Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(1):19-28. doi:10.7150/jca.16655 This issue Cite
Research Paper
Histone Deacetylase Inhibitor SAHA as Potential Targeted Therapy Agent for Larynx Cancer Cells
Aneta Grabarska1 ![]() , Jarogniew J. Łuszczki2,3, Ewa Nowosadzka1, Ewelina Gumbarewicz1, Witold Jeleniewicz1, Magdalena Dmoszyńska-Graniczka1, Krystyna Kowalczuk1, Krzysztof Kupisz5, Krzysztof Polberg4, Andrzej Stepulak1,4
, Jarogniew J. Łuszczki2,3, Ewa Nowosadzka1, Ewelina Gumbarewicz1, Witold Jeleniewicz1, Magdalena Dmoszyńska-Graniczka1, Krystyna Kowalczuk1, Krzysztof Kupisz5, Krzysztof Polberg4, Andrzej Stepulak1,4
1. Department of Biochemistry and Molecular Biology, Medical University of Lublin, Poland.
2. Department of Pathophysiology, Medical University of Lublin, Lublin, Poland.
3. Isobolographic Analysis Laboratory, Institute of Rural Health, Lublin, Poland.
4. Department of Otolaryngology, MSWiA Hospital, Poland.
5. Department of Otolaryngology and Head and Neck Surgery, Specialist District Hospital, Lublin, Poland.
Received 2016-6-30; Accepted 2016-9-19; Published 2017-1-1
Citation:
Grabarska A, Łuszczki JJ, Nowosadzka E, Gumbarewicz E, Jeleniewicz W, Dmoszyńska-Graniczka M, Kowalczuk K, Kupisz K, Polberg K, Stepulak A. Histone Deacetylase Inhibitor SAHA as Potential Targeted Therapy Agent for Larynx Cancer Cells. J Cancer 2017; 8(1):19-28. doi:10.7150/jca.16655. https://www.jcancer.org/v08p0019.htm
Other stylesAbstract
Objective: Laryngeal squamous cell carcinoma is one of the most common malignant tumors in the head and neck region. Due to the poor response to chemotherapeutics in patients and low survival rate, successful treatment of larynx cancer still remains a challenge. Therefore, the identification of novel treatment options is needed. We investigated the anticancer effects of suberoylanilide hydroxamic acid (SAHA), a histone deacetylase inhibitor, on two different laryngeal cancer cell lines RK33 and RK45. We also studied the antiproliferative action of SAHA in combination with cisplatin and defined the type of pharmacological interaction between these drugs. Materials and Methods: Viability and proliferation of larynx cancer cell lines were studied by methylthiazolyldiphenyl-tetrazolium bromide method and 5-bromo-2-deoxyuridine incorporation assay, respectively. The type of interaction between SAHA and cisplatin was determined by an isobolographic analysis. Western blotting, flow cytometry and quantitative polymerase chain reaction method were used to determine acetylation of histone H3, cell cycle progression and genes expression, respectively. Apoptosis was assessed by means of nucleosomes released to cytosol. Results: SAHA alone or in combination with cisplatin inhibited larynx cancer cells proliferation, whereas displayed relatively low toxicity against normal cells - primary cultures of human skin fibroblasts. The mixture of SAHA with cisplatin exerted additive and synergistic interaction in RK33 and RK45 cells, respectively. We showed that SAHA induced hyperacetylation of histone H3 K9, K14 and K23 and triggered apoptosis. SAHA also caused cell cycle arrest by upregulation of CDKN1A and downregulation of CCND1 encoding p21WAF1/CIP1 and cyclin D1 proteins, respectively. Conclusion: Our studies demonstrated that SAHA may be considered as a potential therapeutic agent against larynx tumors.
Keywords: larynx cancer, histone deacetylase inhibitors, suberoylanilide hydroxamic acid (SAHA), cisplatin (CDDP), isobolography.
Introduction
Many of signaling pathways which regulate critical processes such as cell growth, survival, differentiation and migration are altered in cancer cells. Understanding of the molecular changes that accompany cell transformation led to the development of anticancer targeted therapy [1]. The introduction of monoclonal antibodies which can arrest cancer cell growth or promote apoptosis by direct binding with growth factor (bavacizumab Avastin® against vascular endothelial growth factor - VEGF) or extracellular domain of receptor (trastuzumab Herceptin®) used in the treatment of breast cancers overexpressing the human epidermal growth factor receptor 2 (HER2) [2, 3], as well as bcr/abl-activated tyrosine kinase inhibitor imatinib (Glivec®) for the treatment of chronic myelogenous leukemia (CML), are the best examples.
Targeted therapy based on a monoclonal antibody cetuximab (Erbitux®) against epidermal growth factor receptor (EGFR, highly expressed in head and neck squamous cell carcinoma) in combination with radiotherapy has been approved by U S Food and Drug Administration (FDA) as alternative treatment for larynx cancer [4]. Laryngeal squamous cell carcinoma is one of the most common malignant tumors in the head and neck region and has a high mortality rate of 50 % [5]. Despite advancements in the strategies for the treatment of larynx cancer patients, the overall 5-year survival rate and the clinical outcomes are still not satisfactory. [6]. Standard therapeutic procedure of laryngeal cancer-bearing patients depends on the tumor stage, occurrence of local and distant metastases, health status, patient's age, and include surgery, radiotherapy, and docetaxel, cisplatin (CDDP) and 5-fluorouracil based chemotherapy in various combinations [7]. CDDP-based therapy often results in severe side effects induced by high doses of cisplatin and development of drug resistance [8]. Therefore, the identification of novel treatment options and use of new types of chemotherapeutics is needed.
In search of new therapeutic solutions for cancer treatment, an attractive target became proteins involved in the regulation of gene expression - histone acetyltransferases (HATs) and histone deacetylases (HDACs). Alterations in their activity have been reported in many cancers, both solid tumors and hematological malignancies. HDACs are typically overexpressed in cancer cells [9, 10]. HDACs have been found to be aberrant when associated with oncogenic fusion proteins caused by chromosomal translocations in certain forms of leukemia and lymphoma [9]. Such inappropriate requirement of HDACs contributes to constitutive repression of genes involved in cell cycle progression, cellular differentiation, DNA repair and apoptosis [11].
To date, a wide range of natural and synthetic chemical compounds that inhibit HDACs activity have been identified, and became an interesting class of agents for cancer therapy. These substances called histone deacetylase inhibitors (HDIs) are classified into four different groups according to their chemical structures: hydroxamic acids (trichostatin A - TSA, suberoylanilide hydroxamic acid - SAHA, oxamflatin), cyclic peptides (depsipeptide, apicidin), short chain aliphatic acids (sodium butyrate, valproic acid - VPA) and benzamides (entinostat) [12]. HDIs increase histone acetylation of condensed chromatin and thus activate expression of epigenetically silenced regulatory genes and reverse the malignant phenotype [13]. Increasing evidence suggests that HDIs can induce tumor cell apoptosis, growth arrest, senescence, differentiation, immunogenicity, and inhibit angiogenesis [14]. There are a number of HDAC inhibitors being tested in preclinical and clinical trials. Currently, the FDA has approved three types of HDAC inhibitors for cancer therapy, including vorinostat (Zolinza; Merck) for the treatment of refractory cutaneous T-cell lymphoma (CTCL), romidepsin (Istodax; Celgene) for the treatment of CTCL and peripheral-cell lymphoma (PTCL) and belinostat (Beleodaq; Spectrum Pharmaceuticals) for the treatment of PTCL [15].
In this paper, we examined the anticancer effects of suberoylanilide hydroxamic acid (SAHA), a HDAC inhibitor, on two different laryngeal cancer cell lines RK33 and RK45. We also studied the antiproliferative action of SAHA in combination with cisplatin (CDDP) and defined the type of pharmacological interaction between these drugs in examined cell lines.
Materials and Methods
Reagents
Suberoylanilide hydroxamic acid (SAHA) and cisplatin (CDDP) were purchased from Cayman Chemical (San Diego, CA, USA) and Sigma Chemicals (St. Louis, MO, USA), respectively. Stock solution of SAHA (10 μM) and CDDP (1 mg/ml) were prepared in dimethyl sulfoxide (DMSO) and phosphate buffered saline (PBS), respectively.
Cell lines
Human larynx cancer cell lines (RK33 and RK45) were established from laryngeal cancer cells derived from patients with diagnosed laryngeal squamous cell carcinoma. Cancer tissue was removed from the larynx after total laryngectomy. The characterization of RK33 and RK45 cell lines has been published by Rzeski et al. [16]. Normal human primary fibroblast (HSF) culture was generated by the outgrowth technique from skin explants of young persons, used routinely in our laboratory [17], the procedure was approved by local Ethical Committee (KE nr 0254/298/2015). Larynx cancer cell lines and fibroblast cells were grown using RPMI 1640 and Dulbecco's modified Eagle's medium (DMEM) culture media, respectively (Sigma) with 10% Fetal Bovine Serum (FBS) (Sigma), penicillin (100 µg/ml) (Sigma) and streptomycin (100 µg/ml) (Sigma). Cultures were maintained at 37oC in 5% CO2.
Cell viability assessment
RK33 (1 x 104 cells/ml) RK45 (3 x 104 cells/ml) and HSF (5 x 104 cells/ml) cells were incubated in culture medium for 24 hours in 96-well plates (Nunc, Roskilde, Denmark). After that, cells were treated with different concentrations of SAHA and/or CDDP in fresh culture medium. Cell viability was determined after 72 hours by methylthiazolyldiphenyl-tetrazolium bromide (MTT) method as described previously [18].
Cell proliferation assay
Proliferation of RK33 (2 x 104/ml) and RK45 (5 x 104/ml) cells was assessed after cell seeding in 96-well plates (Nunc) and after 72 hours of incubation in selected concentrations of SAHA using Cell Proliferation Elisa, BrdU Kit (Roche Diagnostics, Mannheim, Germany). The procedure was performed according to the manufacturer's instructions as reported previously [18]
Isobolographic analysis of interactions
Log-probit analysis according to Litchfield and Wilcoxon [19], was used to determine the percentage of inhibition of cell viability per dose of CDDP and SAHA when administered singly in the RK33 and RK45 cell lines measured in vitro by the MTT assay. Subsequently, from the log-probit dose-response lines, median inhibitory concentrations (IC50 values) of CDDP and SAHA were calculated as described earlier [20]. Test for parallelism between two dose-response curves (CDDP and SAHA) was performed according to the log-probit method, as described in detail in our previous studies [21-23], and revealed that the dose-response curves for CDDP and SAHA were not parallel to each other in both, RK33 and RK45 cell lines. Interactions between CDDP and SAHA in RK33 and RK45 cancer cell lines were isobolographically analyzed as described elsewhere [21, 24-26]. The median additive inhibitory concentrations (IC50 add) for the mixture of CDDP with SAHA, which theoretically should inhibit 50% of cell viability, were calculated as demonstrated by Tallarida [25, 26]. The assessment of the experimentally-derived IC50 mix at the fixed-ratio of 1:1 was based on the concentration of the mixture of CDDP and SAHA that inhibited 50% of cell viability in both, RK33 and RK45 cancer cell lines measured in vitro by the MTT assay. To calculate the concentrations of particular drugs (CDDP and SAHA) in the mixture, the IC50 mix values were multiplied by the proportions of CDDP and SAHA (denoted for additive mixture). Details concerning the isobolographic analysis have been published elsewhere [20, 21, 25, 26].
Histone extraction and Western blotting analysis
RK33 (2 x 105 cells/ml) and RK45 (3 x 105 cells/ml) cells were incubated in culture medium for 24 hours in 6-well plates (Nunc). Next day, tumor cells were treated with different concentrations of SAHA for 6 hours. After treatment, the cells were lysed in TEB buffer (0.5% Triton X100, 2 mM PMSF and 0.02% NaN3 in PBS, pH 7.4) and centrifuged at 800 x g for 10 minutes at 4oC. Collected nuclear pellet was used for acidic isolation of histones with 0.02 N HCl. The extracted histones were further separated by 15% sodium dodecyl sulphate (SDS)-polyacrylamide gel electrophoresis and transferred to polyvinyl difluoride (PVDF) membrane (Merck Chemicals, Darmstadt, Germany). The membrane was blocked with Tris-buffered saline (TBS), pH 7.5 containing 5% non-fat dry milk and 0.05% Tween-20 and then probed with primary antibody at 4oC overnight. On the following day, the membrane was washed and then incubated with a horseradish peroxidase-labeled secondary antibody (Cell Signaling, Danvers, MA, USA) for 1 hour at room temperature (RT). Finally, the membrane was visualized using a Lumi-Light Western Blotting Substrate (Roche, Mannheim, Germany) according to the manufacturer's instructions The following primary antibodies were used: acetyl-histone H3 (Lys9/14, Upstate Biotechnology, Lake Placid, NY, USA) and acetyl-histone H3 (Lys18 and Lys23, Cell Signaling). Subsequently, stripping buffer (62.5 mM Tris-HCl, pH 6.8 with 100 mM β-mercaptoethanol and 2% SDS) was used to remove bound antibodies and reprobe the membrane with anti-histone H3 (Cell Signaling) recognizing total, acetylated and non-acetylated form of histone H3
Assessment of HDACs activity
Measurement of HDACs activity was performed using HDAC Assay Kit (Upstate Biotechnology). Briefly, RK33 (2 x 105/ml) and RK45 (3 x 105/ml) cells were treated with different concentrations of SAHA for 6 hours. After treatment, the cells were lysed in RIPA buffer (PBS, pH 7.4 with 0.1% SDS, 1 mM EDTA, 1 mM EGTA, 1 mM Na3VO4, 20 mM NaF, 0.5 mM DTT, 1 mM PMSF and protease inhibitor cocktail) for 15 minutes at 4oC and centrifuged at 8000 x g for 10 minutes at 4oC. The determination of HDACs activity in collected supernatant was performed according to the manufacturer's instructions as described previously [27]
Assessment of cell death
Apoptosis of RK33 and RK45 cells following treatment with different concentrations of SAHA (0.5-5 μM) for 24 hours was measured using Cell Death Detection ELISAPLUS kit (Roche). The procedure was performed according to manufacturer instruction as described previously [18].
Flow cytometry analysis
For cell cycle analysis, RK33 and RK45 cells were treated with selected concentrations of SAHA for 24 hours. After treatment, cells were collected, fixed in ice-cold ethanol at -20oC for 24 hours, incubated with Propidium Iodide (PI)/RNase Staining Buffer (Becton Dickinson, San Jose, CA, USA) and then analyzed using the FACSCaliburTM flow cytometer. Details concerning the flow cytometry analysis have been published previously [27]
RNA isolation and quantitative PCR (qPCR)
Total RNA from RK33 and RK45 cells was isolated and DNase treated using High Pure RNA Isolation Kit (Roche) and subsequently reverse transcribed using Transcriptor High Fidelity cDNA Synthesis Kit (Roche) and oligo(dT) primer according to manufacturer's protocols. Amplification with fluorescence detection was performed using LightCycler® 480 II instrument (Roche). Analyzed genes was amplified in duplex with reference gene GAPD utilizing UPL hydrolysis probes (Roche) labelled with FAM and Yellow 555, respectively, in the following steps: 10min at 95°C and 45 cycles of: 10s at 95°C, 30s at 60°C, 10s at 72°C. The following primers and probe sets were used: CCND1 (For: 5'-GAAGATCGTCGCCACCTG-3', Rev: 5'-GACCTCCTCCTCGCACTTCT-3', Probe: 5'-TGCTGGAG-3'); CDKN1A (For: 5'-TCACTGTCTTGTACCCTTGTGC-3', Rev: 5'-GGCGTTTGGAGTGGTAGAAA-3', Probe: 5'-CCTGGAGA-3'); GAPD (For: 5'-CTCTGCTCCTCCTGTTCGAC-3', Rev: 5'-GCCCAATACGACCAAATCC-3', Probe 5'-CTTTTGCGTCGC-3'). Relative quantification expression analysis was made by efficiency method.
Statistics
The calculations were done by means of Student`s t-test and ANOVA test for multiple comparisons. Data is expressed as the mean ± SEM (*p<0.05, **p<0.01, ***p<0.001). The IC50 and IC50 mix values for CDDP and SAHA administered alone or in combination at the fixed-ratio of 1:1 were calculated by computer-assisted log-probit analysis according to Litchfield and Wilcoxon [19]. The experimentally-derived IC50 mix values for the mixture of CDDP with SAHA were statistically compared with their respective theoretical additive IC50 add values by the use of unpaired Student's t-test, according to Tallarida [28].
Results
SAHA decreases HDACs activity and induces histone H3 acetylation in larynx cancer cell lines
Our studies showed that SAHA reduced intracellular HDACs activity in a dose-dependent manner in both cancer cell lines (Figure 1). Inhibition of HDACs activity was accompanied by histone H3 K9, K14, K18 and K23 hyperacetylation as demonstrated by Western-blot analysis. Strong acetylation induction at histone H3 K18 in RK33 cells was observed already after treatment with 1 µM SAHA (Figure 2A). Significant increase in acetylation of histone H3 K9 and K14 was observed when RK33 cells were incubated with 5 and 10 µM SAHA (Figure 2A), and RK45 cells were exposed to 10 µM SAHA (Figure 2B). Pronounced accumulation of acetylated histone H3 K23 was also found in RK45 cells (Figure 2B).
Figure 1
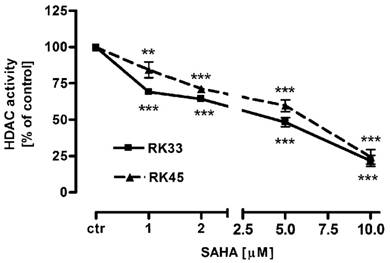
Intracellular HDACs activity in larynx cancer cell lines after treatment with either culture medium alone (control), or SAHA (1-10 µM) for 6 hours. Data are presented as mean ± SEM at each concentration. (**p<0.01; ***p<0.001 by Student's t-test); (***p<0.001 by analysis of variance ANOVA test); n=9 per concentration from three independent experiments.
SAHA, administered singly and in combination with CDDP, shows anti-proliferative action in larynx cancer cell lines
The effect of SAHA on the viability and proliferation of RK33 and RK45 cells was studied by MTT and BrdU assay, respectively. Increasing concentrations of SAHA (0,1-10 µM) significantly reduced the viability of both cancer cell lines and had a low cytotoxic effect on the viability of normal cells (Figure 3A). The reduction in viable cell number was attributed to decreased cell proliferation, as demonstrated by BrdU incorporation assay (Figure 3B). The experimentally denoted IC50 values for SAHA amounted to 0.432 ± 0.059 μg/ml for the RK33 cell line and 0.348 ± 0.074 μg/ml for the RK45 cell line (Table 1). CDDP administered alone also produced anti-proliferative effects in both larynx cancer cell lines. The IC50 values for CDDP were 1.193 ± 0.273 μg/ml (RK33 cell line) and 1.387 ± 0.270 μg/ml (RK45 cell line), respectively (Table 1). The test for parallelism of two dose-response curves between CDDP and SAHA revealed that the curves were non-parallel to each other (Figure 4A-B). The combination of CDDP with SAHA at the fixed-ratio of 1:1 produced the anti-proliferative effects in both larynx cancer cell lines. The experimentally-derived IC50 mix values for the mixture of CDDP and SAHA were 0.967 ± 0.245 μg/ml for RK33 cell line and 0.301 ± 0.051 μg/ml for RK45 cell line, respectively (Table 2).
Table 1
Anti-proliferative effects of CDDP and SAHA administered singly in two cancer cell lines (RK33 and RK45) measured in vitro by the MTT assay.
| Cell line | Drug | IC50 (μg/ml) | n |
|---|---|---|---|
| RK33 | CDDP | 1.193 ± 0.273 | 96 |
| SAHA | 0.432 ± 0.059 | 96 | |
| RK45 | CDDP | 1.387 ± 0.270 | 120 |
| SAHA | 0.348 ± 0.074 | 120 |
Data are median inhibitory concentrations (IC50 values in μg/ml ± S.E.M.) of CDDP and SAHA administered separately in RK33 and RK45 cancer cell lines; n - total number of items used at those concentrations, whose expected anti-proliferative effects in the MTT assay ranged between 16% and 84%.
Table 2
Type I isobolographic analysis of interactions (for non-parallel DRRCs) between CDDP and SAHA at the fixed-ratio combination of 1:1 in two cancer cell lines (RK33 and RK45) measured in vitro by the MTT assay.
| Cell line | Combination | IC50 mix (μg/ml) | n mix | #IC50 add (μg/ml) | n add | &IC50 add (μg/ml) | n add |
|---|---|---|---|---|---|---|---|
| RK33 | CDDP+SAHA | 0.967 ± 0.245 | 96 | 0.735 ± 0.266 | 188 | 0.890 ± 0.323 | 188 |
| RK45 | CDDP+SAHA | 0.301 ± 0.051* | 80 | 0.850 ± 0.250 | 236 | 0.884 ± 0.251 | 236 |
Data are median inhibitory concentrations (IC50 values in μg/ml ± S.E.M.) for two-drug mixtures (CDDP+SAHA), determined either experimentally (IC50 mix) or theoretically calculated (IC50 add) that blocked proliferation in 50% of tested cells in two larynx cancer cell lines (RK33 and RK45) measured in vitro by the MTT assay. n mix - total number of items used at those concentrations whose expected anti-proliferative effects ranged between 16% and 84% for the experimental mixture; n add - total number of animals calculated for the additive mixture of the drugs examined (n add = n_CDDP + n_SAHA - 4); # IC50 add - value for the lower line of additivity; & IC50 add value for the upper line of additivity. Statistical evaluation of data was performed with unpaired Student's t-test according to Tallarida [28]. *P<0.05 vs. both additive IC50 add values indicates supra-additive (synergistic) interaction.
Figure 2
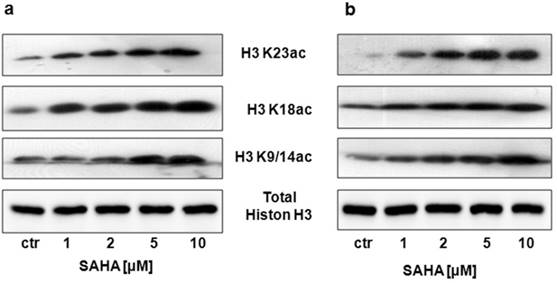
Detection of the histone H3 acetylation level at K9/14, K18 and K23 in RK33 (A) and RK45 (B) cells after treatment with either culture medium alone (control), or SAHA (1-10 µM) for 6 hours by Western blotting analysis. The membranes were re-probed with anti-H3 antibody of following incubation of larynx cancer cells with.
Figure 3
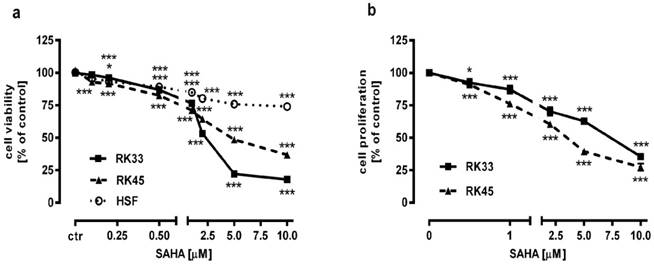
Antiproliferative activity of SAHA in larynx cancer cell lines (RK33 and RK45) and normal human skin fibroblasts (HSF). Larynx cancer cell lines or fibroblasts cells were treated with either culture medium alone (control), or SAHA for 72 hours. The viability and proliferation of cancer cells were measured by the MTT assay (A) and by means of BrdU incorporation (B), respectively. Results are presented as mean ± SEM at each concentration. (*p<0.05; ***p<0.001 by Student's t-test); (***p<0.001 by analysis of variance ANOVA test), n=40 per concentration from five independent experiments.
Figure 4
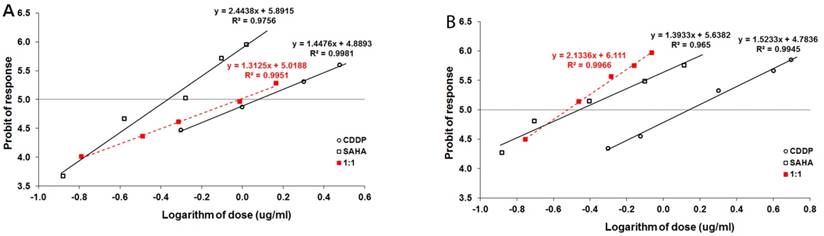
Log-probit analysis of dose-response curves of cisplatin (CDDP) and suberoylanilide hydroxamic acid (SAHA), administered alone and in combination at the fixed-ratio of 1:1, illustrating the anti-proliferative effects of the drugs in the RK33 (A) and RK45 (B) larynx cancer cell lines measured in vitro by the MTT assay. Doses of CDDP and SAHA were transformed into logarithms and the anti-proliferative effects produced by the drugs into probits according to Litchfield and Wilcoxon [19]. Linear regression equations of dose-response curves are presented on the graph, where: y - is the probit of response, and x - is the logarithm (to the base 10) of a drug dose, R2 - coefficient of determination. According to the test for parallelism, the experimentally determined dose-response curves for CDDP and SAHA were not parallel to one another.
Figure 5
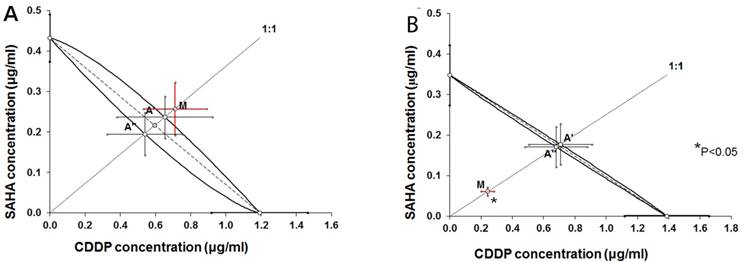
Isobolograms illustrating types of interactions between cisplatin (CDDP) and suberoylanilide hydroxamic acid (SAHA) with respect to their anti-proliferative effects in the RK33 (A) and RK45 (B) larynx cancer cell lines measured in vitro by the MTT assay. The IC50 ± S.E.M. of CDDP and SAHA are plotted graphically on the X- and Y-axes, respectively. The lower and upper isoboles of additivity represent the curves connecting the IC50 values. The dotted line originating from the point (0, 0) corresponds to the fixed-ratio of 1:1 for the combination of CDDP with SAHA. Points A' and A” correspond to the theoretically calculated IC50 add values for both, lower and upper isoboles of additivity. The point M illustrates the experimentally determined IC50 mix value producing a 50% anti-proliferative effect in two cancer cell lines; RK33 (A) and RK45 (B). The S.E.M. values are presented on each graph as horizontal and vertical error bars for every IC50 value. The experimentally-derived IC50 mix value is placed close to the point A', indicating additive interaction between CDDP and SAHA in the cancer cell line RK33 (A), whereas in the cancer cell line RK45 (B), the IC50 mix value is placed significantly (P<0.05) below the point A”, indicating supra-additive (synergistic) interaction between CDDP and SAHA.
Isobolographic analysis of interaction between CDDP and SAHA in larynx cancer cell lines
Type I isobolographic analysis of interaction for non-parallel dose-response curves revealed that the mixture of CDDP with SAHA at the fixed-ratio of 1:1 exerted additive interaction in the RK33 cell line (Figure 5A) and supra-additive (synergistic) interaction in the RK45 cell line (Figure 5B). The experimentally derived IC50 mix value for fixed-ratio combination of 1:1 in the RK33 cell line did not significantly differ from the IC50 add values (Table 2). In contrast, the experimentally derived IC50 mix value for fixed-ratio combination of 1:1 in the RK45 cell line was significantly lower than the additively calculated IC50 add values (P<0.05; Table 2).
SAHA induces apoptosis in larynx cancer cell lines
To determine the mechanism by which SAHA inhibits larynx cancer cells proliferation, we studied the ability of this histone deacetylase inhibitor to induce apoptosis in RK33 and RK45 cells. Both cancer cell lines were treated with 0,5 - 5 µM SAHA for 24 hours, followed by colorimetric detection of mono- and oligonucleosomes released to cytosol by apoptotic cells. As shown in the Figure 6, SAHA triggered apoptotic cell death in a dose-dependent manner. We observed low (not more than 2-fold) increase in apoptosis after treatment with SAHA in the concentration range from 0,5 - 2 µM. Higher concentration of SAHA (5 µM) resulted in 37-fold (***p<0,001) enhance of apoptosis in RK33 cells, compared to only 3-fold (***p<0,001) apoptosis in RK45 cells.
SAHA influences cell cycle progression in larynx cancer cells
Besides induction of apoptosis, we also examined cell cycle progression as another possible mechanism confirming antiproliferative effect of SAHA against larynx cancer cells. FACS analysis revealed that cell cycle distribution changed in a dose-dependent manner in RK33 (Figure 7A) and RK45 cells (Figure 7B) treated with SAHA for 24 hours. We observed an accumulation of cells in G1 phase of the cell cycle, with a concomitant reduction of cells number in S/G2 phase. As shown in Table 3, the cell cycle arrest by SAHA was stronger in RK33 cells. After RK33 cells exposure to 2 µM SAHA, cell population in G1 and S phases reached 89% (***p<0,001) and 3% (***p<0,001), respectively. Similar results were obtained in RK45 cells, when 5 µM of SAHA concentration was applied.
SAHA altered expression of CCND1 and CDKN1A genes in larynx cancer cells
We analyzed changes in expression of CCND1 and CDKN1A encoding proteins engaged in block of cell cycle progression at G1/S phase transition in larynx cancer cell lines after treatment with SAHA (1-5 µM) for 24 hours. We found a dose-dependent downregulation of cyclin D1 and upregulation of p21Waf1/Cip1 mRNA. There were not significant differences in CCND1 expression between RK33 and RK45 cells. In both cell lines exposed to 5 µM SAHA cyclin D1 mRNA decreased 2,5-fold compared with control (untreated cells) (Figure 8A). CDKN1A expression increased 6-fold (***p<0,001) and 10-fold (**p<0,01) in RK33 and RK45 cells, respectively, when treated with 5 µM SAHA (Figure 8B).
Figure 6
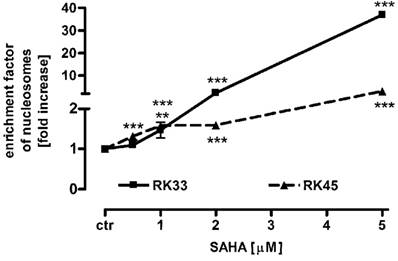
Effects of SAHA on apoptosis in RK33 and RK45 cells. SAHA induces a concentration-dependent increase in apoptotic cells as measured using the Cell Death Detection ELISAPLUS kit (Roche). The rate of apoptosis was expressed as an enrichment factor of oligonucleosome fragments released into the cytoplasm in the absence (control) and after treatment with SAHA (0.5-5 µM) for 24 hours. N=9 per concentration from three independent experiments, **p<0.01; ***p<0.001 versus control, Student's t-test.
Table 3
Cell cycle analysis of larynx cancer cell lines treatment with SAHA.
| Cell line | SAHA [µM] | G1 phase | S phase | G2 phase |
|---|---|---|---|---|
| RK33 | Control | 70.67 ± 0.456 | 10.22 ± 0.115 | 18.96 ± 0.567 |
| 0.5 | 75.55** ± 0.825 | 9.16** ± 0.245 | 15.72* ± 0.575 | |
| 1 | 82.17*** ± 0.855 | 6.91*** ± 0.666 | 11.16*** ± 0.219 | |
| 2 | 89.51*** ± 1.394 | 3.51*** ± 0.846 | 7.03*** ± 0.574 | |
| RK45 | Control | 60.57 ± 0.683 | 16.30 ± 0.583 | 23.54 ± 0.286 |
| 0.5 | 64.11*** ± 0.171 | 13.65** ± 0.216 | 22.32* ± 0.300 | |
| 1 | 67.09** ± 1.225 | 11.45*** ± 0.564 | 21.65* ± 1.195 | |
| 2 | 71.40** ± 1.834 | 10.92*** ± 0.438 | 14.99** ± 1.447 | |
| 5 | 83.70*** ± 0.157 | 3.01*** ± 0.517 | 13.08*** ± 0.689 |
(*p<0.05; **p<0.01; ***p<0.001; Student's t-test, n=9 per concentration from three independent experiments).
Figure 7
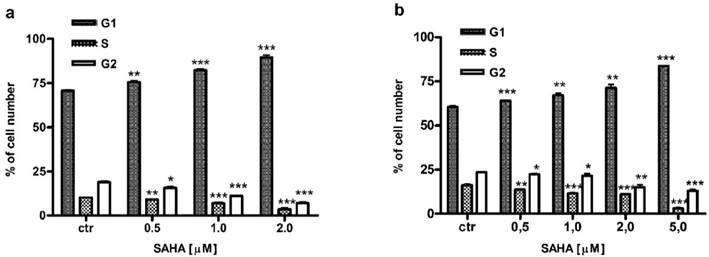
Analysis of the cell cycle in RK33 (A) and RK45 (B) cells treated with SAHA. Larynx cancer cell lines were exposed to either culture medium alone (controls) or SAHA (0.5-5 µM) for 24 hours and cell cycle was assessed by propidium iodide-staining. (*p<0.05; **p<0.01; ***p<0.001; Student's t-test, n=9 per concentration from three independent experiments).
Figure 8
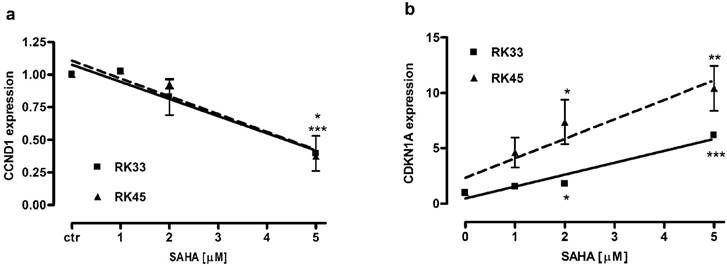
Changes in expression of genes encoding proteins engaged in G1/S transition in larynx cancer cell lines after treatment with SAHA. Expression of CCND1 (A) and CDKN1A (B) was analyzed by qPCR method in RK33 and RK45 cells exposed to either culture medium alone (controls) or SAHA (1-5 µM) for 24 hours. (*p<0.05, **p<0.01, ***p<0.001, n=9 measurements from three separate experiments, Student's t-test).
Discussion
Due to the poor response to chemotherapeutics in patients and low survival rate, successful treatment of larynx cancer still remains a challenge. An interesting therapeutic approach arises with the opportunity to use histone deacetylase inhibitors in solid tumors, especially that overexpression of HDACs has been observed in head and neck cancers [29]. There are limited reports of HDIs action against larynx cancer in comparison with studies on other types of head and neck cancers. Our previous work showed the antiproliferative activity of valproic acid (VPA), a member of the short-chain fatty acid class of HDAC inhibitors, towards both RK33 and RK45 larynx cancer cell lines [27]. Now, we focused on SAHA - the other type of HDI (also known as Vorinostat), which is membrane permeable and thus acts as inhibitor of HDACs in both cytoplasm and nucleus [30]. SAHA is a reversible, competitive inhibitor which binds to the zinc ion located in the catalytic domain of HDACs and thereby suppresses its enzymatic activity. SAHA inhibits the class I HDACs (HDAC1, HDAC2, HDAC3) and the class II HDACs (HDAC6) and under the trade name Zolinza® was the first histone deacetylase inhibitor approved by FDA for clinical use in cutaneous T cell lymphoma (CTCL) [31, 32]. SAHA is a compound with low toxicity in normal cells [33], which was also confirmed in the current study, and high therapeutic potential for different tumors including colon cancer [34], prostate cancer [35], breast cancer [36], non-small cell lung cancer [37], pancreatic cancer [38], glioma cells [39], uterine sarcoma [40], ovarian cancer [41] and oral squamous cell carcinoma [42]. In the present study, we showed that SAHA suppressed the growth of larynx cancer cell lines at micromolar concentrations in a dose-dependent manner. We also observed enhancement of cytotoxic effects, i.e., supra-additive (synergistic) and additive, provided by the combined use of SAHA and cisplatin in larynx cancer cell lines. The mechanism underlying this phenomenon is not clear. It could be related to increased acetylation of lysine in the nucleosomal histone core through SAHA. In consequence, chromatin is more relaxed and allows increased access to CDDP interacting with DNA [43]. In fact, treatment of larynx cancer cell lines with SAHA induced hyperacetylation of histone H3 K9, K14 and K23. Interestingly, although both larynx cancer cell lines represent similar histopathological characteristics, combinatorial use of SAHA with CDDP resulted in different response. It has been shown that HDIs, besides altering chromatin structure, additionally sensitize cancer cells to DNA damaging agents by diminishing DNA repair [44]. In our studies synergy with cisplatin has been observed in RK45 cell line which was more sensitive towards treatment with SAHA. It is likely that strong effects of co-treatment with SAHA and cisplatin in RK45 cells could be linked to increased ability of vorinostat to abrogate the process of repair DNA damage [45]. Histone deacetylases as regulators of chromatin remodeling promote the DNA repair [46]. It is known that SIRT6-dependent global deacetylation of H3K9 is required for the repair of DNA double strand breaks [47] and depletion SIRT6 confers sensitization to DNA-damaging agents [48]. Similarly, deletion of HDAC3 results in the increased H3K9Ac and H3K14Ac, genome instability and reduced ability to DNA damage repair [46, 49]. Since HDAC3 is an important target of SAHA, significant hiperacetylation of H3K9/14 in RK45 cells could be related to abolishment of DNA repair process and therefore higher response of RK45 larynx cancer cells to cisplatin. Increased cytotoxicity of cisplatin resulting from concurrently applied SAHA has been observed in different cancer cells [50-53]. Some of these combinations are now tested in preclinical studies, as well as in clinical trials [54]. We also found that SAHA concentrations, necessary to obtain 50% inhibition of both RK33 and RK45 cells viability, caused the blockade of the cell cycle progression in the G1/S phase and statistically relevant induction of apoptosis. HDAC inhibitors have been shown to cause growth arrest of cancer cells by inducing changes in the expression of key regulatory proteins of cell cycle progression. HDIs upregulate antiproliferative cyclin-dependent kinase inhibitors such as p16, p19, p21WAF1/CIP1, p27Kip1 and p57Kip2, while they decrease the level of pro-growth cyclin- dependent kinases (Cdk2 and Cdk4) and various cyclins (cyclin A, B1, D1, E) which are frequently overexpressed in cancer cells [33, 55]. In our studies, treatment of RK33 and RK45 cell lines with SAHA resulted in significant decrease in CCND1 gene expression and increase in CDKN1A gene expression encoding cyclin D1 and p21WAF1/CIP1 proteins, respectively. The observed cell cycle arrest at G1/S phase in larynx cancer cells may be associated with the loss of Cdk4 activity mediated by repression of cyclin D [9] and/or the binding of p21 inhibitor into Cdk4/cyclin D and Cdk2/cyclin E complexes [56]. Some reports have demonstrated that p21 expression can also determine the sensitivity of cancer cells to apoptosis induced by HDIs, and these pro- or anti-apoptotic responses depend on the type of cancer cells and HDIs-type used [57-59]. In our study SAHA also affected apoptosis in larynx cancer in the cell-type specific manner. The sharp increase of apoptotic RK33 cells correlates with the reduction of p21. This could confirm earlier data that p21 seems to be an important factor preventing cancer cells from HDIs-mediated apoptosis. However, the molecular mechanism by which p21 inhibits apoptosis is not well understood [59].
In conclusion, it should be noted that SAHA sensitized larynx cancer cells to cisplatin as well as suppressed cell proliferation, induced cell cycle arrest and apoptosis at clinically permitted and safe concentration range (0.43-5µM). Previous clinical studies have shown that these levels of SAHA can be achieved in human serum by systemic administration of this drug [60]. Very recently, results presented by Valdez et al. [61] explored the efficiency of SAHA to overcome chemoresistance - the major impediment in medical oncology limiting the success of multidrug regimens in the treatment of most advanced solid tumors and some hematological malignancies. They show that in human clinical trials (clinical trial NCT01983969) SAHA reduced the expression of drug transporter MRP1 at the mRNA and protein levels in monuclear blood cells, thereby decreasing efflux of chemotherapeutic drugs from the cells and thus enhancing their cytotoxicity. Since MRP1 exports also cisplatin/glutathione conjugates, these observations could suggest a potential mechanism of CDDP/SAHA synergism observed in our experiments. Our study demonstrated that the combination of SAHA with CDDP could have clinically relevance because it significantly reduces the dose of cisplatin while maintaining the same therapeutic effect (a 50% proliferation inhibition). Our findings indicate that SAHA could be used to optimize a combined therapy based on cisplatin against laryngeal cancer. Given that cisplatin induces multiple side effects, the use of less toxic doses of this conventional chemotherapeutic in combination with clinically available doses of SAHA seems to be an interesting therapeutic alternative. Moreover, concurrent administration of both agents may be a novel strategy to enhance the efficacy of current anticancer therapy in patients with larynx cancer.
Acknowledgements
This study was supported by Medical University of Lublin DS 440/2012-2013 and DS 440/2014-2015 grants and The Polish Ministry of Science and Higher Education Grant No NN405 617538
Competing Interests
The authors have declared that no competing interest exists.
References
1. Baker SJ, Reddy EP. Targeted inhibition of kinases in cancer therapy. Mt Sinai J Med. 2010;77:573-86
2. Peterson C. Drug therapy of cancer. Eur J Clin Pharmacol. 2011;67:437-47
3. Widmer N, Bardin C, Chatelut E, Paci A, Beijnen J, Leveque D. et al. Review of therapeutic drug monitoring of anticancer drugs part two-targeted therapies. Eur J Cancer. 2014;50:2020-36
4. Jenckel F, Knecht R. State of the art in the treatment of laryngeal cancer. Anticancer Res. 2013;33:4701-10
5. Hunter KD, Parkinson EK, Harrison PR. Profiling early head and neck cancer. Nat Rev Cancer. 2005;5:127-35
6. Xiang C, Lv Y, Wei Y, Wei J, Miao S, Mao X. et al. Effect of EphA7 Silencing on Proliferation, Invasion and Apoptosis in Human Laryngeal Cancer Cell Lines Hep-2 and AMC-HN-8. Cell Physiol Biochem. 2015;36:435-45
7. Marur S, Forastiere AA. Head and Neck Squamous Cell Carcinoma: Update on Epidemiology, Diagnosis, and Treatment. Mayo Clin Proc. 2016;91:386-96
8. Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O. et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869-83
9. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769-84
10. Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26:5420-32
11. Mercurio C, Minucci S, Pelicci PG. Histone deacetylases and epigenetic therapies of hematological malignancies. Pharmacol Res. 2010;62:18-34
12. Zhang J, Zhong Q. Histone deacetylase inhibitors and cell death. Cell Mol Life Sci. 2014;71:3885-901
13. Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules. 2015;20:3898-941
14. West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124:30-9
15. Zwergel C, Valente S, Jacob C, Mai A. Emerging approaches for histone deacetylase inhibitor drug discovery. Expert Opin Drug Discov. 2015;10:599-613
16. Rzeski W, Paduch R, Klatka J, Kandefer-Szerszen M, Stepulak A, Pozarowski P. et al. Establishment and preliminary characterization of two cell lines derived from larynx carcinoma. Folia Histochem Cytobiol. 2002;40:195-6
17. Rzeski W, Stepulak A, Szymanski M, Juszczak M, Grabarska A, Sifringer M. et al. Betulin elicits anti-cancer effects in tumour primary cultures and cell lines in vitro. Basic Clin Pharmacol Toxicol. 2009;105:425-32
18. Jarzab A, Grabarska A, Kielbus M, Jeleniewicz W, Dmoszynska-Graniczka M, Skalicka-Wozniak K. et al. Osthole induces apoptosis, suppresses cell-cycle progression and proliferation of cancer cells. Anticancer Res. 2014;34:6473-80
19. Litchfield JT Jr, Wilcoxon F. A simplified method of evaluating dose-effect experiments. J Pharmacol Exp Ther. 1949;96:99-113
20. Wawruszak A, Luszczki JJ, Grabarska A, Gumbarewicz E, Dmoszynska-Graniczka M, Polberg K. et al. Assessment of Interactions between Cisplatin and Two Histone Deacetylase Inhibitors in MCF7, T47D and MDA-MB-231 Human Breast Cancer Cell Lines - An Isobolographic Analysis. PLoS One. 2015;10:e0143013
21. Luszczki JJ. Isobolographic analysis of interaction between drugs with nonparallel dose-response relationship curves: a practical application. Naunyn Schmiedebergs Arch Pharmacol. 2007;375:105-14
22. Luszczki JJ, Czuczwar SJ. Isobolographic profile of interactions between tiagabine and gabapentin: a preclinical study. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:434-46
23. Luszczki JJ, Czuczwar SJ. Biphasic characteristic of interactions between stiripentol and carbamazepine in the mouse maximal electroshock-induced seizure model: a three-dimensional isobolographic analysis. Naunyn Schmiedebergs Arch Pharmacol. 2006;374:51-64
24. Grabovsky Y, Tallarida RJ. Isobolographic analysis for combinations of a full and partial agonist: curved isoboles. J Pharmacol Exp Ther. 2004;310:981-6
25. Tallarida RJ. An overview of drug combination analysis with isobolograms. J Pharmacol Exp Ther. 2006;319:1-7
26. Tallarida RJ. Interactions between drugs and occupied receptors. Pharmacol Ther. 2007;113:197-209
27. Grabarska A, Dmoszyńska-Graniczka M, Jeleniewicz W, Kiełbus M, Nowosadzka E, Rivero-Muller A. et al. Valproic acid suppresses growth and enhances cisplatin cytotoxicity to larynx cancer cells. Head and Neck Oncology. 2014;6:7
28. Tallarida RJ. Drug Synergism and Dose-Effect Data Analysis: Chapman and Hall/CRC; 2000.
29. Kumar B, Yadav A, Lang JC, Teknos TN, Kumar P. Suberoylanilide hydroxamic acid (SAHA) reverses chemoresistance in head and neck cancer cells by targeting cancer stem cells via the downregulation of nanog. Genes Cancer. 2015;6:169-81
30. Olson DE, Sleiman SF, Bourassa MW, Wagner FF, Gale JP, Zhang YL. et al. Hydroxamate-based histone deacetylase inhibitors can protect neurons from oxidative stress via a histone deacetylase-independent catalase-like mechanism. Chem Biol. 2015;22:439-45
31. Iwamoto M, Friedman EJ, Sandhu P, Agrawal NG, Rubin EH, Wagner JA. Clinical pharmacology profile of vorinostat, a histone deacetylase inhibitor. Cancer Chemother Pharmacol. 2013;72:493-508
32. Slingerland M, Guchelaar HJ, Gelderblom H. Histone deacetylase inhibitors: an overview of the clinical studies in solid tumors. Anticancer Drugs. 2014;25:140-9
33. Emanuele S, Lauricella M, Tesoriere G. Histone deacetylase inhibitors: apoptotic effects and clinical implications (Review). Int J Oncol. 2008;33:637-46
34. Sun PC, Tzao C, Chen BH, Liu CW, Yu CP, Jin JS. Suberoylanilide hydroxamic acid induces apoptosis and sub-G1 arrest of 320 HSR colon cancer cells. J Biomed Sci. 2010;17:76
35. Butler LM, Agus DB, Scher HI, Higgins B, Rose A, Cordon-Cardo C. et al. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res. 2000;60:5165-70
36. Chiaradonna F, Barozzi I, Miccolo C, Bucci G, Palorini R, Fornasari L. et al. Redox-Mediated Suberoylanilide Hydroxamic Acid Sensitivity in Breast Cancer. Antioxid Redox Signal. 2015;23:15-29
37. Komatsu N, Kawamata N, Takeuchi S, Yin D, Chien W, Miller CW. et al. SAHA, a HDAC inhibitor, has profound anti-growth activity against non-small cell lung cancer cells. Oncol Rep. 2006;15:187-91
38. Xu XD, Yang L, Zheng LY, Pan YY, Cao ZF, Zhang ZQ. et al. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses vasculogenic mimicry and proliferation of highly aggressive pancreatic cancer PaTu8988 cells. BMC Cancer. 2014;14:373
39. Xu J, Sampath D, Lang FF, Prabhu S, Rao G, Fuller GN. et al. Vorinostat modulates cell cycle regulatory proteins in glioma cells and human glioma slice cultures. J Neurooncol. 2011;105:241-51
40. Hrzenjak A, Moinfar F, Kremser ML, Strohmeier B, Petru E, Zatloukal K. et al. Histone deacetylase inhibitor vorinostat suppresses the growth of uterine sarcomas in vitro and in vivo. Mol Cancer. 2010;9:49
41. Chen S, Zhao Y, Gou WF, Zhao S, Takano Y, Zheng HC. The anti-tumor effects and molecular mechanisms of suberoylanilide hydroxamic acid (SAHA) on the aggressive phenotypes of ovarian carcinoma cells. PLoS One. 2013;8:e79781
42. Nagumo T, Takaoka S, Yoshiba S, Ohashi M, Shirota T, Hatori M. et al. Antitumor activity of suberoylanilide hydroxamic acid against human oral squamous cell carcinoma cell lines in vitro and in vivo. Oral Oncol. 2009;45:766-70
43. Diyabalanage HV, Granda ML, Hooker JM. Combination therapy: histone deacetylase inhibitors and platinum-based chemotherapeutics for cancer. Cancer Lett. 2013;329:1-8
44. Thurn KT, Thomas S, Raha P, Qureshi I, Munster PN. Histone deacetylase regulation of ATM-mediated DNA damage signaling. Mol Cancer Ther. 2013;12:2078-87
45. Richon VM, Garcia-Vargas J, Hardwick JS. Development of vorinostat: current applications and future perspectives for cancer therapy. Cancer Lett. 2009;280:201-10
46. Li Z, Zhu WG. Targeting histone deacetylases for cancer therapy: from molecular mechanisms to clinical implications. Int J Biol Sci. 2014;10:757-70
47. Etchegaray JP, Zhong L, Mostoslavsky R. The histone deacetylase SIRT6: at the crossroads between epigenetics, metabolism and disease. Curr Top Med Chem. 2013;13:2991-3000
48. Cea M, Cagnetta A, Adamia S, Acharya C, Tai YT, Fulciniti M. et al. Evidence for a role of the histone deacetylase SIRT6 in DNA damage response of multiple myeloma cells. Blood. 2016;127:1138-50
49. Gong F, Miller KM. Mammalian DNA repair: HATs and HDACs make their mark through histone acetylation. Mutat Res. 2013;750:23-30
50. Jin KL, Park JY, Noh EJ, Hoe KL, Lee JH, Kim JH. et al. The effect of combined treatment with cisplatin and histone deacetylase inhibitors on HeLa cells. J Gynecol Oncol. 2010;21:262-8
51. Shen J, Huang C, Jiang L, Gao F, Wang Z, Zhang Y. et al. Enhancement of cisplatin induced apoptosis by suberoylanilide hydroxamic acid in human oral squamous cell carcinoma cell lines. Biochem Pharmacol. 2007;73:1901-9
52. Suzuki M, Endo M, Shinohara F, Echigo S, Rikiishi H. Enhancement of cisplatin cytotoxicity by SAHA involves endoplasmic reticulum stress-mediated apoptosis in oral squamous cell carcinoma cells. Cancer Chemother Pharmacol. 2009;64:1115-22
53. Kim MS, Blake M, Baek JH, Kohlhagen G, Pommier Y, Carrier F. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res. 2003;63:7291-300
54. Giannini G, Cabri W, Fattorusso C, Rodriquez M. Histone deacetylase inhibitors in the treatment of cancer: overview and perspectives. Future Med Chem. 2012;4:1439-60
55. Dashwood RH, Myzak MC, Ho E. Dietary HDAC inhibitors: time to rethink weak ligands in cancer chemoprevention? Carcinogenesis. 2006;27:344-9
56. He G, Kuang J, Koomen J, Kobayashi R, Khokhar AR, Siddik ZH. Recruitment of trimeric proliferating cell nuclear antigen by G1-phase cyclin-dependent kinases following DNA damage with platinum-based antitumour agents. Br J Cancer. 2013;109:2378-88
57. Kwon SH, Ahn SH, Kim YK, Bae GU, Yoon JW, Hong S. et al. Apicidin, a histone deacetylase inhibitor, induces apoptosis and Fas/Fas ligand expression in human acute promyelocytic leukemia cells. J Biol Chem. 2002;277:2073-80
58. Vrana JA, Decker RH, Johnson CR, Wang Z, Jarvis WD, Richon VM. et al. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene. 1999;18:7016-25
59. Wu X, Yang N, Zhou WH, Xu J, Chen JJ, Zheng FM. et al. Up-regulation of P21 inhibits TRAIL-mediated extrinsic apoptosis, contributing resistance to SAHA in acute myeloid leukemia cells. Cell Physiol Biochem. 2014;34:506-18
60. Kelly WK, O'Connor OA, Krug LM, Chiao JH, Heaney M, Curley T. et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23:3923-31
61. Valdez BC, Li Y, Murray D, Brammer JE, Liu Y, Hosing C. et al. Differential effects of histone deacetylase inhibitors on cellular drug transporters and their implications for using epigenetic modifiers in combination chemotherapy. Oncotarget. 2016 ahead of print
Author contact
![]() Corresponding author: Aneta Grabarska, Ph.D., Department of Biochemistry and Molecular Biology, Medical University of Lublin, Chodzki 1, 20-093 Lublin, Poland,Tel./Fax: +48 817423793, e-mail: anetagrabarskapl.
Corresponding author: Aneta Grabarska, Ph.D., Department of Biochemistry and Molecular Biology, Medical University of Lublin, Chodzki 1, 20-093 Lublin, Poland,Tel./Fax: +48 817423793, e-mail: anetagrabarskapl.
Citation styles
APA
Grabarska, A., Łuszczki, J.J., Nowosadzka, E., Gumbarewicz, E., Jeleniewicz, W., Dmoszyńska-Graniczka, M., Kowalczuk, K., Kupisz, K., Polberg, K., Stepulak, A. (2017). Histone Deacetylase Inhibitor SAHA as Potential Targeted Therapy Agent for Larynx Cancer Cells. Journal of Cancer, 8(1), 19-28. https://doi.org/10.7150/jca.16655.
ACS
Grabarska, A.; Łuszczki, J.J.; Nowosadzka, E.; Gumbarewicz, E.; Jeleniewicz, W.; Dmoszyńska-Graniczka, M.; Kowalczuk, K.; Kupisz, K.; Polberg, K.; Stepulak, A. Histone Deacetylase Inhibitor SAHA as Potential Targeted Therapy Agent for Larynx Cancer Cells. J. Cancer 2017, 8 (1), 19-28. DOI: 10.7150/jca.16655.
NLM
Grabarska A, Łuszczki JJ, Nowosadzka E, Gumbarewicz E, Jeleniewicz W, Dmoszyńska-Graniczka M, Kowalczuk K, Kupisz K, Polberg K, Stepulak A. Histone Deacetylase Inhibitor SAHA as Potential Targeted Therapy Agent for Larynx Cancer Cells. J Cancer 2017; 8(1):19-28. doi:10.7150/jca.16655. https://www.jcancer.org/v08p0019.htm
CSE
Grabarska A, Łuszczki JJ, Nowosadzka E, Gumbarewicz E, Jeleniewicz W, Dmoszyńska-Graniczka M, Kowalczuk K, Kupisz K, Polberg K, Stepulak A. 2017. Histone Deacetylase Inhibitor SAHA as Potential Targeted Therapy Agent for Larynx Cancer Cells. J Cancer. 8(1):19-28.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.