Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(8):740-749. doi:10.7150/jca.11691 This issue Cite
Research Paper
Methylation Profiling of Multiple Tumor Suppressor Genes in Hepatocellular Carcinoma and the Epigenetic Mechanism of 3OST2 Regulation
Haiyan Chen1, 2, #, Tingguo Zhang1, #, Yan Sheng1, #, Cheng Zhang1, Yunfei Peng1, Xiao Wang1, Cuijuan Zhang1, ![]()
1. Institute of Pathology and Pathophysiology, Shandong University School of Medicine, Jinan 250012, P. R. China
2. Department of Pathology, Shandong Provincial Chest Hospital, Jinan 250012, P. R. China
#These authors contributed equally to this work.
Received 2015-1-25; Accepted 2015-5-12; Published 2015-6-25
Citation:
Chen H, Zhang T, Sheng Y, Zhang C, Peng Y, Wang X, Zhang C. Methylation Profiling of Multiple Tumor Suppressor Genes in Hepatocellular Carcinoma and the Epigenetic Mechanism of 3OST2 Regulation. J Cancer 2015; 6(8):740-749. doi:10.7150/jca.11691. https://www.jcancer.org/v06p0740.htm
Other stylesAbstract
DNA methylation is considered as a significant mechanism that silences tumor suppressor genes (TSGs) and could be used in the early diagnosis of cancer. Histone modifications often work together with DNA methylation; however, how these epigenetic alterations regulate TSGs remains unclear. Here, we determined the methylation status of ten TSGs (3OST2, ppENK, CHFR, LKB1, THBS1, HIC1, SLIT2, EDNRB, COX2, and CLDN7) in hepatocellular carcinoma (HCC) and corresponding noncancerous tissues. Methylation profiling revealed that four genes had very high frequencies of methylation in HCCs, but interestingly, similar high frequencies were also detected in corresponding noncancerous tissues (97.9% vs 95.8% for SLIT2, 93.8% vs 81.3% for EDNRB, 66.7% vs 85.4% for HIC1, and 56.3% vs 56.3% for ppENK, P > 0.05). Only the 3OST2 gene was frequently methylated in HCCs and there was significant difference between HCCs and corresponding noncancerous tissues (68.8% vs 37.5%, P < 0.05). 5-aza-2'-deoxycytidine (5-Aza-CdR) or trichostatin A (TSA) alone could partially reverse 3OST2 methylation, and their combination resulted in complete reversal. UHRF1 and histone H3R8me2s were both enriched on the hypermethylated 3OST2 promoter, but H3R8me2a was not. After 5-Aza-CdR or TSA treatment, the UHRF1 and H3R8me2s enrichment was decreased, while H3R8me2a enrichment increased. We demonstrated that 3OST2 methylation may play a critical role in the earliest steps of hepatocarcinogenesis and is directly regulated by UHRF1. Furthermore, H3R8me2s acted as a repressive mark, while H3R8me2a was correlated with 3OST2 transcriptional activity.
Keywords: Hepatocellular carcinoma, DNA methylation, Histone modification, Tumor suppressor gene, Gene expression
Introduction
Hepatocellular carcinoma (HCC) is one of the most common human cancers worldwide, and it is the second leading cause of cancer-related death in China. Although several novel therapeutic methods have been developed in recent years, the 5-year survival rate of HCC remains poor, with currently 7% of survival rate due to delayed diagnosis. The majority of patients with advanced HCC do not survive for longer than 6 months from the time of diagnosis. Over the last two decades, many genetic alterations in cancer-related genes have been implicated in hepatocarcinogenesis, including mutations of p53, Rb1, β-catenin and DNA amplification [1-3]. However, these alterations more likely occurred in advanced HCC. Thus, markers for the early diagnosis of HCC are urgently needed.
Epigenetics has emerged recently as one of the most exciting frontiers in the study of human carcinogenesis [4,5]. Tumor suppressor genes (TSGs) that are hypermethylated in tumor cells have the potential to be diagnostic markers or prognostic indicators, and the functional pathways in which they participate might represent potential targets for therapeutic exploitation. The tumor- and tissue-specific TSGs methylation profiling had been made in a number of cancers, such as multiple myeloma, gallbladder cancer, ovarian cancer, etc. [6-8]. Frequent hypermethylation of several TSGs such as p16, GSTP1, E-cadherin, SOCS1 and RASSF1A have been implicated in hepatocarcinogenesis [9-11]. However, hypermethylation of TSGs is not only observed in HCC tissues, but also present in premalignant conditions such as dysplastic nodules or cirrhotic liver [12,13]. This generates confusion whether these genes could serve as epigenetic biomarkers for the early detection of liver cancer from premalignant liver diseases. Therefore, tumor-specific biomarkers for the early diagnosis of HCC are particularly important.
Another epigenetic phenomenon is post-translational modification of the N-terminal tails of core histones, including methylation, acetylation, ubiquitylation and phosphorylation. Histone modifications and DNA methylation represent two layers of epigenetic information that regulate chromatin structure and gene transcription. Methyl-CpG binding domain (MBD) proteins play important roles in the interactions of DNA methylation and histone modifications by either recognizing methylated DNA and recruiting histone modifiers, or recognizing histone modification status and recruiting DNA methyltransferases (DNMTs) [14]. In contrast to these proteins, UHRF1 (also known as ICBP90 or Np95) specifically recognizes both methylated DNA and trimethylated H3K9, and links them [15-17]. Our previous data showed that UHRF1 was overexpressed in HCC tissues but not in corresponding noncancerous tissues (data not shown); however, how it regulates HCC-related TSGs is unknown. It is now well established that there is a cross-talk between DNA methylation and histone lysine methylation. However, we currently know much less about the interactions between DNA methylation and arginine methylation, and the mechanisms by which histone arginine methylation regulates transcription remain poorly understood. More recently, it has been reported that, in addition to histone lysines, UHRF1 can bind to histone arginine H3R2 to regulate gene expression [18]. The new structure of the tandem tudor domain of UHRF1 bound with histone H3 tail reveals that it contains three structured H3 residues, H3R8-H3K9me3-H3S10 [19].
In the present study, we first made the promoter methylation profile of ten TSGs (3OST2, ppENK, CHFR, LKB1, THBS1, HIC1, SLIT2, EDNRB, COX2, and CLDN7) in 48 paired HCCs and corresponding noncancerous tissues. Subsequently, we selected the most valuable biomarker to explore its epigenetic regulation mechanisms, using a demethylating agent, 5-aza-2-deoxycytidine (5-Aza-CdR), and a histone deacetylase (HDAC) inhibitor, trichostatin A (TSA), alone or in combination in HCC cells. Whether UHRF1 or the different methylation states of H3R8 are associated with the hypermethylated biomarker was also analyzed. Our present data may provide useful clues for the relationship between DNA methylation and histone modifications in the regulation of DNA methylation-induced gene silencing.
Materials and Methods
Patients and tissue specimens
Forty-eight cases of HCC and matched corresponding noncancerous liver tissues with confirmed histopathological diagnosis were obtained immediately after hepatectomies at the Prince of Wales Hospital in Hong Kong. Both the HCC and noncancerous liver tissues were immediately snapped frozen in liquid nitrogen and stored at -80ºC in a freezer before DNA extraction. The median age of the patients was 54.3 years (range of 17 to 75). The patients received clinical follow-up for up to 80.3 months, and the clinical characteristics of the patients are shown in Supplementary Table 1. The Committee of Ethical Research at Shandong University and the Ethics Committee of Chinese University of Hong Kong, China provided ethical approval.
Cell lines and inhibitors treatment
Human normal liver cell line QSG-7701 and HCC cell line BEL-7402 were obtained from the Chinese Academy of Sciences Cell Bank, Shanghai. Both cell lines were maintained in RPMI 1640 with 10% fetal bovine serum at 37°C and 5% CO2. BEL-7402 cells were plated and incubated with 5-Aza-CdR and TSA (Sigma, St. Louis, MO, USA) either alone or in combination. Cells were exposed to 5-Aza-CdR (5 μmol/L) for 96 h or to TSA (300 ng/ml) for 48 h. For combined treatment, cells were initially cultured with 5-Aza-CdR (5 μmol/L) for 48 h and subsequently co-treated with TSA (300 ng/ml) for an additional 48 h.
Table 1
The Methylation-specific PCR primers of ten tumor suppressor genes
| Gene | Sense 5'-3' | Antisense 5'-3' |
|---|---|---|
| 3OST2(M) | CGGTTGTTCGGAGTTTTATC | GTAACGCTACCACGACCACG |
| 3OST2(U) | TGGAGTTTTATTGTTTAGGATT | AAAACTCACATAACACTACCACA |
| ppENK(M) | TGTGGGGAGTTATCGAGC | GCCTTCGCGAAAAAAATCG |
| ppENK(U) | TTGTGTGGGGAGTTATTGAGT | CACCTTCACAAAAAAAATCAATC |
| CHFR(M) | TTTCGTGATTCGTAGGCGAC | GCGATTAACTAACGACGACG |
| CHFR(U) | TTTTGTGATTTGTAGGTGAT | ACAATTAACTAACAACAACA |
| LKB1(M) | ACGAAGTTGATTTTGATCGGGTC | CGATACAAAATCTACGAACCGACG |
| LKB1(U) | GGATGAAGTTGATTTTGATTGGGTT | ACCCAATACAAAATCTACAAACCAACA |
| THBS1(M) | TGCGAGCGTTTTTTTAAATGC | TAAACTCGCAAACCAACTCG |
| THBS1(U) | GTTTGGTTGTTGTTTATTGGTTG | CCTAAACTCACAAACCAACTCA |
| HIC1(M) | TCGGTTTTCGCGTTTTGTTCGT | AACCGAAAACTATCAACCCTCG |
| HIC1(U) | TTGGGTTTGGTTTTTGTGTTTTG | CACCCTAACACCACCCTAAC |
| SLIT2(M) | CGGTTTAGGTTGCGGCGGAGTCGAGGGC | CGCGAAAACCCAACGAACCCGTAACAAAACGCG |
| SLIT2(U) | TGGTTTAGGTTGTGGTGGAGTTGAGGGT | CACAAAAACCCAACAAACCCATAACAAAACACA |
| EDNRB(M) | AAATTGCGGAGCGGTTATC | CCAAATCCGCGACAAACCG |
| EDNRB(U) | GGTTTTGAAATTGTGGAGTGG | TCCCCAAATCCACAACAAAC |
| COX2 (M) | TTAGATACGGCGGCGGCGGC | TCTTTACCCGAACGCTTCCG |
| COX2 (U) | ATAGATTAGATATGGTGGTGGTGGT | CACAATCTTTACCCAAACACTTCCA |
| CLDN-7(M) | GACGTTAGGTTATTTTCGGTC | AAACGCGTTTCTAAACGCCG |
| CLDN-7(U) | TGGGGAAAGGGTGGTGTTG | TTACCCAATTTTAACCACCAC |
Table 2
Methylation profiling of tumor suppressor genes in primery HCCs and the matched corresponding noncancerous tissues
| Gene | Annealing temperature (℃) | Located chromosome | Methylated rate in HCCs | Methylated rate in non-HCCs |
|---|---|---|---|---|
| 3OST2 (M) | 60 | 16p12 | 68.8%* | 37.5% |
| 3OST2 (U) | 60 | |||
| ppENK(M) | 62 | 8q23-q24 | 56.3% | 56.3% |
| ppENK(U) | 62 | |||
| CHFR (M) | 58 | 12q24.33 | 20.8%** | 2.1% |
| CHFR (U) | 53 | |||
| LKB1(M) | 60 | 19p13.3 | 4.2% | 0% |
| LKB1(U) | 60 | |||
| THBS1(M) | 62 | 15q15 | 16.7% | 22.9% |
| THBS1(U) | 62 | |||
| HIC1(M) | 60 | 17p13.3 | 66.7% | 85.4% |
| HIC1 (U) | 60 | |||
| SLIT2 (M) | 68 | 4p15.2 | 97.9% | 95.8% |
| SLIT2 (U) | 59 | |||
| EDNRB(M) | 58 | 13q22 | 93.8% | 81.3% |
| EDNRB(U) | 58 | |||
| COX2 (M) | 60 | 1q25.2- q25.3 | 25%** | 4.2% |
| COX2 (U) | 60 | |||
| CLDN7 (M) | 56 | 17p13 | 10.4% | 14.6% |
| CLDN7 (U) | 56 |
HCC, Hepatocellular carcinoma.
P value is relative to non-HCCs ( χ2 tests). *P < 0.05; ** P < 0.01.
Methylation-specific PCR (MSP)
Genomic DNA was extracted from tissue samples and liver cell lines using a Genomic DNA Purification Kit (QIAGEN, Hilden, Germany). Bisulfite modification of the DNA was carried out by the Intergen CpGenome DNA modification Kit (Intergen Company, New York, NY) according to the manufacturer's protocol. Methylation-specific PCR (MSP) was performed with methylation-specific primers (sequences are shown in Table 1). Each PCR reaction was performed with a total volume of 12.5 μL, which contained 0.05 μL of Hot-StarTaq Master Mix (Qiagen), 1 μg of bisulfite-treated DNA template, and 0.2 μM of each primer pair. Annealing temperature of PCR amplification for each gene is list in Table 2. The reaction mixture was incubated at 95°C for 12min, followed by 40 cycles of 94°C for 30 sec, X°C for 30 sec (X= annealing temperature), 72°C for 45 sec, and a final extension at 72°C for 10 min. PCR products were analyzed on 3% TBE agarose gels, and subsequently visualized by ethidium bromide staining.
Real-time reverse transcription PCR (qRT-PCR)
Total RNA was extracted from HCC cells using TriZol reagent (Invitrogen) according to the manufacturer's instructions. First-strand cDNA was synthesized from 1 μg of total RNA with Primer Mix and RT Enzyme Mix (Toyobo, Japan). qRT-PCR was performed in a 20 μl reaction, which included 3 μl of cDNA template, 5 μM each of forward and reverse primer, and 10 μL of SYBR Green I (Bio-Rad, Hercules, CA, USA). The primer sequences for 3OST2 and UHRF1 amplification were: 3OST2 (F, 5'-CGGCGAGATGGGGCGAGTCC-3' and R, 5'-TCGGAGCTGGTCTATCACTT-3'), and UHRF1 (F, 5'-CCAGCAGAGCAGCCTCATC-3' and R, 5'-TCCTTGAGTGACGCCAGGA-3'). The transcription of β-actin was used as an internal control for normalization. PCR amplification was under the following conditions: 35 cycles of 95°C for 30s, 60°C for 60s. The Ct value was defined as the number of PCR cycles in which the fluorescence signal exceeded the detection threshold value. Firstly, ΔCt = Ct Gene - Ct β-actin. Then, ΔΔCt = ΔCt treated - ΔCt control. Lastly, 2-ΔΔCt was calculated to represent the relative mRNA expression of target genes.
Immunohistochemistry
Cells were seeded in a six-well plate that contained chamber slides and were exposed to inhibitors after the cells had adhered. To ensure that the antibody could enter the nuclei, the slides were incubated with 0.5% TritonX-100 for 30 min. The cells were incubated with antibodies against UHRF1 (1:100) overnight at 4°C, followed by incubation with secondary antibody for 1 hour at room temperature, and visualized using diaminobenzidine. Only distinct nuclear staining was considered as positive. Immunohistochemical scoring was completed using a semiquantitative assessment of both the intensity of staining (intensity score: absent, 0; weak, 1; moderate, 2; strong, 3) and the percentage of positive cells (extent score: absent, 0; <10%, 1; 10-50%, 2; >50%, 3). From multiplying the intensity score by the extent score, a score index was derived, which ranged from 0 to 9.
Western blot analysis
Equal quantities (50μg) of whole-cell protein lysates from untreated and 5-Aza-CdR/TSA treated cells were separated on 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes. The membranes were blocked with 5% non-fat dry milk in TBST for 2h, and then incubated with the primary antibodies specific for UHRF1 (1:100; Active Motif), H3R8me1 (1:200; Active Motif), H3R8me2s (1:200; Novus Biologicals) and H3R8me2a (1:100; Novus B iologicals) at 4°C overnight, followed by incubation with secondary antibodies. Bands were visualized using the ECL Western blotting system (Amersham Biosciences, Buckingham, UK). An anti-β-actin antibody was used as an internal control.
Chromatin immunoprecipitation (ChIP) assay
In brief, 2×106 cells were cross-linked in 1% formaldehyde, followed by sonication to fragment the DNA into 200 to 1000 bp fragments. 20μl of the supernatant was saved as the input DNA, and the remainder was diluted 1:10 in ChIP dilution buffer. The chromatin fraction was pre-cleared with a salmon sperm DNA/protein agarose as a 50% gel slurry for 3 hours, and then immunoprecipitated overnight at 4°C with antibodies for UHRF1, H3R8me2s and H3R8me2a. A non-specific IgG (Santa Cruz) was used as a control. Incubating chromatin at 65°C for 2 hours reversed the cross-links, and DNA was extracted using the phenol-chloroform-isoamyl alcohol method. Purified DNA was analyzed by real time PCR (qPCR) to determine fold enrichment relative to input DNA. The primers used to amplify specific region of the 3OST2 promoter were: F, 5'-GTTCCAACCACTCCGGCTCA-3' and R, 5'-CGTCCGGGTGTACTCGGATAA-3'. Thermal cycler conditions were followed by 35 cycles of 95°C for 30 s and 60°C for 60 s. A Ct value was calculated for each sample: ΔCt = Ct (sample) - Ct (Input). Next, a ΔΔCt value was calculated by ΔΔCt = ΔCt (IP sample) - ΔCt (IP control). The fold difference between the IP sample and IP control was then calculated as 2-ΔΔCt.
Statistical analysis
Statistical analysis was performed using SPSS 16.0 for windows (SPSS Inc, Chicago, IL). Frequencies of methylation were compared using chi-square test and Fisher's exact test. The chi-square test and Fisher's exact test were also applied to examine the correlation between promoter methylation status and clinicopathological features. The methylation status associated with patients survival was detected by Kaplan-Meier analysis. A P-value ≤ 0.05 was considered statistically significant.
Results
Methylation profiling of TSGs in primary HCCs and matched corresponding noncancerous tissues
We detected 48 pairs of HCCs and matched corresponding noncancerous liver tissues. Representative examples of methylation-specific PCR (MSP) analysis on primary HCCs are shown in Fig. 1. Table 2 summarizes the methylation frequency of each gene in the HCC samples and corresponding noncancerous tissues. The results showed that five genes had very high frequencies of methylation in HCC tissues; however, four of them had similar high frequencies (P > 0.05) in corresponding noncancerous tissues (56.3% vs. 56.3% for ppENK, 66.7% vs. 85.4% for HIC1, 97.9% vs. 95.8% for SLIT2, and 93.8% vs. 81.3% for EDNRB, P > 0.05). Only the 3OST2 gene was frequently methylated in HCCs but less frequently methylated in corresponding noncancerous tissues (68.8% vs. 37.5%, P < 0.05). Moderate frequencies of CHFR and COX2 methylation were found in HCC tissues while very low frequency was detected in corresponding noncancerous tissues (20.8% vs. 2.1%, P < 0.01 and 25% vs. 4.2%, P < 0.01, respectively). CLDN7, THBS1, and LKB1 had low frequencies of methylation in HCC tissues and corresponding noncancerous tissues (10.4% vs. 14.6%, P > 0.05; 16.7% vs. 22.9%, P > 0.05; and 4.2% vs. 0%, P > 0.05, respectively).
The 3OST2 gene was the best biomarker studied in the present data for the early diagnosis of HCC; thus, it was selected for further analysis. To evaluate the biological significance of 3OST2 methylation in hepatocarcinogenesis, we initially assessed the associations between 3OST2 methylation and clinicopathological parameters, such as sex, age, cirrhosis, HBV-infection, tumor size, and tumor differentiation. Subsequently, we also investigated whether the methylation of 3OST2 was associated with patient survival. However, the data revealed no correlation between 3OST2 methylation and any of the clinicopathological parameters, and there was no significant association between 3OST2 methylation and patient survival.
Transcriptional repression of 3OST2 is associated with promoter hypermethylation and histone modifications
To determine whether 3OST2 is directly regulated by promoter hypermethylation in HCC, we investigated the changes in the 3OST2 promoter methylation status and mRNA expression in HCC cells before and after treatment with epigenetic inhibitors. The MSP results showed that the 3OST2 promoter was completely methylated in HCC cells but not in normal liver cells. 5-Aza-CdR partially reversed the 3OST2 promoter methylation. Interestingly, TSA alone could also alter the 3OST2 promoter methylation (Fig. 2A). After treatment by both inhibitors in combination, 3OST2 promoter methylation was completely reversed. Accordingly, the real-time reverse transcription PCR (qRT-PCR) results revealed that 3OST2 was downregulated in HCC cells compared to normal liver cells. Either 5-Aza-CdR or TSA treatment dramatically increased the expression of 3OST2 mRNA (by 2.97- or 4.43-fold, P < 0.01, respectively) (Fig. 2B). In addition, combining 5-Aza-CdR with TSA showed additive effects (3OST2 expression increased by 8.21-fold, P < 0.01).
UHRF1 expression was downregulated by 5-Aza-CdR and/or TSA in HCC cells and was involved in the regulation of 3OST2
Several studies have indicated that UHRF1 is a conductor of the replication process by ensuring the cross-talk between DNA methylation and histone modifications [20,21]. This cross-talk allows cancer cells to maintain the repression of TSGs during cell proliferation [22]. However, whether UHRF1 participates in HCC and how it regulates TSG has not been investigated. In this study, we detected its expression in HCC and normal liver cell lines. The results showed that UHRF1 was also highly expressed in HCC cells compared to normal liver cells, in which UHRF1 was not expressed. 5-Aza-CdR treatment alone slightly downregulated its expression, while TSA caused a sharp decrease (Fig. 3A).
Figure 1
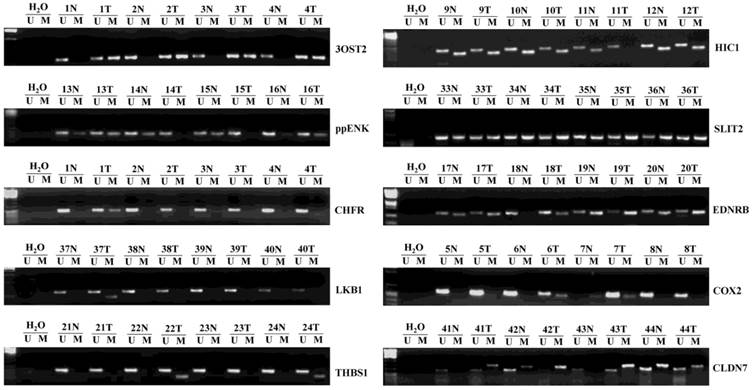
Representative samples (n=3) of the methylation status of ten umor suppressor genes (TSGs) in 48 hepatocellular carcinomas (HCCs) and matched corresponding noncancerous tissues. The methylation-specific PCR (MSP) products in the M lanes indicate the presence of methylated alleles, and those in the U lanes indicate the presence of unmethylated allele; N, non-tumor; T, tumor.
Figure 2
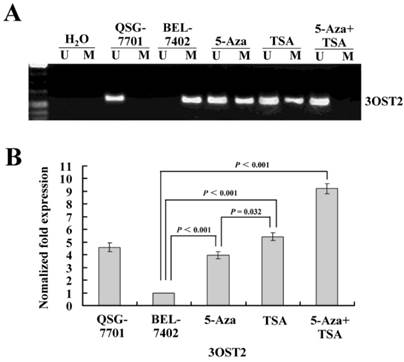
(A) The methylation status of the 3OST2 promoter in normal liver cells and in HCC cells with or without 5-Aza-CdR and/or TSA treatment. The 3OST2 promoter was not methylated in normal liver cells and completely methylated (only methylated alleles were present) in HCC cells. This was partially reversed by 5-Aza-CdR or TSA treatment (both unmethylated and methylated alleles were present). When 5-Aza-CdR was combined with TSA, the promoter was completely reversed (only unmethylated alleles were present). (B) Real-time reverse transcription PCR (qRT-PCR) analysis of 3OST2 mRNA expression in HCC cells. Expression was normalized using β-actin as an internal control.
To measure the levels of UHRF1 expression quantitatively after treatment with 5-Aza-CdR and/or TSA, we performed qRT-PCR and western blot assays. The results demonstrated very high levels of UHRF1 mRNA and protein in HCC cells, and a significant decrease was observed after treatment with 5-Aza-CdR. TSA caused a larger decrease, which was consistent with the results of immunohistochemistry. After treatment with both inhibitors, UHRF1 expression was decreased in both transcript and protein levels (Fig. 3B-D).
Next, we investigated whether UHRF1 is directly involved in the regulation of 3OST2 using a chromatin immunoprecipitation (ChIP) assay. The results revealed enrichment of UHRF1 at the 3OST2 promoter region in HCC cells but very low level in normal liver cells. After exposure to either or both of the inhibitors, the amount of UHRF1 binding to the 3OST2 promoter significantly decreased. Accordingly, the 3OST2 expression was increased (Fig. 2B), indicating that UHRF1 directly regulates 3OST2 expression (Fig. 3E).
Three methylation states of histone H3R8 have different functions in HCC cells
To understand whether H3R8 is methylated in HCC cells and whether the three methylation states would exert similar or opposite effects, we performed western blot analysis on the whole-cell extracts. The results showed that H3R8me1 and H3R8me2s were prominently present in the untreated HCC cell line BEL-7402 but were at low levels in human normal liver cell line QSG-7701. By contrast, H3R8me2a was present at a high level in QSG-7701 cells, but not in BEL-7402 cells (Fig. 4). After treatment with TSA, the levels of H3R8me2a increased; accordingly, H3R8me1 and H3R8me2s levels were decreased. Although the effect of 5-Aza-CdR was relatively small, it was a statistically significant change. The combination of 5-Aza-CdR and TSA produced additive effects.
H3R8 is symmetrically dimethylated in the methylated promoter of 3OST2
Subsequently, to investigate whether methylated H3R8 could directly regulate hypermethylated 3OST2, we performed ChIP assays in QSG-7701 and BEL-7402 cell lines using anti-H3R8me2s and anti-H3R8me2a antibodies (no ChIP-grade H3R8me1 antibody was available). In the QSG-7701 cell line, the promoter region of 3OST2 lacked methylated H3R8me2s, while this epigenetic modification was enriched in the methylated 3OST2 promoter in the BEL-7402 cell line (Fig. 5). Interestingly, the H3R8me2s on the promoter region of 3OST2 in HCC cells was not inhibited by 5-Aza-CdR or TSA treatment alone; whereas it was 46.12% inhibited by the combination of these two inhibitors. In contrast, the amount of H3R8me2a in the 3OST2 promoter was very low in BEL-7402 cells compared to QSG-7701 cells, and was accumulated after 5-Aza-CdR or/and TSA treatment. Based on these data combined with the results of Fig.2, it indicated that the level of H3R8me2s correlated with repression of 3OST2, while the level of H3R8me2a was associated with activated transcription.
Figure 3
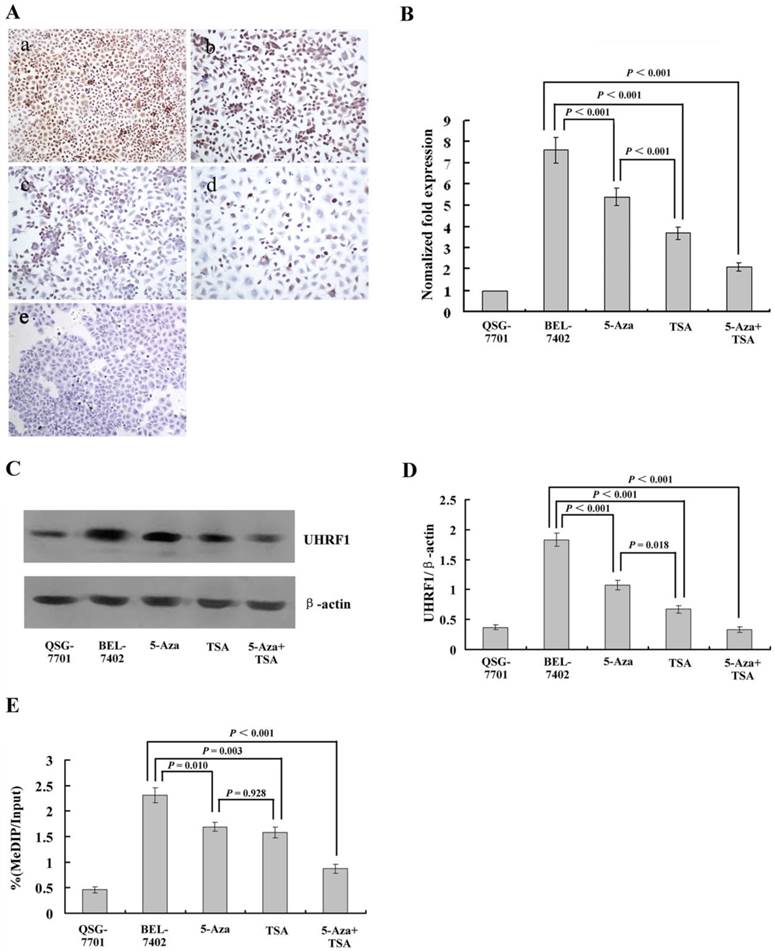
(A) Immunohistochemical analysis of UHRF1 protein expression in HCC cells with or without 5-Aza-CdR and/or TSA treatment. (a) Untreated HCC cells; (b) after 5-Aza-CdR treatment; (c) after TSA treatment; (d) after 5-Aza-CdR and TSA combined treatment; (e) Untreated normal liver cell line QSG-7701 cells. (B) qRT-PCR analysis of UHRF1 mRNA expression (C, D) Western blot analysis of UHRF1 protein expression; the ratio of UHRF1 to β-actin is shown on the y-axis. (E) Chromatin immunoprecipitation (ChIP)-quantitative real time PCR (qPCR) assay for UHRF1 on the 3OST2 promoter. Shown is the enrichment, relative to the input obtained in each ChIP sample.
Figure 4
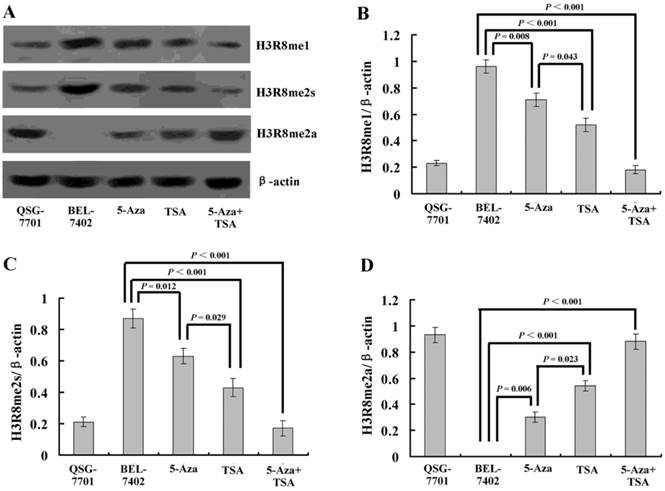
(A) Western blot analysis of three methylation states of H3R8 (H3R8me1, H3R8me2s and H3R8me2a) in normal liver cells and HCC cells with or without 5-Aza-CdR and/or TSA treatment. (B-E) The ratio of H3R8me1/H3R8me2s/H3R8me2a to β-actin is shown on the y-axis. Note that H3R8me2a was not present in the HCC cells.
Figure 5
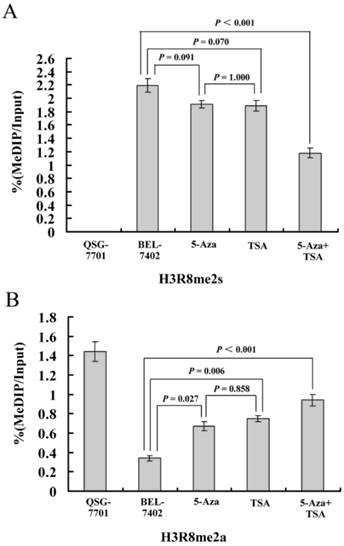
ChIP-qPCR assays for H3R8me2s and H3R8me2a on the 3OST2 promoter in normal liver cells and HCC cells with or without 5-Aza-CdR and/or TSA treatment. Cross-linked chromatin from HCC cells treated or untreated with 5-Aza-CdR and/or TSA underwent immunoprecipitation using control IgG, anti-UHRF1, anti-H3R8me2s or anti-H3R8me2a antibodies; the immunoprecipitated DNA was analyzed by qPCR using primers specific for 3OST2. Shown is the enrichment, relative to the input obtained in each ChIP samples.
Discussion
Methylation of promoter has recently emerged as potential diagnostic markers in virtually all tumor types and plays a significant role in the early screening of cancer [23-25]. To identify critical tumor-specific events in hepatocellular carcinogenesis, we profiled the methylation status of ten tumor-related genes in primary HCC tissues and matched noncancerous tissues. A good marker for early diagnostic of HCC should be more easily detected in HCCs and the difference between HCCs and noncancerous tissues should be statistically significant. Our results showed that five genes (3OST2, SLIT2, EDNRB, HIC1, and ppENK) were very frequently methylated in HCC tissues. However, all of them, except 3OST2, had similar frequencies of methylation in corresponding noncancerous tissues (P > 0.05); therefore, the diagnostic value of these biomarkers for HCC is debatable. For the CHFR and COX2 gene, there were significant differences in their methylation frequencies between HCC tissues and corresponding noncancerous tissues; however, the frequency in HCC tissues was not high (20.8% and 25%, respectively), thus limiting their diagnostic value in the clinic. The LKB1, THBS1, and CLDN7 genes were rarely methylated in HCC. DNA methylation has been involved in virtually every step of tumor development and progression, and some TSGs methylation is believed to occur early in tumor development. Our data indicated that during the transformation of HCC, methylation of some TSGs might occur in the early stage (preceding the malignant process), while methylation of other TSGs might occur in the later stage. Among the ten TSGs tested, only the 3OST2 gene methylation had relatively high sensitivity (68.8%) and specificity (62.5%). It also should be taken into account that the corresponding noncancerous liver tissues from HCC patients are usually accompanied by HBV infection and cirrhosis; therefore, the 3OST2 gene methyaltion might serve as a good biomarker for the early diagnosis of HCC, and further experiments conducted to detect the 3OST2 gene methylation in donated normal liver tissues may confirm its higher specificity for early diagnosis.
The 3OST2 gene encodes an O-sulfotransferase that is involved in the final modification step of glycosaminoglycan chains of heparin sulfate proteoglycans (HSPGs). HSPGs are important for migration, cell growth and adhesion because of their interactions with a wide range of growth factors, morphogens, cytokines and the extracellular matrix. Methylation-associated silencing of 3OST2 has been demonstrated in human breast, gastric, colon, lung and pancreatic cancers [26-28]; however, the methylation status was not clear in HCC. Our data indicated that 3OST2 promoter methylation may play an important role in hepatocellular carcinogenesis. We examined the relationship between 3OST2 methylation and the clinicopathological features; however, 3OST2 methylation could not act as a prognostic marker or a recurrence indicator, but only as a valuable diagnostic marker.
Unlike genetic alterations, all epigenetic changes are reversible. Many DNMT inhibitors and HDAC inhibitors are currently in clinical trials for their anti-cancer activities [29,30]. Some TSGs can also be re-expressed through inhibition of HDAC inhibitors without affecting the hypermethylation status [31]. Interestingly, in our study, not only 5-Aza-CdR but also TSA could partially reverse the 3OST2 promoter methylation, and after combined treatment with 5-Aza-CdR and TSA, 3OST2 promoter methylation was completely reversed. Accordingly, mRNA expression additively increased, demonstrating the cross-talk between DNA methylation and histone modifications, and implying that HDAC inhibitors may regulate gene expression in a DNA methylation-dependent manner.
UHRF1, an essential cofactor for the maintenance of genomic DNA methylation, has multiple domains that confer a variety of functions to this protein. The SRA domain of UHRF1 can specifically recognize hemi-methylated DNA [15-17]. On the other hand, the PHD domain of UHRF1 recognizes methylated H3K9 [32]. Further studies indicated that UHRF1 often formed a complex with DNMT1 and HDAC1, and could bind to the methylated promoter of tumor suppressors such as p14 and p16 [33], to suppress their expression in cancer cells. Our results indicated that, for the first time, UHRF1 was overexpressed in HCC cells but not in normal liver cells, and could be downregulated by 5-Aza-CdR or TSA. Besides DNMT1 and HDAC1, Kim et al. reported that UHRF1 interacted with G9a (a histone methyltransferase) to enhance p21 transcriptional repression. These observations implied that UHRF1 may have an extended role in recruiting a broad range of chromatin modification enzymes during epigenetic regulation [34]. The structure of UHRF1 reveals that it contains H3R8-H3K9me3-H3S10 binding sites and our previous studies had demonstrated that the hypoacetylation and trimethylation of H3K9 were involved in the regulation of HCC-related TSGs [35,36]. In the present study, we wanted to determine whether, in addition to UHRF1, histone H3R8 methylation was involved in the regulation of 3OST2. Arginine methylation is catalyzed by arginine methyltransferases (PRMTs) and can be monomethylated (me1) or di-methylated (me2), with the latter in a symmetrical or an asymmetrical configuration (me2s or me2a). Pal et al. reported that PRMT5 could dimethylate H3R8 to negatively regulate expression of ST7 and NM23 tumor suppressor genes [37]. However, our results revealed that H3R8me2s was a repressive mark, while H3R8me2a was linked to 3OST2 activation, which was opposite to the function of H3R8me2s. Although the western blotting results showed that H3R8me1 was present in HCC cells and was inhibited by 5-Aza-CdR and/or TSA, its regulatory role of 3OST2 remains unexplored. In our data, all three methylation states of H3R8 could be changed by 5-Aza-CdR and/or TSA, suggesting that the two inhibitors may affect not only DNMTs/HDACs, but also PRMTs, although which PRMT is involved requires further investigation. It should be noted that the methylation state of H3R82s in the 3OST2 promoter could only be changed by 5-Aza-CdR combined with TSA, implying that the enzymes involved might function as complexes.
In conclusion, our results provide a specific methylation profile for the diagnosis of HCC. 3OST2 methylation may play a critical role in the earliest steps of hepatocarcinogenesis and is directly regulated by UHRF1 protein. Therefore, 3OST2 may be considered as a novel biomarker for early diagnosis of HCC and a molecular target for gene therapy. We also describe that, for the first time, H3R8me2s acts as a repressive mark, while H3R8me2a was correlated with 3OST2 transcriptional activity in HCC.
Supplementary Material
Supplementary Table 1.
Acknowledgements
We thank Dr. Hiuming Li of the Department of Anatomical and Cellular Pathology of Chinese University of Hong Kong for providing the materials of HCC patients. This work was supported by grants from the National Natural Science Foundation of China (No. 81272277) and the China Postdoctoral Science Foundation (No. 2013M540554).
Conflict of Interest
There are no conflicts of interest.
References
1. Edamoto Y, Hara A, Biernat W, Terracciano L, Cathomas G, Riehle HM, Matsuda M, Fujii H, Scoazec JY, Ohgaki H. Alterations of RB1, p53 and Wnt pathways in hepatocellular carcinomas associated with hepatitis C, hepatitis B and alcoholic liver cirrhosis. Int J Cancer. 2003;106:334-41
2. Yuan RH, Jeng YM, Chen HL, Lai PL, Pan HW, Hsieh FJ, Lin CY, Lee PH, Hsu HC. Stathmin overexpression cooperates with p53 mutation and osteopontin overexpression, and is associated with tumour progression, early recurrence, and poor prognosis in hepatocellular carcinoma. J Pathol. 2006;209:549-58
3. Du GS, Wang JM, Lu JX, Li Q, Ma CQ, Du JT, Zou SQ. Expression of P-aPKC-iota, E-cadherin, and beta-catenin related to invasion and metastasis in hepatocellular carcinoma. Ann Surg Oncol. 2009;16:1578-86
4. Selaru FM, David S, Meltzer SJ, Hamilton JP. Epigenetic events in gastrointestinal cancer. Am J Gastroenterol. 2009;104:1910-2
5. Graves BT, Munro CL. Epigenetics in critical illness: a new frontier. Nurs Res Pract. 2013;2013:503686
6. Kaiser MF, Johnson DC, Wu P, Walker BA, Brioli A, Mirabella F, Wardell CP, Melchor L, Davies FE, Morgan GJ. Global methylation analysis identifies prognostically important epigenetically inactivated tumor suppressor genes in multiple myeloma. Blood. 2013;122:219-26
7. Roa JC, Anabalon L, Roa I, Melo A, Araya JC, Tapia O, de Aretxabala X, Munoz S, Schneider B. Promoter methylation profile in gallbladder cancer. J Gastroenterol. 2006;41:269-75
8. Jin C, Yu W, Lou X, Zhou F, Han X, Zhao N, Lin B. UCHL1 Is a Putative Tumor Suppressor in Ovarian Cancer Cells and Contributes to Cisplatin Resistance. J Cancer. 2013;4:662-70
9. Nishida N, Nagasaka T, Nishimura T, Ikai I, Boland CR, Goel A. Aberrant methylation of multiple tumor suppressor genes in aging liver, chronic hepatitis, and hepatocellular carcinoma. Hepatology. 2008;47:908-18
10. Lehmann U, Wingen LU, Brakensiek K, Wedemeyer H, Becker T, Heim A, Metzig K, Hasemeier B, Kreipe H, Flemming P. Epigenetic defects of hepatocellular carcinoma are already found in non-neoplastic liver cells from patients with hereditary haemochromatosis. Hum Mol Genet. 2007;16:1335-42
11. Li B, Liu W, Wang L, Li M, Wang J, Huang L, Huang P, Yuan Y. CpG island methylator phenotype associated with tumor recurrence in tumor-node-metastasis stage I hepatocellular carcinoma. Ann Surg Oncol. 2010;17:1917-26
12. Kanai Y. Genome-wide DNA methylation profiles in precancerous conditions and cancers. Cancer Sci. 2010;101:36-45
13. Roncalli M, Bianchi P, Bruni B, Laghi L, Destro A, Di Gioia S, Gennari L, Tommasini M, Malesci A, Coggi G. Methylation framework of cell cycle gene inhibitors in cirrhosis and associated hepatocellular carcinoma. Hepatology. 2002;36:427-32
14. Unoki M. Current and potential anticancer drugs targeting members of the UHRF1 complex including epigenetic modifiers. Recent Pat Anticancer Drug Discov. 2011;6:116-30
15. Arita K, Ariyoshi M, Tochio H, Nakamura Y, Shirakawa M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 2008;455:818-21
16. Avvakumov GV, Walker JR, Xue S, Li Y, Duan S, Bronner C, Arrowsmith CH, Dhe-Paganon S. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature. 2008;455:822-5
17. Hashimoto H, Horton JR, Zhang X, Bostick M, Jacobsen SE, Cheng X. The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature. 2008;455:826-9
18. Rajakumara E, Wang Z, Ma H, Hu L, Chen H, Lin Y, Guo R, Wu F, Li H, Lan F, Shi YG, Xu Y, Patel DJ, Shi Y. PHD finger recognition of unmodified histone H3R2 links UHRF1 to regulation of euchromatic gene expression. Mol Cell. 2011;43:275-84
19. Hashimoto H, Horton JR, Zhang X, Cheng X. UHRF1, a modular multi-domain protein, regulates replication-coupled crosstalk between DNA methylation and histone modifications. Epigenetics. 2009;4:8-14
20. Sabatino L, Fucci A, Pancione M, Carafa V, Nebbioso A, Pistore C, Babbio F, Votino C, Laudanna C, Ceccarelli M, Altucci L, Bonapace IM, Colantuoni V. UHRF1 coordinates peroxisome proliferator activated receptor gamma (PPARG) epigenetic silencing and mediates colorectal cancer progression. Oncogene. 2012;31:5061-72
21. Arita K, Isogai S, Oda T, Unoki M, Sugita K, Sekiyama N, Kuwata K, Hamamoto R, Tochio H, Sato M, Ariyoshi M, Shirakawa M. Recognition of modification status on a histone H3 tail by linked histone reader modules of the epigenetic regulator UHRF1. Proc Natl Acad Sci U S A. 2012;109:12950-5
22. Alhosin M, Sharif T, Mousli M, Etienne-Selloum N, Fuhrmann G, Schini-Kerth VB, Bronner C. Down-regulation of UHRF1, associated with re-expression of tumor suppressor genes, is a common feature of natural compounds exhibiting anti-cancer properties. J Exp Clin Cancer Res. 2011;30:41
23. Dai W, Teodoridis JM, Zeller C, Graham J, Hersey J, Flanagan JM, Stronach E, Millan DW, Siddiqui N, Paul J, Brown R. Systematic CpG islands methylation profiling of genes in the wnt pathway in epithelial ovarian cancer identifies biomarkers of progression-free survival. Clin Cancer Res. 2011;17:4052-62
24. Gloss BS, Patterson KI, Barton CA, Gonzalez M, Scurry JP, Hacker NF, Sutherland RL, O'Brien PM, Clark SJ. Integrative genome-wide expression and promoter DNA methylation profiling identifies a potential novel panel of ovarian cancer epigenetic biomarkers. Cancer Lett. 2012;318:76-85
25. Summers T, Langan RC, Nissan A, Brucher BL, Bilchik AJ, Protic M, Daumer M, Avital I, Stojadinovic A. Serum-based DNA methylation biomarkers in colorectal cancer: potential for screening and early detection. J Cancer. 2013;4:210-6
26. Dauksa A, Gulbinas A, Barauskas G, Pundzius J, Oldenburg J, El-Maarri O. Whole blood DNA aberrant methylation in pancreatic adenocarcinoma shows association with the course of the disease: a pilot study. PLoS ONE. 2012;7:e37509
27. Tokuyama Y, Takahashi T, Okumura N, Nonaka K, Kawaguchi Y, Yamaguchi K, Osada S, Gazdar A, Yoshida K. Aberrant methylation of heparan sulfate glucosamine 3-O-sulfotransferase 2 genes as a biomarker in colorectal cancer. Anticancer Res. 2010;30:4811-8
28. Miyamoto K, Asada K, Fukutomi T, Okochi E, Yagi Y, Hasegawa T, Asahara T, Sugimura T, Ushijima T. Methylation-associated silencing of heparan sulfate D-glucosaminyl 3-O-sulfotransferase-2 (3-OST-2) in human breast, colon, lung and pancreatic cancers. Oncogene. 2003;22:274-80
29. Fandy TE. Development of DNA methyltransferase inhibitors for the treatment of neoplastic diseases. Curr Med Chem. 2009;16:2075-85
30. Griffiths EA, Gore SD. DNA methyltransferase and histone deacetylase inhibitors in the treatment of myelodysplastic syndromes. Semin Hematol. 2008;45:23-30
31. Pruitt K, Zinn RL, Ohm JE, McGarvey KM, Kang SH, Watkins DN, Herman JG, Baylin SB. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet. 2006;2:e40
32. Karagianni P, Amazit L, Qin J, Wong J. ICBP90, a novel methyl K9 H3 binding protein linking protein ubiquitination with heterochromatin formation. Mol Cell Biol. 2008;28:705-17
33. Unoki M, Nishidate T, Nakamura Y. ICBP90, an E2F-1 target, recruits HDAC1 and binds to methyl-CpG through its SRA domain. Oncogene. 2004;23:7601-10
34. Kim JK, Esteve PO, Jacobsen SE, Pradhan S. UHRF1 binds G9a and participates in p21 transcriptional regulation in mammalian cells. Nucleic Acids Res. 2009;37:493-505
35. Zhang C, Li H, Wang Y, Liu W, Zhang Q, Zhang T, Zhang X, Han B, Zhou G. Epigenetic inactivation of the tumor suppressor gene RIZ1 in hepatocellular carcinoma involves both DNA methylation and histone modifications. J Hepatol. 2010;53:889-95
36. Zhang C, Li H, Zhou G, Zhang Q, Zhang T, Li J, Zhang J, Hou J, Liew CT, Yin D. Transcriptional silencing of the TMS1/ASC tumour suppressor gene by an epigenetic mechanism in hepatocellular carcinoma cells. J Pathol. 2007;212:134-42
37. Pal S, Vishwanath SN, Erdjument-Bromage H, Tempst P, Sif S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol Cell Biol. 2004;24:9630-45
Author contact
![]() Corresponding author: Cuijuan Zhang, Institute of Pathology and Pathophysiology, Shandong University School of Medicine, Wenhuaxi Road 44#, Jinan 250012, P. R. China. E-mail: cuijuanzhangedu.cn; Tel.: 86-531-88382574; Fax numbler: 86-531-88383168
Corresponding author: Cuijuan Zhang, Institute of Pathology and Pathophysiology, Shandong University School of Medicine, Wenhuaxi Road 44#, Jinan 250012, P. R. China. E-mail: cuijuanzhangedu.cn; Tel.: 86-531-88382574; Fax numbler: 86-531-88383168
Citation styles
APA
Chen, H., Zhang, T., Sheng, Y., Zhang, C., Peng, Y., Wang, X., Zhang, C. (2015). Methylation Profiling of Multiple Tumor Suppressor Genes in Hepatocellular Carcinoma and the Epigenetic Mechanism of 3OST2 Regulation. Journal of Cancer, 6(8), 740-749. https://doi.org/10.7150/jca.11691.
ACS
Chen, H.; Zhang, T.; Sheng, Y.; Zhang, C.; Peng, Y.; Wang, X.; Zhang, C. Methylation Profiling of Multiple Tumor Suppressor Genes in Hepatocellular Carcinoma and the Epigenetic Mechanism of 3OST2 Regulation. J. Cancer 2015, 6 (8), 740-749. DOI: 10.7150/jca.11691.
NLM
Chen H, Zhang T, Sheng Y, Zhang C, Peng Y, Wang X, Zhang C. Methylation Profiling of Multiple Tumor Suppressor Genes in Hepatocellular Carcinoma and the Epigenetic Mechanism of 3OST2 Regulation. J Cancer 2015; 6(8):740-749. doi:10.7150/jca.11691. https://www.jcancer.org/v06p0740.htm
CSE
Chen H, Zhang T, Sheng Y, Zhang C, Peng Y, Wang X, Zhang C. 2015. Methylation Profiling of Multiple Tumor Suppressor Genes in Hepatocellular Carcinoma and the Epigenetic Mechanism of 3OST2 Regulation. J Cancer. 6(8):740-749.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.