Molecular & cellular proteomics: MCP. 2012;11:1404-15 doi:10.1074/mcp.M112.020933
27. Bastea LI, Doppler H, Balogun B, Storz P. Protein kinase D1 maintains the epithelial phenotype by inducing a DNA-bound, inactive SNAI1 transcriptional repressor complex. PloS one. 2012;7:e30459. doi:10.1371/journal.pone.0030459
28. Parashar G, Capalash N. Expression of the TIMP2 gene is not regulated by promoter hypermethylation in the Caski cell line. Oncology letters. 2012;3:1079-82 doi:10.3892/ol.2012.608
29. Allott EH, Oliver E, Lysaght J, Gray SG, Reynolds JV, Roche HM. et al. The SGBS cell strain as a model for the in vitro study of obesity and cancer. Clinical & translational oncology: official publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico. 2012;14:774-82 doi:10.1007/s12094-012-0863-6
30. Yamasaki T, Seki N, Yamada Y, Yoshino H, Hidaka H, Chiyomaru T. et al. Tumor suppressive microRNA138 contributes to cell migration and invasion through its targeting of vimentin in renal cell carcinoma. International journal of oncology. 2012;41:805-17 doi:10.3892/ijo.2012.1543
31. Wu K, Fan J, Zhang L, Ning Z, Zeng J, Zhou J. et al. PI3K/Akt to GSK3beta/beta-catenin signaling cascade coordinates cell colonization for bladder cancer bone metastasis through regulating ZEB1 transcription. Cellular signalling. 2012;24:2273-82 doi:10.1016/j.cellsig.2012.08.004
32. Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic acids research. 2001;29:e45
33. Calmon MF, Colombo J, Carvalho F, Souza FP, Filho JF, Fukuyama EE. et al. Methylation profile of genes CDKN2A (p14 and p16), DAPK1, CDH1, and ADAM23 in head and neck cancer. Cancer genetics and cytogenetics. 2007;173:31-7 doi:10.1016/j.cancergencyto.2006.09.008
34. Chakarun CJ, Forrester DM, Gottsegen CJ, Patel DB, White EA, Matcuk GR Jr. Giant cell tumor of bone: review, mimics, and new developments in treatment. Radiographics: a review publication of the Radiological Society of North America, Inc. 2013;33:197-211 doi:10.1148/rg.331125089
35. Lee CH, Espinosa I, Jensen KC, Subramanian S, Zhu SX, Varma S. et al. Gene expression profiling identifies p63 as a diagnostic marker for giant cell tumor of the bone. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2008;21:531-9 doi:10.1038/modpathol.3801023
36. de Silva MV, Reid R. Chondroblastoma: varied histologic appearance, potential diagnostic pitfalls, and clinicopathologic features associated with local recurrence. Annals of diagnostic pathology. 2003;7:205-13
37. Lersundi A, Mankin HJ, Mourikis A, Hornicek FJ. Chondromyxoid fibroma: a rarely encountered and puzzling tumor. Clinical orthopaedics and related research. 2005;439:171-5
38. Horvai AE, Kramer MJ, Garcia JJ, O'Donnell RJ. Distribution and prognostic significance of human telomerase reverse transcriptase (hTERT) expression in giant-cell tumor of bone. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2008;21:423-30 doi:10.1038/modpathol.3801015
39. Okubo T, Saito T, Mitomi H, Takagi T, Torigoe T, Suehara Y. et al. p53 mutations may be involved in malignant transformation of giant cell tumor of bone through interaction with GPX1. Virchows Archiv: an international journal of pathology. 2013;463:67-77 doi:10.1007/s00428-013-1435-z
40. Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nature reviews Cancer. 2006;6:107-16 doi:10.1038/nrc1799
41. Kuroda A, Rauch TA, Todorov I, Ku HT, Al-Abdullah IH, Kandeel F. et al. Insulin gene expression is regulated by DNA methylation. PloS one. 2009;4:e6953. doi:10.1371/journal.pone.0006953
42. Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:9821-6
43. Fellenberg J, Sahr H, Liu L, Schonsiegel F, Depeweg D, Lehner B. et al. Rescue of silenced UCHL1 and IGFBP4 expression suppresses clonogenicity of giant cell tumor-derived stromal cells. Cancer letters. 2013;336:61-7 doi:10.1016/j.canlet.2013.04.011
44. Han YX, Liang DY. The role of the tumor suppressor RUNX3 in giant cell tumor of the bone. International journal of oncology. 2012;40:673-8 doi:10.3892/ijo.2011.1249
45. Malhotra P, Kochhar R, Vaiphei K, Wig JD, Mahmood S. Aberrant promoter methylation of p16 in colorectal adenocarcinoma in North Indian patients. World journal of gastrointestinal oncology. 2010;2:295-303 doi:10.4251/wjgo.v2.i7.295
46. Takada H, Imoto I, Tsuda H, Nakanishi Y, Ichikura T, Mochizuki H. et al. ADAM23, a possible tumor suppressor gene, is frequently silenced in gastric cancers by homozygous deletion or aberrant promoter hypermethylation. Oncogene. 2005;24:8051-60 doi:10.1038/sj.onc.1208952
47. Costa FF, Colin C, Shinjo SM, Zanata SM, Marie SK, Sogayar MC. et al. ADAM23 methylation and expression analysis in brain tumors. Neuroscience letters. 2005;380:260-4 doi:10.1016/j.neulet.2005.01.050
48. Costa FF, Verbisck NV, Salim AC, Ierardi DF, Pires LC, Sasahara RM. et al. Epigenetic silencing of the adhesion molecule ADAM23 is highly frequent in breast tumors. Oncogene. 2004;23:1481-8 doi:10.1038/sj.onc.1207263
49. Kim JH, Jung EJ, Lee HS, Kim MA, Kim WH. Comparative analysis of DNA methylation between primary and metastatic gastric carcinoma. Oncology reports. 2009;21:1251-9
50. Verbisck NV, Costa ET, Costa FF, Cavalher FP, Costa MD, Muras A. et al. ADAM23 negatively modulates alpha(v)beta(3) integrin activation during metastasis. Cancer research. 2009;69:5546-52 doi:10.1158/0008-5472.CAN-08-2976
51. Cal S, Freije JM, Lopez JM, Takada Y, Lopez-Otin C. ADAM 23/MDC3, a human disintegrin that promotes cell adhesion via interaction with the alphavbeta3 integrin through an RGD-independent mechanism. Molecular biology of the cell. 2000;11:1457-69
52. Foulkes WD, Flanders TY, Pollock PM, Hayward NK. The CDKN2A (p16) gene and human cancer. Mol Med. 1997;3:5-20
53. Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian SV. et al. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994;264:436-40
54. Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC. et al. 5' CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nature medicine. 1995;1:686-92
55. Gonzalez-Zulueta M, Bender CM, Yang AS, Nguyen T, Beart RW, Van Tornout JM. et al. Methylation of the 5' CpG island of the p16/CDKN2 tumor suppressor gene in normal and transformed human tissues correlates with gene silencing. Cancer research. 1995;55:4531-5
56. Reed AL, Califano J, Cairns P, Westra WH, Jones RM, Koch W. et al. High frequency of p16 (CDKN2/MTS-1/INK4A) inactivation in head and neck squamous cell carcinoma. Cancer research. 1996;56:3630-3
57. Poetsch M, Hemmerich M, Kakies C, Kleist B, Wolf E, vom Dorp F. et al. Alterations in the tumor suppressor gene p16(INK4A) are associated with aggressive behavior of penile carcinomas. Virchows Archiv: an international journal of pathology. 2011;458:221-9 doi:10.1007/s00428-010-1007-4
58. Yuan D, Ye S, Pan Y, Bao Y, Chen H, Shao C. Long-term cadmium exposure leads to the enhancement of lymphocyte proliferation via down-regulating p16 by DNA hypermethylation. Mutation research. 2013;757:125-31 doi:10.1016/j.mrgentox.2013.07.007
59. Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature. 1994;368:753-6 doi:10.1038/368753a0
60. van Oosterwijk JG, de Jong D, van Ruler MA, Hogendoorn PC, Dijkstra PD, van Rijswijk CS. et al. Three new chondrosarcoma cell lines: one grade III conventional central chondrosarcoma and two dedifferentiated chondrosarcomas of bone. BMC cancer. 2012;12:375. doi:10.1186/1471-2407-12-375
61. van Beerendonk HM, Rozeman LB, Taminiau AH, Sciot R, Bovee JV, Cleton-Jansen AM. et al. Molecular analysis of the INK4A/INK4A-ARF gene locus in conventional (central) chondrosarcomas and enchondromas: indication of an important gene for tumour progression. The Journal of pathology. 2004;202:359-66 doi:10.1002/path.1517
62. Hayslip J, Montero A. Tumor suppressor gene methylation in follicular lymphoma: a comprehensive review. Molecular cancer. 2006;5:44. doi:10.1186/1476-4598-5-44
63. Veganzones-de-Castro S, Rafael-Fernandez S, Vidaurreta-Lazaro M, de-la-Orden V, Mediero-Valeros B, Fernandez C. et al. p16 gene methylation in colorectal cancer patients with long-term follow-up. Revista espanola de enfermedades digestivas: organo oficial de la Sociedad Espanola de Patologia Digestiva. 2012;104:111-7
64. Lee JJ, Ko E, Cho J, Park HY, Lee JE, Nam SJ. et al. Methylation and Immunoexpression of p16(INK4a) Tumor Suppressor Gene in Primary Breast Cancer Tissue and Their Quantitative p16(INK4a) Hypermethylation in Plasma by Real-Time PCR. Korean journal of pathology. 2012;46:554-61 doi:10.4132/KoreanJPathol.2012.46.6.554
65. Piaton E, Carre C, Advenier AS, Decaussin-Petrucci M, Mege-Lechevallier F, Lantier P. et al. p16 INK4a overexpression and p16/Ki-67 dual labeling versus conventional urinary cytology in the evaluation of urothelial carcinoma. Cancer cytopathology. 2014;122:211-20 doi:10.1002/cncy.21376
66. Yang HJ, Liu VW, Wang Y, Tsang PC, Ngan HY. Differential DNA methylation profiles in gynecological cancers and correlation with clinico-pathological data. BMC cancer. 2006;6:212. doi:10.1186/1471-2407-6-212
67. Xiao P, Chen JR, Zhou F, Lu CX, Yang Q, Tao GH. et al. Methylation of P16 in exhaled breath condensate for diagnosis of non-small cell lung cancer. Lung Cancer. 2014;83:56-60 doi:10.1016/j.lungcan.2013.09.008
68. Fu HY, Wu DS, Zhou HR, Shen JZ. CpG island methylator phenotype and its relationship with prognosis in adult acute leukemia patients. Hematology. 2013 doi:10.1179/1607845413Y.0000000137
69. Cao J, Zhou J, Gao Y, Gu L, Meng H, Liu H. et al. Methylation of p16 CpG island associated with malignant progression of oral epithelial dysplasia: a prospective cohort study. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:5178-83 doi:10.1158/1078-0432.CCR-09-0580
70. Herman JG, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE. et al. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer research. 1995;55:4525-30
71. Wettergren Y, Odin E, Nilsson S, Carlsson G, Gustavsson B. p16INK4a gene promoter hypermethylation in mucosa as a prognostic factor for patients with colorectal cancer. Mol Med. 2008;14:412-21 doi:10.2119/2007-00096.Wettergren
72. Sato F, Meltzer SJ. CpG island hypermethylation in progression of esophageal and gastric cancer. Cancer. 2006;106:483-93 doi:10.1002/cncr.21657
73. Koontongkaew S. The tumor microenvironment contribution to development, growth, invasion and metastasis of head and neck squamous cell carcinomas. Journal of Cancer. 2013;4:66-83 doi:10.7150/jca.5112
74. Rao VH, Singh RK, Bridge JA, Neff JR, Schaefer GB, Delimont DC. et al. Regulation of MMP-9 (92 kDa type IV collagenase/gelatinase B) expression in stromal cells of human giant cell tumor of bone. Clinical & experimental metastasis. 1997;15:400-9
75. Matsui H, Yamasaki M, Nakata K, Amano K, Nakamura H. Expression of MMP-8 and MMP-13 in the development of periradicular lesions. International endodontic journal. 2011;44:739-45 doi:10.1111/j.1365-2591.2011.01880.x
76. Ozeki N, Kawai R, Yamaguchi H, Hiyama T, Kinoshita K, Hase N. et al. IL-1beta-induced matrix metalloproteinase-13 is activated by a disintegrin and metalloprotease-28-regulated proliferation of human osteoblast-like cells. Experimental cell research. 2014;323:165-77 doi:10.1016/j.yexcr.2014.02.018
77. Mak IW, Cowan RW, Popovic S, Colterjohn N, Singh G, Ghert M. Upregulation of MMP-13 via Runx2 in the stromal cell of Giant Cell Tumor of bone. Bone. 2009;45:377-86 doi:10.1016/j.bone.2009.04.253
78. Yoshida S, Takahashi H. Expression of extracellular matrix molecules in brain metastasis. Journal of surgical oncology. 2009;100:65-8 doi:10.1002/jso.21296
79. Laudanski P, Swiatecka J, Kozlowski L, Lesniewska M, Wojtukiewicz M, Wolczynski S. Increased serum level of membrane type 1-matrix metalloproteinase (MT1-MMP/MMP-14) in patients with breast cancer. Folia histochemica et cytobiologica / Polish Academy of Sciences, Polish Histochemical and Cytochemical Society. 2010;48:101-3 doi:10.2478/v10042-009-0085-0
80. Wei J, Xu G, Wu M, Zhang Y, Li Q, Liu P. et al. Overexpression of vimentin contributes to prostate cancer invasion and metastasis via src regulation. Anticancer research. 2008;28:327-34
81. Zhou C, Nitschke AM, Xiong W, Zhang Q, Tang Y, Bloch M. et al. Proteomic analysis of tumor necrosis factor-alpha resistant human breast cancer cells reveals a MEK5/Erk5-mediated epithelial-mesenchymal transition phenotype. Breast cancer research: BCR. 2008;10:R105. doi:10.1186/bcr2210
82. Chen YR, Juan HF, Huang HC, Huang HH, Lee YJ, Liao MY. et al. Quantitative proteomic and genomic profiling reveals metastasis-related protein expression patterns in gastric cancer cells. Journal of proteome research. 2006;5:2727-42 doi:10.1021/pr060212g
83. Wang JW, Peng SY, Li JT, Wang Y, Zhang ZP, Cheng Y. et al. Identification of metastasis-associated proteins involved in gallbladder carcinoma metastasis by proteomic analysis and functional exploration of chloride intracellular channel 1. Cancer letters. 2009;281:71-81 doi:10.1016/j.canlet.2009.02.020
84. Jung S, Yi L, Kim J, Jeong D, Oh T, Kim CH. et al. The role of vimentin as a methylation biomarker for early diagnosis of cervical cancer. Molecules and cells. 2011;31:405-11 doi:10.1007/s10059-011-0229-x
85. Iwatsuki M, Mimori K, Fukagawa T, Ishii H, Yokobori T, Sasako M. et al. The clinical significance of vimentin-expressing gastric cancer cells in bone marrow. Annals of surgical oncology. 2010;17:2526-33 doi:10.1245/s10434-010-1041-0
86. Behzatoglu K, Durak H, Canberk S, Aydin O, Huq GE, Oznur M. et al. Giant cell tumor-like lesion of the urinary bladder: a report of two cases and literature review; giant cell tumor or undifferentiated carcinoma? Diagnostic pathology. 2009;4:48. doi:10.1186/1746-1596-4-48
87. Zhao Y, Yan Q, Long X, Chen X, Wang Y. Vimentin affects the mobility and invasiveness of prostate cancer cells. Cell biochemistry and function. 2008;26:571-7 doi:10.1002/cbf.1478
88. Kumar B, Koul S, Petersen J, Khandrika L, Hwa JS, Meacham RB. et al. p38 mitogen-activated protein kinase-driven MAPKAPK2 regulates invasion of bladder cancer by modulation of MMP-2 and MMP-9 activity. Cancer research. 2010;70:832-41 doi:10.1158/0008-5472.CAN-09-2918
89. Behren A, Muhlen S, Acuna Sanhueza GA, Schwager C, Plinkert PK, Huber PE. et al. Phenotype-assisted transcriptome analysis identifies FOXM1 downstream from Ras-MKK3-p38 to regulate in vitro cellular invasion. Oncogene. 2010;29:1519-30 doi:10.1038/onc.2009.436
90. Junttila MR, Ala-Aho R, Jokilehto T, Peltonen J, Kallajoki M, Grenman R. et al. p38alpha and p38delta mitogen-activated protein kinase isoforms regulate invasion and growth of head and neck squamous carcinoma cells. Oncogene. 2007;26:5267-79 doi:10.1038/sj.onc.1210332
91. Demuth T, Reavie LB, Rennert JL, Nakada M, Nakada S, Hoelzinger DB. et al. MAP-ing glioma invasion: mitogen-activated protein kinase kinase 3 and p38 drive glioma invasion and progression and predict patient survival. Molecular cancer therapeutics. 2007;6:1212-22 doi:10.1158/1535-7163.MCT-06-0711
92. Johansson E, Komuro A, Iwata C, Hagiwara A, Fuse Y, Watanabe A. et al. Exogenous introduction of tissue inhibitor of metalloproteinase 2 reduces accelerated growth of TGF-beta-disrupted diffuse-type gastric carcinoma. Cancer science. 2010;101:2398-403 doi:10.1111/j.1349-7006.2010.01688.x
93. Giannopoulos G, Pavlakis K, Parasi A, Kavatzas N, Tiniakos D, Karakosta A. et al. The expression of matrix metalloproteinases-2 and -9 and their tissue inhibitor 2 in pancreatic ductal and ampullary carcinoma and their relation to angiogenesis and clinicopathological parameters. Anticancer research. 2008;28:1875-81
Author contact
![]() Corresponding author: Paula Rahal, PhD, Department of Biology, Instituto de Biociências, Letras e Ciências Exatas - IBILCE/UNESP, Street: Cristóvão Colombo, 2265, Zip/Postal Code: 15054-000 - City: São José do Rio Preto, SP, Brazil. E-mail address: rahalpcom.br, Phone: +55 17 3221-2379, Fax: +55 17 3221-2390.
Corresponding author: Paula Rahal, PhD, Department of Biology, Instituto de Biociências, Letras e Ciências Exatas - IBILCE/UNESP, Street: Cristóvão Colombo, 2265, Zip/Postal Code: 15054-000 - City: São José do Rio Preto, SP, Brazil. E-mail address: rahalpcom.br, Phone: +55 17 3221-2379, Fax: +55 17 3221-2390.
Citation styles
APA
Conceição, A.L.G., Babeto, E., Candido, N.M., Franco, F.C., de Campos Zuccari, D.A.P., Bonilha, J.L., Cordeiro, J.A., Calmon, M.F., Rahal, P. (2015). Differential Expression of ADAM23, CDKN2A (P16), MMP14 and VIM Associated with Giant Cell Tumor of Bone. Journal of Cancer, 6(7), 593-603. https://doi.org/10.7150/jca.11238.
ACS
Conceição, A.L.G.; Babeto, E.; Candido, N.M.; Franco, F.C.; de Campos Zuccari, D.A.P.; Bonilha, J.L.; Cordeiro, J.A.; Calmon, M.F.; Rahal, P. Differential Expression of ADAM23, CDKN2A (P16), MMP14 and VIM Associated with Giant Cell Tumor of Bone. J. Cancer 2015, 6 (7), 593-603. DOI: 10.7150/jca.11238.
NLM
Conceição ALG, Babeto E, Candido NM, Franco FC, de Campos Zuccari DAP, Bonilha JL, Cordeiro JA, Calmon MF, Rahal P. Differential Expression of ADAM23, CDKN2A (P16), MMP14 and VIM Associated with Giant Cell Tumor of Bone. J Cancer 2015; 6(7):593-603. doi:10.7150/jca.11238. https://www.jcancer.org/v06p0593.htm
CSE
Conceição ALG, Babeto E, Candido NM, Franco FC, de Campos Zuccari DAP, Bonilha JL, Cordeiro JA, Calmon MF, Rahal P. 2015. Differential Expression of ADAM23, CDKN2A (P16), MMP14 and VIM Associated with Giant Cell Tumor of Bone. J Cancer. 6(7):593-603.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.