Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(8):694-700. doi:10.7150/jca.11709 This issue Cite
Review
Genes Regulating Epithelial Polarity Are Critical Suppressors of Esophageal Oncogenesis
Xiu-Min Li1, Hui Wang2, Li-Li Zhu1, Run-Zhen Zhao3, Hong-Long Ji3,4 ![]()
1. Center for Cancer Research, Xinxiang Medical University, Xinxiang Henan, 453003, China.
2. Ontario Cancer Institute, Campbell Family Institute for Breast Cancer Research, University of Toronto, Toronto, Ontario, M5G 2M9, Canada.
3. Department of Cellular and Molecular Biology, University of Texas Health Science Center at Tyler, Tyler, Texas, 75708, USA
4. Texas Lung Injury Institute, University of Texas Health Science Center at Tyler, Tyler, Texas, 75708, USA.
Received 2015-1-27; Accepted 2015-5-5; Published 2015-6-11
Citation:
Li XM, Wang H, Zhu LL, Zhao RZ, Ji HL. Genes Regulating Epithelial Polarity Are Critical Suppressors of Esophageal Oncogenesis. J Cancer 2015; 6(8):694-700. doi:10.7150/jca.11709. https://www.jcancer.org/v06p0694.htm
Other stylesAbstract
Esophageal cancer is an aggressive disease featured by early lymphatic and hematogenous dissemination, and is the sixth leading cause of cancer-related deaths worldwide. The proper formation of apicobasal polarity is essential for normal epithelium physiology and tissue homeostasis, while loss of polarity is a hallmark of cancer development including esophageal oncogenesis. In this review, we summarized the stages of esophageal cancer development associated with the loss or deregulation of epithelial cell apicobasal polarity. Loss of epithelial apicobasal polarity exerts an indispensable role in the initiation of esophageal oncogenesis, tumor progression, and the advancement of tumors from benign to malignant. In particular, we reviewed the involvement of several critical genes, including Lkb1, claudin-4, claudin-7, Par3, Lgl1, E-cadherin, and the Scnn1 gene family. Understanding the role of apicobasal regulators may lead to new paradigms for treatment of esophageal tumors, including improvement of prognostication, early diagnosis, and individually tailored therapeutic interventions in esophageal oncology.
Keywords: apicobasal polarity, esophageal adenocarcinoma, esophageal squamous cell carcinoma, LKB1, LGL1, SCNN1
Introduction
Esophageal cancer is characterized by early metastatic spread and intrinsic resistance to current systemic therapies, and although considerable progress has been made in both basic and clinical studies, the disease remains a major public health threat and the prognosis at the advanced stages is still very poor (1, 2). The two major types of esophageal cancer, adenocarcinoma and squamous cell carcinoma, have significantly distinct features in both etiology and epidemiology. Esophageal adenocarcinoma is more prevalent in Western countries, with a dramatic increase in incidence observed in the last two decades. The precursor condition for esophageal adenocarcinoma is Barrett's esophagus, which can proceed from dysplastic stages to adenocarcinoma at an overall rate of 0.33% per year (3). In sharp contrast, esophageal squamous cell carcinoma (ESCC) is more prevalent in the developing world, with very high incidences in East Asia and the Caspian belt (also known as the central Asian esophageal cancer belt) (4, 5).
The proper establishment and maintenance of cellular polarity (apicobasal combined with planar cell polarity) play an indispensable role in epithelium formation. Conversely, disruption of cell polarity and tissue organization is a hallmark of epithelial tissue-orientated cancer (6). In this review, following an overview of epithelial cell polarity changes in cancer development, we highlight the latest advancements in our understanding of how the misexpression of polarity genes contributes to the progress of esophageal cancer. The authors regret that, due to page limitations, not all applicable studies could be included in this review.
Epithelial polarity and oncogenesis
Epithelial cells comprise the foundation for the majority of organs in the mammalian body, and are the origin of approximately 90% of all human cancers. Characteristically, epithelial cells form intercellular adhesions, exhibit apicobasal polarity, and orient their mitotic spindles in the plane of the epithelial sheet (6, 7). Defects in these attributes result in the tissue disorganization associated with cancer (8). Therefore, the dysregulation of genes governing the establishment or maintenance of epithelium polarity is likely to be essential during oncogenesis.
A distinctive feature of polarized epithelia is the presence of adherens and tight junctions, which control the segregation of the plasma membrane biochemically and functionally into distinct apical and basolateral domains (Figure 1). In vertebrate epithelial cells, the adherens and tight junctions are localized to the apex of the lateral membrane, at the boundary between the apical and basolateral plasma membrane domains (9). The basolateral domains are enriched in cell-cell adhesion molecules and receptors for the extracellular matrix (Figure 1). Three groups of proteins act together to generate and maintain apicobasal polarity: 1) the PAR complex, which is composed of Par proteins, CDC42 proteins, and atypical protein kinase C (aPKC); 2) the Scrib complex, comprising Scrib, Dlg, and LGL1 proteins; and 3) the Crumbs (Crb) complex, consisting of PALS1 and PATJ, in addition to Crb molecules (8). These polarity proteins contribute to the establishment of epithelial apicobasal polarity and were first identified using model organisms, such as Caenorhabditis elegans and Drosophila melanogaster (10-13). Additionally, most of these polarity proteins are evolutionarily highly conserved with pivotal roles in several other critical cellular biological processes, for example, proliferation, differentiation, and migration (8). Drosophila studies have clearly demonstrated the tumor suppressive function of the polarity complexes (14). Moreover, ours and other groups found that epithelium polarity acts as an independent tumor suppressor through participating in the establishment and maintenance of the three-dimensional organization of epithelial tissue (6, 15, 16).
In the following sections, we summarize recent progress towards understanding the involvement of epithelial cell polarity genes in the development of esophageal cancer. The crucial roles of two other key genes, namely, CRB3 and DLG5, in maintaining epithelial polarity and oncogenesis were reviewed very recently (17, 18), and we have not duplicated related material in this review.
Figure 1
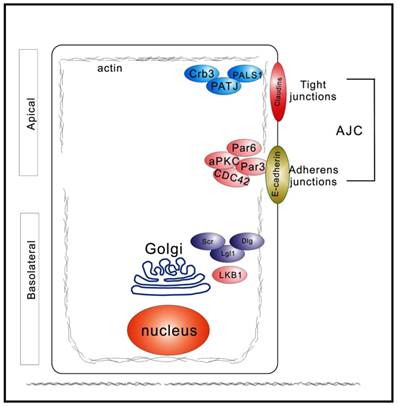
Diagram depicting spatial distribution of polarity regulators in epithelial cells polarized along the apicobasal axis. Three major polarity networks participate in the formation and maintenance of apicobasal polarity. The Crumbs complex (Crb3, PALS1, and PATJ) is required to establish the apical membrane. The PAR complex (Par3, Par6, aPKC, and CDC42) promotes the establishment of the apicolateral membrane border. The Scrib, or Scribble, complex (Scr, Dlg, and Lgl1) defines the basolateral plasma membrane domain. These three complexes act antagonistically and spatiotemporally to regulate epithelial polarization.
LKB1
The tumor suppressor protein liver kinase B1 (LKB1), also named serine/threonine kinase 11 or Par4, is a serine/threonine kinase. It has a critical role in cell proliferation, polarity, and spindle orientation. Inactivation of LKB1 has been linked to tumorigenesis in various types of cancer (19, 20). For example, loss of LKB1 induces a progrowth metabolic program in proliferating A549 cells, which were derived from non-small cell lung cancer tissue, and cells lacking LKB1 display increased uptake and utilization of glucose and glutamine. This effect is dependent on the hypoxia-inducible factor-1 alpha (HIF-1 alpha) (21). In addition, mouse germline of LKB1 mutation displays an impaired spindle orientation in the upper gastrointestinal tract. This observation is validated by spindle misorientation in three-dimensional MDCK cell cysts caused by RNAi for LKB1 (PMID:22815934). Apparently, LKB1-mediated spindle orientation could be an important tumor suppressor. Classic studies from Liu and coworkers regarding the relationship between LKB1 and the WNT signaling pathway in esophageal carcinoma suggest that LKB1 transcription was significantly lower in cancerous tissues than in adjacent normal epithelium (22). The reduced expression of LKB1 correlated tightly with progression stages, and furthermore, the expression of WNT-targeted downstream genes, e.g., cyclin D1, c-Myc, MMP2, and FZD2, was significantly upregulated in esophageal cancer tissues. LKB1 overexpression in TE10 cells inhibited the expression of WNT target genes even in the presence of WNT3A. Conversely, LKB1 knockdown enhanced WNT signaling activity. Moreover, LKB1 antagonized the WNT signaling pathway, through interaction with GSK3beta, to downregulate beta-catenin expression. Therefore, LKB1 may antagonize WNT-induced cell proliferation (22).
Another pioneer study investigated the regulation of CREB-regulated transcription co-activators (CRTCs) through phosphorylation by LKB1, and the roles of this regulation in the migration and invasion of esophageal cancer cells (23). The authors reported altered LKB1-CRTC signaling in a subset of cell lines and biopsy specimens of esophageal cancer, and LKB1 negatively regulated cell migration and invasion in vitro. Furthermore, CRTC signaling was activated subsequent to the loss of LKB1 expression, which results in the transcriptional activation of specific downstream molecules, such as LYPD3 (23). Reduction in LKB1 activity leads to nuclear abnormalities through impairing mitotic spindle integrity. Mitotic delay, binuclear, enlarged, and micronuclear are found in LKB1 deficiency. In esophageal cancerous tissues, phospho-PLK1 is negatively correlated with LKB1 when an H-score analysis was applied. Intriguingly, LKB1 deficiency-induced centrosome amplification depends on Polo-like kinase 1 but not AMP-activated protein kinase (PMID: 24722282). Conclusively, dysregulation of LKB1 is a crucial step in the progression of esophageal cancer, including proliferation, migration, and invasion (Figure 2).
Claudins
Claudins belong to a family of adhesion proteins with sizes of 22-27 kDa, and have four transmembrane domains, with both the N-terminus and C-terminus located in the cytoplasm (24). Claudins are the most important components of tight junctions, which establish a paracellular epithelial barrier that controls transport of electrolytes and biomolecules across the epithelium. In normal esophagi, claudin-7 was confined to the cell membrane of differentiated keratinocytes. In tumor samples, however, claudin-7 expression was either not detectable or mislocalized in the cytosol (25). Knockdown of this gene in squamous cell carcinoma cell lines led to reduced expression of E-cadherin, increased cell growth, and enhanced invasion into a three-dimensional matrix. In contrast, cells overexpressing claudin-7 became more adhesive and less invasive, accompanied by increased E-cadherin expression. Mislocalized claudin-7, therefore, apparently promotes transformation and regulates E-cadherin expression during oncogenesis of esophageal keratinocytes (25).
Figure 2
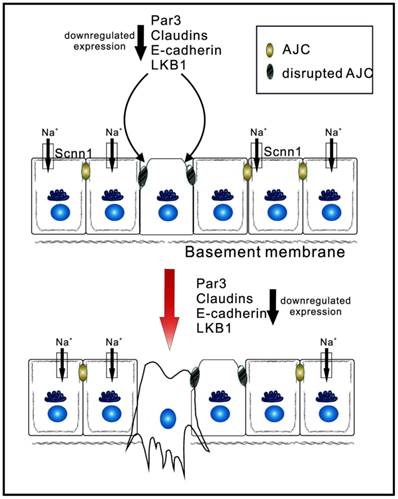
Epithelial apicobasal polarity suppresses the development of tumor to later stages. Apicobasal polarity is involved in the establishment and maintenance of the apical junction complex (AJP). Downregulation of core polarity proteins (Par3, claudins, E-cadherin, and LKB1) weakens the AJC and disrupts the basolateral membrane. Eventually, stromal invasion, as well as lymphatic and hematogenous dissemination, takes place, allowing the spread of carcerous cells.
The prognostic significance of claudin-4 was investigated in 164 cases of ESCC using immunohistochemistry (26). Decreased claudin-4 expression at the mRNA level was found to be significantly associated with invasion depth, lymph node metastasis, and poor overall survival. Furthermore, this study demonstrated that loss of claudin-4 was associated with promoter hypermethylation (26). One transcriptional regulator of claudin-4, termed Twist1, was involved in epithelial-mesenchymal transition by suppressing intercellular adhesion. Upregulation of Twist1 decreased claudin-4 expression levels, and this suppression was mediated by direct binding of Twist1 to the canonical E-box in the promoter region of claudin-4. Greater expression of Twist1 and reduced levels of claudin-4 were both associated with the poorest prognosis, and were also closely correlated with adverse outcomes. Therefore, Twist1 can induce the repression of claudin-4 during epithelial-mesenchymal transition of esophageal carcinoma (27). In other words, factors such as Twist1 that induce epithelial-mesenchymal transition enhance oncogenic cell invasiveness and metastatic ability by repressing claudins (Figure 2).
PARD-3
The partition-defective 3 (PARD-3) proteins are implicated in the formation of tight junctions at epithelial cell-cell contacts (16, 28). Aberrant DNA copy number in human ESCC cells was investigated using a high-density oligonucleotide microarray, and a homozygous deletion of PARD3 gene, encoding the PAR-3 protein, was detected. Exogenous expression of PARD3 gene in PARD3-deficient ESCC cells enhanced the recruitment of zonula occludens-1 (ZO-1, a marker of tight junctions) to cell-cell contact sites (29). Conversely, knockdown of PARD3 caused a disrupted localization of ZO-1 protein at cell-cell borders. In addition, the copy number of PARD3 was reduced by 15% in freshly harvested ESCC cells. Consistent with in vitro analysis, a significant difference in the expression of PARD3 was observed between ESCC tumor tissues and normal counterparts from the same patients. It is noteworthy that the reduction in PARD3 expression was associated with both detectable lymph node metastasis and poor differentiation. Mitotic spindle pole orientation could be affected by PARD3, for spindle pole orientation during epithelial cell division by excluding Pins from the apical cortex is regulated by PARD3. Compromised PARD3 activity causally disrupts spindle orientation in MDCK three-dimensional cultures (PMID: 20933426). Therefore, these observations raise the intriguing possibility that abnormality in PARD3 could be a novel mechanism for the progression of ESCC (Figure 2) (29).
LGL1
Lethal giant larvae (LGL) protein has been implicated in the maintenance of cell polarity in Drosophila and cultured mammalian cells. Downregulation of LGL1 has been detected in approximately 50% of primary cultures of human ESCC, and a reverse correlation was described between suppressed LGL1 and clinical characteristics, including severity, lymph node metastasis, tumor invasion, and survival (30). Furthermore, this group reported that LGL1 could inhibit cell growth, clonogenicity, cell motility, and tumor formation in nude mice. It is likely that under physiological conditions, LGL1 activates extracellular signal-regulated kinase (ERK1/2) to downregulate matrix metalloproteinase (MMP)-3, and subsequently to suppress tumor metastasis (30). These observations were substantiated by another study (31), in which overexpression of LGL1 decreased growth of human ESCC cells through induction of G0/G1 cell-cycle arrest. Moreover, LGL1 promoted apoptosis by modulating Bcl-2 and Bax expression and elevating both caspase-3 and caspase-9 activation. The growth arrest and apoptosis induction were confirmed in vivo in a xenograft model with hyperactivated LGL1 (31). It can be postulated that upregulation of LGL1 would suppress esophageal cancer via inhibiting proliferation and promoting apoptosis.
E-cadherin
E-cadherin is a transmembrane calcium-dependent cell adhesion protein of 120 kDa. Upon proteolytic cleavage, a soluble peptide of 80 kDa, known as soluble E-cadherin (s-Ecad), is present in circulation. A meta-analysis found that reduced E-cadherin expression was markedly associated with poor differentiation, indicating that attenuated expression of E-cadherin could be a potent biomarker in the prognosis of esophageal cancer, especially for squamous cell carcinoma (32). The clinical observations were subsequently corroborated by research demonstrating that the absence of E-cadherin strongly contributed to the tumorigenesis and metastasis of ESCC (33).
What are the underlying mechanisms for the repressed expression of E-cadherin in esophageal cancer? miR-92a, a microRNA, could be a key player. Metastasis to lymph nodes and the TNM stage of ESCC were associated with miR-92a significantly, and this may result from the direct targeting of the 3'-UTR of E-cadherin by miR-92a and the repression in E-cadherin expression. On the other hand, restoring the expression of E-cadherin (an miR-92a-resistant version) to normal levels in miR-92a-overexpressing cells recovered the pro-metastasis activity of miR-92a (34). Impaired mitotic spindle is critically associated with decreased E-cadherin activity. Furthermore, a reduction in E-cadherin expression following application of either RNAi or dominant-negative peptide also disorientates mitotic spindle (PMID: 19553471). Thus, in addition to impaired cell-cell junction and disrupted E-cadherin function facilitates, disoriented cell division may be involved in the development of esophageal cancer. However, whether s-Ecad can be used as an independent prognostic marker for esophageal cancer, as in other cancers (35, 36), needs further investigation (Figure 2).
Scnn1
Scnn1 genes encode four subunits of epithelial sodium channels (ENaC), namely, α, β, δ, and γ ENaC. One of the major physiological functions of tight polarized epithelial cell monolayers is to actively adjust the turnover and resolution of luminal fluid. The machinery of transepithelial ion salt and fluid transport is distributed in a polarity-dependent manner. For example, ENaC is located in the apical membrane, while Na+/K+-ATPase is expressed in the basolateral membrane. ENaC proteins are expressed in esophageal epithelial cells (37-39), and their expression and activity take place in a polarization-dependent manner (40, 41). Importantly, Scnn1 genes have been implicated in oncogenesis, based on their broad contributions to cell volume (42, 43), migration (44-48), proliferation (49-51), apoptosis (52, 53), neoplasm (44), metastasis (54), and epithelial-mesenchymal transformation (55). Oncogenic signal pathways could be involved in the aforementioned processes, including EGFR/MEK/ERK, PI3K/AKT, RTKs, WNT, and hedgehog signaling cascades (56). In addition to serving as a biomarker of the apical membrane for polarization, it is possible that ENaC proteins serve as critical mediators of the oncogenesis associated with the polarity-related genes reviewed above (Figure 2). From the technical aspect to study the polarity of cultured epithelial cells, attention should be paid to the culture mode used in studies involving cell polarity. The main drawback of two-dimensional (2-D) culturing methodologies is that cells grow as monolayers, bearing little to no structural resemblance to actual cancer tissue in situ. This fault in methodology could be overcome using 3-D ex vivo culture systems, which recapitulate numerous features of in vivo tumor tissues and which are gaining increasing popularity in the cancer research community (57, 58). Although this novel technique is still in its infancy (59), we believe that it will greatly accelerate studies in esophageal cancer.
Concluding remarks and future perspectives
A wealth of knowledge about the mechanisms that regulate cell polarity formation and maintenance has tremendously enriched our understanding of the tumorigensis of esophageal cancer as well as other tumors. In addition to disrupted epithelium organization, alterations in genes regulating epithelial polarization have been implicated in the development of esophageal cancer, including enhanced cell proliferation, abnormal metabolism, and reduced apoptosis. Therefore, these polarity-related genes could have potential applications as biomarkers in clinical prognosis. Scribble and Par6 are polarity regulators in other types of cancer progression (60, 61), but their roles in esophageal cancer remain obscure. Finally, understanding whether ENaC and membrane proteins, which are distributed in a polarity-dependent manner, mediate the development of esophageal cancer by these polarity-determinant genes awaits additional studies.
Abbreviations
CRTC, CREB-regulated transcription co-activator; AJC, apical junction complex; ESCC, esophageal squamous cell carcinoma; LKB1, liver kinase B1; Par3, partition-defective 3; Par6, partition-defective 6; LGL1, lethal giant larvae 1; Dlg 1, polarity protein disc large 1; Scr, scribble; CDC42, cell division control 42; PATJ, PALS1-associated tight junction protein; ENaC, epithelial sodium channel; Scnn1, sodium channel non-neuronal 1; CRB3, Crumbs homolog 3; DLG5, polarity protein discs large homolog 5.
Acknowledgements
This work was supported by the American Heart Association (Award 14GRNT20130034 to HLJ and RZ), the National High Technology Research and Development Program of China (Award 2012AA02A201-1 to XL) and the Science and Technology Development Plan of China (Awards 112102310283 and 122102310198 to XL).
Authors Contributions
HLJ and HW conceived the idea. XL, HW, and RZ searched the scientific literature for the most relevant articles on the topic. PW and HLJ integrated different sections of articles and drafted the manuscript. HW, XL, RZ and HLJ discussed the conclusions and designed the figures. All authors read and approved the final manuscript.
Author Details
XL is a Full Professor and LLZ is an instructor at the Center for Cancer Research, Xinxiang Medical University, 601 Jinsui Road, Xinxiang Henan, China, 453003. HW is a Senior Postdoctoral Research Scientist at the Ontario Cancer Institute, University of Toronto, Canada. RZ is an Instructor and HLJ is an Associate Professor in the Department of Cellular and Molecular Biology, University of Texas Health Science Center at Tyler, 11937 US Highway 271, Tyler, Texas 75708, USA.
Competing Interests
The authors declare that they have no competing interests.
References
1. Sakai NS, Samia-Aly E, Barbera M, Fitzgerald RC. A review of the current understanding and clinical utility of miRNAs in esophageal cancer. Semin Cancer Biol. 2013;23:512-21
2. Hong L, Han Y, Brain L. Epidermal growth factor receptor: an important target in esophageal cancer. Expert Opin Ther Targets. 2013;17:1179-85
3. Desai TK, Krishnan K, Samala N, Singh J, Cluley J, Perla S, Howden CW. The incidence of oesophageal adenocarcinoma in non-dysplastic Barrett's oesophagus: a meta-analysis. Gut. 2012;61:970-6
4. Abrahamsen B, Pazianas M, Eiken P, Russell RG, Eastell R. Esophageal and gastric cancer incidence and mortality in alendronate users. J Bone Miner Res. 2012;27:679-86
5. Kaz AM, Grady WM. Epigenetic biomarkers in esophageal cancer. Cancer Lett. 2014;342:193-9
6. Martin-Belmonte F, Perez-Moreno M. Epithelial cell polarity, stem cells and cancer. Nat Rev Cancer. 2012;12:23-38
7. McCaffrey LM, Macara IG. Epithelial organization, cell polarity and tumorigenesis. Trends Cell Biol. 2011;21:727-35
8. Etienne-Manneville S. Polarity proteins in migration and invasion. Oncogene. 2008;27:6970-80
9. Zegers MMP, O'Brien LE, Yu W, Datta A, Mostov KE. Epithelial polarity and tubulogenesis in vitro. Trends Cell Biol. 2003;13:169-76
10. Kemphues KJ, Priess JR, Morton DG, Cheng NS. Identification of genes required for cytoplasmic localization in early C. elegans embryos. Cell. 1988;52:311-20
11. Tepass U, Theres C, Knust E. crumbs encodes an EGF-like protein expressed on apical membranes of Drosophila epithelial cells and required for organization of epithelia. Cell. 1990;61:787-99
12. Knust E, Tepass U, Wodarz A. crumbs and stardust, two genes of Drosophila required for the development of epithelial cell polarity. Dev Suppl. 1993:261-8
13. Bilder D, Li M, Perrimon N. Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science. 2000;289:113-6
14. Royer C, Lu X. Epithelial cell polarity: a major gatekeeper against cancer? Cell Death Differ. 2011;18:1470-7
15. Hinck L, Nathke I. Changes in cell and tissue organization in cancer of the breast and colon. Curr Opin Cell Biol. 2014;26C:87-95
16. Wang H, Lacoche S, Huang L, Xue B, Muthuswamy SK. Rotational motion during three-dimensional morphogenesis of mammary epithelial acini relates to laminin matrix assembly. Proc Ntl Acad Sci U S A. 2013;110:163-8
17. Liu J, Li J, Ren Y, Liu P. DLG5 in cell polarity maintenance and cancer development. Int J Biol Sci. 2014;10:543-9
18. Li P, Mao X, Ren Y, Liu P. Epithelial Cell Polarity Determinant CRB3 in Cancer Development. Int J Biol Sci. 2015;11:31-7
19. Han X, Li F, Fang Z, Gao Y, Li F, Fang R, Yao S, Sun Y, Li L, Zhang W, Ma H, Xiao Q. et al. Transdifferentiation of lung adenocarcinoma in mice with Lkb1 deficiency to squamous cell carcinoma. Nat Commun. 2014;5:3261
20. Taliaferro-Smith L, Nagalingam A, Zhong D, Zhou W, Saxena NK, Sharma D. LKB1 is required for adiponectin-mediated modulation of AMPK-S6K axis and inhibition of migration and invasion of breast cancer cells. Oncogene. 2009;28:2621-33
21. Faubert B, Vincent EE, Griss T, Samborska B, Izreig S, Svensson RU, Mamer OA, Avizonis D, Shackelford DB, Shaw RJ, Jones RG. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1alpha. Proc Natl Acad Sci U S A. 2014;111:2554-9
22. Liu K, Luo Y, Tian H, Yu KZ, He JX, Shen WY. The tumor suppressor LKB1 antagonizes WNT signaling pathway through modulating GSK3beta activity in cell growth of esophageal carcinoma. Tumour Biol. 2014;35:995-1002
23. Gu Y, Lin S, Li JL, Nakagawa H, Chen Z, Jin B, Tian L, Ucar DA, Shen H, Lu J, Hochwald SN, Kaye FJ. et al. Altered LKB1/CREB-regulated transcription co-activator (CRTC) signaling axis promotes esophageal cancer cell migration and invasion. Oncogene. 2012;31:469-79
24. Findley MK, Koval M. Regulation and roles for claudin-family tight junction proteins. IUBMB Life. 2009;61:431-7
25. Lioni M, Brafford P, Andl C, Rustgi A, El-Deiry W, Herlyn M, Smalley KS. Dysregulation of claudin-7 leads to loss of E-cadherin expression and the increased invasion of esophageal squamous cell carcinoma cells. Am J Pathol. 2007;170:709-21
26. Sung CO, Han SY, Kim SH. Low expression of claudin-4 is associated with poor prognosis in esophageal squamous cell carcinoma. Annals of surgical oncology. 2011;18:273-81
27. Lee KW, Lee NK, Kim JH, Kang MS, Yoo HY, Kim HH, Um SH, Kim SH. Twist1 causes the transcriptional repression of claudin-4 with prognostic significance in esophageal cancer. Biochem Biophys Res Commun. 2012;423:454-60
28. Hao Y, Du Q, Chen X, Zheng Z, Balsbaugh JL, Maitra S, Shabanowitz J, Hunt DF, Macara IG. Par3 controls epithelial spindle orientation by a PKC-mediated phosphorylation of apical Pins. Curr Biol. 2010;20:1809-18
29. Zen K, Yasui K, Gen Y, Dohi O, Wakabayashi N, Mitsufuji S, Itoh Y, Zen Y, Nakanuma Y, Taniwaki M, Okanoue T, Yoshikawa T. Defective expression of polarity protein PAR-3 gene (PARD3) in esophageal squamous cell carcinoma. Oncogene. 2009;28:2910-8
30. Zhu YH, Liu H, Zhang LY, Zeng T, Song Y, Qin YR, Li L, Liu L, Li J, Zhang B, Guan XY. Downregulation of LGI1 promotes tumor metastasis in esophageal squamous cell carcinoma. Carcinogenesis. 2014;35:1154-61
31. Song J, Peng XL, Ji MY, Ai MH, Zhang JX, Dong WG. Hugl-1 induces apoptosis in esophageal carcinoma cells both in vitro and in vivo. World J Gastroenterol. 2013;19:4127-36
32. Xu XL, Ling ZQ, Chen SZ, Li B, Ji WH, Mao WM. The impact of E-cadherin expression on the prognosis of esophageal cancer: a meta-analysis. Dis Esophagus. 2014;27:79-86
33. Zhai JW, Yang XG, Yang FS, Hu JG, Hua WX. Expression and clinical significance of Ezrin and E-cadherin in esophageal squamous cell carcinoma. Chin J Cancer. 2010;29:317-20
34. Chen ZL, Zhao XH, Wang JW, Li BZ, Wang Z, Sun J, Tan FW, Ding DP, Xu XH, Zhou F, Tan XG, Hang J. et al. microRNA-92a promotes lymph node metastasis of human esophageal squamous cell carcinoma via E-cadherin. J Biol Chem. 2011;286:10725-34
35. Chan AO, Lam SK, Chu KM, Lam CM, Kwok E, Leung SY, Yuen ST, Law SY, Hui WM, Lai KC, Wong CY, Hu HC. et al. Soluble E-cadherin is a valid prognostic marker in gastric carcinoma. Gut. 2001;48:808-11
36. Katayama M, Hirai S, Kamihagi K, Nakagawa K, Yasumoto M, Kato I. Soluble E-cadherin fragments increased in circulation of cancer patients. Br J Cancer. 1994;69:580-5
37. Awayda MS, Bengrine A, Tobey NA, Stockand JD, Orlando RC. Nonselective cation transport in native esophageal epithelia. Am J Physiol Cell Physiol. 2004;287:C395-402
38. Tobey NA, Argote CM, Vanegas XC, Barlow W, Orlando RC. Electrical parameters and ion species for active transport in human esophageal stratified squamous epithelium and Barrett's specialized columnar epithelium. Am J Physiol Gastroint Liver Physiol. 2007;293:G264-70
39. Tobey NA, Argote CM, Awayda MS, Vanegas XC, Orlando RC. Effect of luminal acidity on the apical cation channel in rabbit esophageal epithelium. Am J Physiol Gastroint Liver Physiol. 2007;292:G796-805
40. Yue G, Hu P, Oh Y, Jilling T, Shoemaker RL, Benos DJ, Cragoe EJ Jr, Matalon S. Culture-induced alterations in alveolar type II cell Na+ conductance. Am J Physiol. 1993;265:C630-40
41. Jain L. et al. Expression of highly selective sodium channels in alveolar type II cells is determined by culture conditions. Am J Physiol. Lung Cell Mol. Physiol. 2001;280:L646-58
42. Wehner F. Cell volume-regulated cation channels. Contrib Nephrol. 2006;152:25-53
43. Fillon S, Warntges S, Matskevitch J, Moschen I, Setiawan I, Gamper N, Feng YX, Stegen C, Friedrich B, Waldegger S, Broer S, Wagner CA. et al. Serum- and glucocorticoid-dependent kinase, cell volume, and the regulation of epithelial transport. Comp Biochem Physiol A Mol Integr Physiol. 2001;130:367-76
44. Rooj AK, McNicholas CM, Bartoszewski R, Bebok Z, Benos DJ, Fuller CM. Glioma-specific cation conductance regulates migration and cell cycle progression. J Biol Chem. 2012;287:4053-65
45. Kapoor N, Bartoszewski R, Qadri YJ, Bebok Z, Bubien JK, Fuller CM, Benos DJ. Knockdown of ASIC1 and epithelial sodium channel subunits inhibits glioblastoma whole cell current and cell migration. J Biol Chem. 2009;284:24526-41
46. Grifoni SC, McKey SE, Drummond HA. Hsc70 regulates cell surface ASIC2 expression and vascular smooth muscle cell migration. Am J Physiol Heart Circ Physiol. 2008;294:H2022-30
47. Grifoni SC, Gannon KP, Stec DE, Drummond HA. ENaC proteins contribute to VSMC migration. Am J Physiol Heart Circ Physiol. 2006;291:H3076-86
48. Vila-Carriles WH, Kovacs GG, Jovov B, Zhou ZH, Pahwa AK, Colby G, Esimai O, Gillespie GY, Mapstone TB, Markert JM, Fuller CM, Bubien JK. et al. Surface expression of ASIC2 inhibits the amiloride-sensitive current and migration of glioma cells. J Biol Chem. 2006;281:19220-32
49. Zheleznova NN, Wilson PD, Staruschenko A. Epidermal growth factor-mediated proliferation and sodium transport in normal and PKD epithelial cells. Biochim Biophys Acta. 2011;1812:1301-13
50. Bondarava M, Li T, Endl E, Wehner F. alpha-ENaC is a functional element of the hypertonicity-induced cation channel in HepG2 cells and it mediates proliferation. Pflugers Arch. 2009;458:675-87
51. Lang F, Henke G, Embark HM, Waldegger S, Palmada M, Bohmer C, Vallon V. Regulation of channels by the serum and glucocorticoid-inducible kinase - implications for transport, excitability and cell proliferation. Cell Physiol Biochem. 2003;13:41-50
52. Pan JA, Fan Y, Gandhirajan RK, Madesh M, Zong WX. Hyperactivation of the mammalian degenerin MDEG promotes caspase-8 activation and apoptosis. J Biol Chem. 2013;288:2952-63
53. Hu W, Chen FH, Yuan FL, Zhang TY, Wu FR, Rong C, Jiang S, Tang J, Zhang CC, Lin MY. Blockade of acid-sensing ion channels protects articular chondrocytes from acid-induced apoptotic injury. Inflamm Res. 2012;61:327-35
54. Gusev Y, Riggins RB, Bhuvaneshwar K, Gauba R, Sheahan L, Clarke R, Madhavan S. In silico discovery of mitosis regulation networks associated with early distant metastases in estrogen receptor positive breast cancers. Cancer Inform. 2013;12:31-51
55. Aldehni F, Spitzner M, Martins JR, Barro-Soria R, Schreiber R, Kunzelmann K. Bestrophin 1 promotes epithelial-to-mesenchymal transition of renal collecting duct cells. J Am Soc Nephrol. 2009;20:1556-64
56. Ji HL, Zhao RZ, Chen ZX, Shetty S, Idell S, Matalon S. δ ENaC: a novel divergent amiloride-inhibitable sodium channel. Am J Physiol Lung Cell Mol Physiol. 2012;303:L1013-26
57. Yamada KM, Cukierman E. Modeling tissue morphogenesis and cancer in 3D. Cell. 2007;130:601-10
58. Geraldo S, Simon A, Elkhatib N, Louvard D, Fetler L, Vignjevic DM. Do cancer cells have distinct adhesions in 3D collagen matrices and in vivo? Eur J Cell Biol. 2012;91:930-7
59. Fujiwara D, Kato K, Nohara S, Iwanuma Y, Kajiyama Y. The usefulness of three-dimensional cell culture in induction of cancer stem cells from esophageal squamous cell carcinoma cell lines. Biochem Biophys Res Commun. 2013;434:773-8
60. Gunaratne A, Thai BL, Di Guglielmo GM. Atypical protein kinase C phosphorylates Par6 and facilitates transforming growth factor beta-induced epithelial-to-mesenchymal transition. Mol Cell Biol. 2013;33:874-86
61. Elsum IA, Yates LL, Pearson HB, Phesse TJ, Long F, O'Donoghue R, Ernst M, Cullinane C, Humbert PO. Scrib heterozygosity predisposes to lung cancer and cooperates with KRas hyperactivation to accelerate lung cancer progression in vivo. Oncogene. 2013;33:5523-33
Author contact
![]() Corresponding author: Dr. James (Hong-Long) Ji, MD, Department of Cellular and Molecular Biology and Department of Medicine, University of Texas Health Science Center at Tyler, Tyler, Texas, 75708, USA, Email: james.jiedu
Corresponding author: Dr. James (Hong-Long) Ji, MD, Department of Cellular and Molecular Biology and Department of Medicine, University of Texas Health Science Center at Tyler, Tyler, Texas, 75708, USA, Email: james.jiedu
Citation styles
APA
Li, X.M., Wang, H., Zhu, L.L., Zhao, R.Z., Ji, H.L. (2015). Genes Regulating Epithelial Polarity Are Critical Suppressors of Esophageal Oncogenesis. Journal of Cancer, 6(8), 694-700. https://doi.org/10.7150/jca.11709.
ACS
Li, X.M.; Wang, H.; Zhu, L.L.; Zhao, R.Z.; Ji, H.L. Genes Regulating Epithelial Polarity Are Critical Suppressors of Esophageal Oncogenesis. J. Cancer 2015, 6 (8), 694-700. DOI: 10.7150/jca.11709.
NLM
Li XM, Wang H, Zhu LL, Zhao RZ, Ji HL. Genes Regulating Epithelial Polarity Are Critical Suppressors of Esophageal Oncogenesis. J Cancer 2015; 6(8):694-700. doi:10.7150/jca.11709. https://www.jcancer.org/v06p0694.htm
CSE
Li XM, Wang H, Zhu LL, Zhao RZ, Ji HL. 2015. Genes Regulating Epithelial Polarity Are Critical Suppressors of Esophageal Oncogenesis. J Cancer. 6(8):694-700.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.