Journal of Cancer 3.2
Impact Factor
ISSN: 1837-9664
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(3):332-344. doi:10.7150/jca.17547 This issue Cite
Review
Surveilling the Potential for Precision Medicine-driven PD-1/PD-L1-targeted Therapy in HNSCC
JE Mann1,2*, R Hoesli 1*, NL Michmerhuizen 1,3, SN Devenport 1,6, ML Ludwig 1,4, TR Vandenberg 1, C Matovina 1, N Jawad 1, M Mierzwa 5,6, AG Shuman 1,6#, ME Spector 1,6#, JC Brenner 1,6# ![]()
1. Department of Otolaryngology - Head and Neck Surgery, University of Michigan Medical School, Ann Arbor, MI.
2. Department of Pathology, University of Michigan Medical School, Ann Arbor, MI.
3. Department of Pharmacology, University of Michigan Medical School, Ann Arbor, MI.
4. Program in Cellular and Molecular Biology, University of Michigan Medical School, Ann Arbor, MI.
5. Department of Radiation Oncology, University of Michigan Medical School, Ann Arbor, MI.
6. Comprehensive Cancer Center, University of Michigan Medical School, Ann Arbor, MI.
*Authors contributed equally.
#Authors are co-senior on this paper.
Received 2016-9-12; Accepted 2016-11-14; Published 2017-2-9
Citation:
Mann JE, Hoesli R, Michmerhuizen NL, Devenport SN, Ludwig ML, Vandenberg TR, Matovina C, Jawad N, Mierzwa M, Shuman AG, Spector ME, Brenner JC. Surveilling the Potential for Precision Medicine-driven PD-1/PD-L1-targeted Therapy in HNSCC. J Cancer 2017; 8(3):332-344. doi:10.7150/jca.17547. https://www.jcancer.org/v08p0332.htm
Other stylesAbstract
Immunotherapy is becoming an accepted treatment modality for many patients with cancer and is now approved for use in platinum-refractory recurrent or metastatic head and neck squamous cell carcinoma (HNSCC). Despite these successes, a minority of patients with HNSCC receiving immunotherapy respond to treatment, and few undergo a complete response. Thus, there is a critical need to identify mechanisms regulating immune checkpoints in HNSCC such that one can predict who will benefit, and so novel combination strategies can be developed for non-responders. Here, we review the immunotherapy and molecular genetics literature to describe what is known about immune checkpoints in common genetic subsets of HNSCC. We highlight several highly recurrent genetic lesions that may serve as biomarkers or targets for combination immunotherapy in HNSCC.
Keywords: Genetics, Immunotherapy, EGFR, personalized medicine, HNSCC.
Introduction
Over the last decade, research in head and neck squamous cell carcinoma (HNSCC) has shown that suppression of the host immune system plays a key role in the development and progression of HNSCC. Many critical components of both the innate and adaptive immune systems are dysfunctional in patients with HNSCC, including the activity of natural killer cells, the function of antigen presenting machinery, and the maturation of dendritic cells [1-4]. Additionally, in the composition of functioning cells such as T lymphocytes, there is a shift towards immunosuppression, with higher numbers of the immunosuppressive regulatory T cells and immunosuppressive cytokines, while the overall number of lymphocytes is decreased [5-9]. Many of these defects feedback amongst themselves, resulting in the release of cytokines and recruitment of immunosuppressive cells, further promoting the immunosuppressive environment. Importantly, this immunosuppression appears to play pivotal roles in both HNSCCs driven by high risk human papilloma virus (HPV) and HPV negative disease.
In addition to these defects, HNSCCs, like other immunosuppressive cancers, have co-opted beneficial physiologic signaling pathways to aid in immune evasion. In the intact immune system, there is a necessary equilibrium between activation and suppression of the immune system. This balance prevents excessive activation of the immune system resulting in autoimmune diseases, as well as pathologic suppression resulting in opportunistic infections. Various co-stimulatory and co-inhibitory signaling pathways are involved in maintaining this equilibrium, including the CTLA-4 and programmed death-1 (PD-1):programmed death ligand-1 (PD-L1) pathways, which serve as checkpoints to mitigate excessive inflammation. Both of these pathways are thought to have been exploited by HNSCC in order to enhance the immunosuppressive environment, preventing immune surveillance and tumor destruction [10, 11]. Therapies targeting the co-inhibitory receptor CTLA-4 were among the earliest immunotherapies for cancer, and following their success in the clinic, additional targets, including the PD-1:PD-L1 pathway, were quickly advanced [10]. This review will focus on the various genetic alterations and molecular pathways that may contribute to dysregulation of the PD-1:PD-L1 pathway.
Immune Checkpoints and PD1/PDL1 Signaling
The PD-1/PD-L1 pathway is an important co-inhibitory pathway involved in the regulation of the human immune response. This pathway serves as an immune checkpoint, providing protection against excessive tissue damage induced by inflammation [10], and is especially important in regulating antigen-specific effector T-cell activity in peripheral tissues. The PD-1 receptor is a transmembrane protein expressed by T cells, B cells, and many types of tumor-infiltrating lymphocytes (TILs). PD-1 can bind either of two ligands: PD-L1 or PD-L2, both of which are cell surface proteins of the B7 family [12, 13]. Upon ligand binding, generation and activation of effector T cells, particularly CD8+ T cells, is dramatically suppressed [14].
Expression of PD-L1 can be stimulated by interferon-γ (IFN-γ), a cytokine produced primarily by effector lymphocytes [15]. In general, interferons function as lines of communication between the innate and adaptive immune responses by activating immature dendritic cells and CD8+ T cells. IFN-γ is largely responsible for inducing inflammation, a critical component of the immune response. Cancer cells are thought to induce an immune response, and inflammation is common within the tumor microenvironment. However, it is hypothesized that cancer cells can develop an “adaptive immune resistance” to increased levels of inflammation by upregulating PD-L1 in response to IFN-γ, thereby protecting themselves against immune attack by promoting T cell anergy and apoptosis [16, 17]. An interesting connection has also emerged between HPV infection, now recognized as a common initiator of oropharyngeal cancer, and immunosuppression. Major sites of HPV infection, such as the tonsillar crypts, may be prone to harboring high levels of foreign antigens [18]. It is plausible that activation of immunosuppressive mechanisms to stifle excessive inflammation also creates a permissive environment for both persistent HPV and associated tumors to flourish [18]. It has also been postulated that immunosuppressive pathways initiated by HPV itself allow for malignant transformation at the site of infection [19].
Although the PD-1/PD-L1 pathway was discovered more than two decades ago, the therapeutic potential associated with targeting this pathway in cancer was not immediately recognized. As the immunosuppressive function of the PD-1/PD-L1 interaction was revealed and PD-L1 expression was observed in tumor cells, researchers hypothesized that blockade of this pathway could help facilitate eradication of tumors and impede tumor metastasis [20]. This hypothesis was first tested in melanoma, in which treatment with monoclonal antibodies (mAbs) directed against PD-1 showed promising results. In a meta-analysis of 5 multi-center randomized control trials including 2,828 patients, patients treated with an anti- PD-1 mAb experienced superior progression free survival (PFS) ranging from 3.7 to 6.9 months as compared with 2.2 to 4.2 months in the control group. Six-month PFS was significantly improved for patients treated with anti-PD-1 therapy, with a hazard ratio (HR) of 0.55 [21]. There was also a statistically significant improvement in overall response rate (ORR) with an odds ratio (OR) of 3.89 [21]. Two anti-PD-1 antibodies, pembrolizumab and nivolumab, have since been approved by the FDA for the treatment of advanced melanoma due to convincing data demonstrating increased ORR, as well as improved PFS and OS [11].
Currently, five mAb therapies targeting the PD-1/PD-L1 pathway are under investigation for use in HNSCC. Pembrolizumab and Nivolumab target PD-1, while MEDI4736, Atezolizumab, and Avelumab target PD-L1. Pembrolizumab has shown considerable promise for use in patients with recurrent and/or metastatic HNSCC. An 18.2% ORR has been reported with a severe adverse reaction rate of 7.6%, leading to the August 2016 approval of Pembrolizumab for platinum-refractory recurrent or metastatic HNSCC patients [22]. Interestingly, ORR was independent of HPV status and PD-L1 status [23]. A phase 3 trial is also currently underway for Nivolumab versus either Cetuximab, Docetaxel, or Methotrexate for recurrent/metastatic HNSCC (Clinical Trial: NCT02105636). Preliminary results show a doubling in the one year overall survival rate to 36% in Nivolumab treated patients. Median overall survival was 7.5 months in the Nivolumab group as compared to 5.1 months in the standard treatment group. Interestingly, both HPV positive and HPV negative patients benefit with the median survival for HPV positive patients increasing from 4.4 to 9.1 months, and that of HPV negative patients increasing from 5.8 to 7.5 months [24]. Due to its early success, additional trials evaluating the efficacy of Nivolumab in combination with chemotherapy and/or radiation for definitive therapy in locoregionally advanced HNSCC are currently underway (NCT02764593).
Of the mAbs against PD-L1, MEDI4736 or Durvalumab, is currently being evaluated in a phase 3 trial of Durvalumab alone versus Durvalumab plus an anti-CTLA-4 inhibitor (tremelimumab) versus standard treatment (NCT02369874) [25]. Atezolizumab and Avelumab are currently in several Phase 1b/2 trials evaluating the safety and efficacy of these inhibitors in advanced solid tumor malignancies, as listed in Table 1. Several ongoing trials are investigating their use alone and in combination with other targeted therapies.
Table 1
List of current trials evaluating the use of targeted therapies towards the PD-1/PD-L1 pathway in combination with other immunotherapy treatments in HNSCC.
| Trial | PD-1/PD-L1 Inhibitors + Immunotherapies | ||||
|---|---|---|---|---|---|
| Phase | Therapy | Description | Target | Condition | |
| NCT02655822 | Phase I/Ib | CPI-444 + Atezolizumab | Evaluate the safety and preliminary efficacy of CPI-444 (small molecule immune checkpoint inhibitor of adenosine A2A receptor) in combination with Atezolizumab | PD-L1 Adenosine A2A receptor | Advanced malignancies (including HNSCC) |
| NCT02543645 | Phase I/II | Atezolizumab + Varlilumab | Evaluate safety and preliminary efficacy of Atezolizumab + Varlilumab (anti-CD27 mAb) | PD-L1 CD27 | Advanced malignancies (including HNSCC) |
| NCT02554812 | Phase 1b/2 | Avelumab + PF-05082566 or PF-04518600 | Evaluate safety, pharmacokinetics, and pharmacodynamics of Avelumab + PF-05082566 (stimulatory mAb towards CD137) versus Avelumab + PF-04518600 (stimulatory mAb towards CD134) | PD-L1 CD-137 CD-134 | Advanced solid malignancies (including HNSCC) |
| NCT02301130 | Phase I | MEDI4736 + Mogamulizumab versus Mogamulizumab + Tremelimumab | Evaluate the safety and preliminary efficacy of MEDI4736 + Mogamulizumab (C-C chemokine receptor 4 (CCR4) inhibitor) versus Mogamulizumab + Tremelimumab (CTLA-4 inhibitor) | PD-L1 C-C chemokine receptor 4 (CCR4) CTLA-4 receptor | Advanced solid tumors (including HNSCC) |
| NCT02658214 | Phase I | MEDI4736 + Tremelimumab + Paclitaxel + Carboplatin | Evaluate the safety, tolerability, and preliminary efficacy of MEDI4736 + Tremelimumab (CTLA-4 inhibitor) + Paclitaxel + Carboplatin | PD-L1 CTLA-4 receptor | Chemotherapy naïve locally advanced unresectable or metastatic HNSCC and other solid tumors |
| NCT02262741 | Phase I | MEDI4736 + Tremelimumab | Evaluate the safety, tolerability, and efficacy of MEDI4736 + Tremelimumab (CTLA-4 inhibitor) | PD-L1 CTLA-4 receptor | Recurrent or metastatic HNSCC |
| NCT02291055 | Phase I/II | MEDI4736 +/- ADXS11-001 | Evaluate safety and tolerability of MEDI4736 +/- ADXS11-001 (Listeria monocytogenes cancer vaccine) | PD-L1 Cancer vaccine against HPV-16 E7 | Recurrent or metastatic HNSCC |
| NCT02643303 | Phase I/II | MEDI4736 + Tremelimumab + PolyICLC | Evaluate the safety and efficacy of MEDI4736 + Tremelimumab (CTLA-4 inhibitor) + PolyICLC (Toll-like receptor agonist) | PD-L1 CTLA-4 receptor Toll-like receptor | Advanced biopsy accessible tumors (including HNSCC) |
| NCT02319044 | Phase II | MEDI4736 versus Tremelimumab versus MEDI4736 + Tremelimumab | Evaluate safety and preliminary efficacy of MEDI4736 versus Tremelimumab (CTLA-4 inhibitor) versus MEDI4736 + Tremelimumab | PD-L1 CTLA-4 receptor | Recurrent or metastatic HNSCC |
| NCT02551159 | Phase III | MEDI4736 + Tremelimumab | Evaluate safety and efficacy of MEDI4736 with and without Tremelimumab (CTLA-4 inhibitor) versus standard of care | PD-L1 CTLA-4 receptor | Recurrent or metastatic HNSCC without prior systemic chemotherapy |
| NCT02369874 | Phase III | MEDI4736 + Tremelimumab | Evaluate safety and efficacy of MEDI4736 + Tremelimumab (CTLA-4 inhibitor) versus MEDI4736 alone versus standard of care | PD-L1 CTLA-4 receptor | Previously treated recurrent or metastatic HNSCC |
| NCT02124850 | Phase Ib | Nivolumab + Cetuximab + Motolimod | Evaluate change in immune biomarkers and tumor markers after treatment with Cetuximab + Motolimod (small molecule agonist of toll-like receptor 8) versus Nivolumab + Cetuximab + Motolimod | PD-1 EGF receptor Toll-like receptor 8 | Stage II, III, or IVa HNSCC |
| NCT02488759 | Phase I/II | Nivolumab +/- Ipilimumab | Evaluate safety and preliminary efficacy of Nivolumab +/- Ipilimumab | PD-1 CTLA-4 receptor | Virus associated malignancy (including HPV+ HNSCC) |
| NCT02335918 | Phase I/II | Nivolumab + Varlilumab | Evaluate safety, tolerability, and preliminary efficacy of Nivolumab + Varlilumab (anti-CD27 mAb) | PD-1 CD27 | Refractory solid tumors (including HNSCC) |
| NCT02741570 | Phase III | Nivolumab + Ipilimumab | Evaluate safety and efficacy of Nivolumab + Ipilimumab versus Cetuximab + Cisplatin/Carboplatin + Fluorouracil | PD-1 CTLA-4 receptor EGF receptor | Recurrent or metastatic HNSCC, without prior treatment with systemic cancer therapy |
| NCT02636036 | Phase I | Pembrolizumab + Enadenotucirev | Evaluate safety of Pembrolizumab + Enadenotucirev (oncolytic virus) | PD-1 Targeted viral therapy | Metastatic or advanced solid tumors (including HNSCC) |
| NCT02475213 | Phase I | Pembrolizumab + Enoblituzumab | Evaluate safety of Pembrolizumab + Enoblituzumab (mAB towards B7-H3) | PD-1 B7-H3 | Refractory malignancies including recurrent or metastatic HNSCC that expresses B7-H3 |
| NCT02626000 | Phase I | Pembrolizumab + Talimogene laherparepvec | Evaluate the safety and preliminary efficacy of pembrolizumab + Talimogene laherparepvec (oncolytic virus) | PD-1 Targeted viral therapy | Recurrent or metastatic HNSCC |
Given the enthusiasm for drugs targeting the PD1/PDL1 pathways in the management of recurrent HNSCC, it is reasonable to expect that new protocols will be developed expanding the application of immunotherapy in this context. An enhanced understanding of PD-1 and PD-L1 expression and modulation, in addition to other factors predicting response to PD-1/PD-L1 blockade, will be essential to the rational design of future trials. Here, we review potential modulators of PD-L1 expression, as well as important considerations associated with targeting PD-L1 in HNSCC patients.
PD-1 and PD-L1 expression in HNSCC
While several studies have evaluated expression of PD-L1 in HNSCC by immunohistochemistry, results are highly variable. Thus, the clinical significance of PD-L1 expression in tumor cells remains unclear. PD-L1 staining may be membranous and/or diffuse, and often appears at the interface of tumor cells and T cells. PD-L1 expression is reported in 51-87% of HNSCC tumors across several reports and criteria for PD-L1 “positivity” are highly variable [18, 26-31]. Small sample size, inconsistent representation of different disease sites, prior therapy, or the antibody used to detect PD-L1 may contribute to discrepancies among these studies. Intratumoral heterogeneity and temporal changes in PD-L1 expression may also lead to false negatives [32]. HPV associated cancers are more likely to express PD-L1, but PD-L1 expression has not been directly correlated with survival despite the better prognosis observed among these cancers. However, tumor infiltration with high numbers of PD-1 expressing CD8+ T cells is associated with longer overall survival; the mechanism and implications thereof remain unclear [29, 33].
Potential for intrinsic modulation of PD-L1 by oncogenic pathways
JAK2/STAT1 mediated induction of PD-L1 expression on the surface of tumor cells has been demonstrated in response to IFN-γ, and, recently, epidermal growth factor (EGF) [34]. The finding that EGF can promote PD-L1 expression was especially interesting given that the EGF receptor (EGFR) is highly overexpressed in HNSCC. This observation highlights the possibility that other signals, including common oncogenic drivers, may contribute to immunosuppression by promoting PD-L1 expression. Elucidating the molecular mechanisms governing PD-L1 expression in the context of HNSCC may provide a compelling rationale for combining immunotherapy with other agents targeting upstream regulators of the PD-L1/PD-1 pathway. While mechanisms regulating PD-L1 have been examined in other cancers, few studies have addressed intrinsic modulation of PD-L1 in HNSCC, especially in the context of emerging precision medicine paradigms that match targeted therapies with specific genetic lesions [35-39]. Interestingly, several potentially targetable pathways including EGFR, phosphatidylinositol 3-kinase (PI3K)/Akt, MAPK, p53, STAT and HIF-1α are commonly deregulated in HNSCC and could potentially influence the expression of PD-L1 (Figure 1).
EGFR
EGFR is overexpressed in >90% of HNSCCs by a variety of genetic mechanisms including amplification (~10% of cases) [40-43]. It is therefore not surprising that targeting EGFR has become an important strategy in the management of HNSCC. Several small molecule inhibitors of EGFR signaling are currently being investigated, and clinical success has been achieved with biologics such as Cetuximab, a chimeric IgG1 mAb against EGFR [44]. As both EGFR- and PD-1/PD-L1- targeted therapies have advanced, evidence has emerged for a potential confluence of these two pathways. In NSCLC, where immune checkpoint inhibitors have been more extensively studied and exploited, an interesting relationship between EGFR and PD-L1 has been noted. Specifically, tumors with activating genetic aberrations to EGFR were more likely to have PD-L1 overexpression than those without [45, 46], and treatment of these tumors with the small molecule EGFR inhibitor gefitinib caused a substantial reduction in PD-L1 expression suggesting a direct mechanistic link between the two molecules [46]. Surprisingly, refractory NSCLC tumors arising after gefitinib treatments also have elevated PD-L1 expression [47], suggesting that immune escape is an important mechanism to overcoming EGFR inhibitor response. More recently, Concha-Benavente et. al. noted a similar relationship in HNSCC models showing that Cetuximab-mediated EGFR inhibition caused a reduction in adaptive PD-L1 expression through modulation of the JAK2/STAT1 effectors [34].
The postulate that EGFR activation can promote immune escape through regulation of PD-L1 expression is especially interesting given the prevalence of Cetuximab-based therapies in HNSCC. The logical hypothesis addresses whether these two therapies can be combined, particularly given the poor observed response to Cetuximab as a single agent. It is unclear if combination Cetuximab + PD1/PDL1 checkpoint inhibitors would provide an incremental effect, as Cetuximab inhibits PD-L1 expression thereby removing the immunotherapy target, perhaps suggesting the need for sequential use of the drugs. However, by activating T-cells to clear any tumor cells that are innately resistant to Cetuximab (or the EGFR-based modulation of PD-L1), this combination may effectively clear heterogeneous tumor cell populations, although this remains to be proven in vivo. Clinical trials assessing these combinations in advanced HNSCC patients are currently being evaluated and sequential trials assessing PD-L1 inhibition following Cetuximab failure are already ongoing (NCT02255097). An additional facet of mAb therapies is the potential for antibody-dependent cellular cytotoxicity (ADCC), a mechanism for the clearance of IgG1-coated target cells. ADCC is triggered by engagement of Fc receptors on immune effector cells with the Fc region of IgG1. Cetuximab induces ADCC in an EGFR-dependent manner in cell culture and in murine models [48-51], and although the contribution of ADCC to the anti-tumor effects of Cetuximab in humans is not fully understood, evidence of increased ADCC has been reported. In a study of 22 HNSCC and colorectal cancer patients, Bertino et al observed more pronounced increases in ADCC in patients who experienced clinical benefit from Cetuximab than in those who did not [52]. ADCC may play a key role in the clinical efficacy of EGFR targeted mAbs, and could have important implications for combination immunotherapeutic strategies. For example, it is possible that the combined effects of relief from T-cell anergy, stimulation of natural killer cells, and inhibition of EGFR signaling in addicted cells may amplify the anti-tumor function of either strategy alone. EGFR-based therapies may therefore have important mechanistic consequences on PD1/PDL1-based therapies in HNSCC, and these molecules may serve as important companion diagnostics for adjunctive immunotherapy.
Figure 1
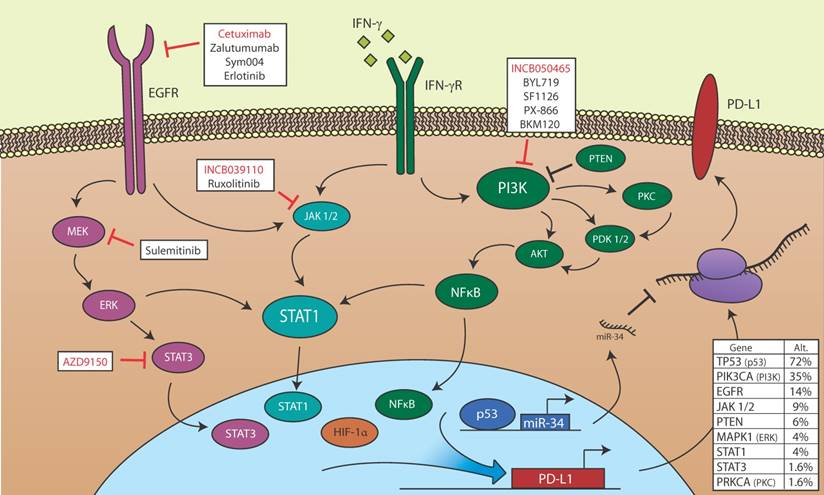
Potential for PD-L1 modulation in HNSCC. Members of the EGFR, STAT, PI3K, p53, and HIF-1α signaling pathways, all of which may be de-regulated in HNSCC, are shown. EGFR is aberrantly activated in HNSCC and may promote PD-L1 transcription via JAK/STAT or MEK/ERK signaling. IFN-γ induces PD-L1 transcription in HSCC via JAK/STAT signaling, and may also activate the PI3K signaling pathway, which regulates PD-L1 transcription in other cancers. HIF-1α binds directly to the PD-L1 promoter and correlates with high PD-L1 expression. p53 loss of function is common in HNSCC, and wild type p53 blocks PD-L1 translation via transcription of miR-34. Also shown are targeted small molecule inhibitors and biologics currently being evaluated in active clinical trials open to HNSCC patients. Red text indicates inhibitors for which efficacy in combination with PD-1/PD-L1 blockade is being evaluated. Table (lower right) is based upon data generated by the TCGA Research Network (http://cancergenome.nih.gov). The middle column denotes the percentage of 504 cases with alternations (mutations or copy number variations) in the indicated gene. Within this dataset, 35 of 106 cases in which p16 was assessed were designated HPV positive. The rightmost column denotes the percentage of HPV positive cases with alterations in the indicated gene [112].
PI3K/AKT
The PI3K pathway, based on data from The Cancer Genome Atlas and other analyses, has been identified as the most frequently mutated of any oncogenic and targetable pathway in HNSCC [53-55]. Activating mutations and/or amplification of PIK3CA, which encodes PI3K's catalytic subunit and alpha isoform, was the most frequent alteration observed in the PI3K pathway. Aberration of this gene was reported in 37% of the TCGA HNSCC cohort [53]. As a whole, alterations in the PI3K pathway contribute to tumor progression and increased cell growth and viability; they are also more commonly observed in advanced stage disease [56, 57].
Various interferons have been shown to activate the PI3K pathway as well as to induce the expression of PD-L1. In response to interferon, PI3K signaling is required for phosphorylation of STAT1 at the serine 727 residue and for the associated increase in gene transcription [58, 59]. PKC (δ and ε isoforms) and/or NF-κB may serve as downstream mediators of this response [15, 60-63]. Chen et al. showed that PDK2 signaling, which can be initiated by the PI3K-PKC pathway, is responsible for interferon-induced PD-L1 expression in oral squamous cell carcinoma [15], and Kondo et al. demonstrated the dependence of interferon-induced PD-L1 expression on NF-κB in myodysplastic syndrome blast cells [64]. Additionally, in dermal fibroblasts, interferon treatment led to translocation of NF-κB to the nucleus and transient phosphorylation of ERK and AKT. This effect was abrogated upon treatment with PI3K and MEK but not PKC inhibitors [60]. BRAF-inhibitor resistant melanoma cells also display inducible PD-L1 expression that is dependent on NF-κB. In contrast to observations in dermal fibroblasts, however, the PI3K and MAPK pathways were not significant mediators of this response [65]. Ferris and colleagues reported that PI3K inhibitors wortmannin and BYL719 blocked AKT phosphorylation but failed to reverse the extrinsic interferon- or intrinsic EGFR-mediated increases in PD-L1 expression in HNSCC cell lines [34]. These data indicate that the role of PI3K, NF-κB, and PKC may be cell-type specific and therefore motivate further studies of the signaling mechanisms in HNSCCs.
The PI3K pathway is also commonly activated in HNSCC via loss of PTEN, which functions as a “brake” on PI3K signaling. The PTEN tumor suppressor has been associated with PD-L1 expression in other cancer types. Transgenic mouse models of lung SCC with loss of both PTEN and Lkb1, for example, developed tumors with elevated levels of PD-L1 [66]. Parsa et al. showed that loss of PTEN in glioblastoma patients correlated with increased PD-L1 expression and that PD-L1 translation was S6K1-mediated [67]. Furthermore, a negative correlation between PTEN and PD-L1 expression has been identified in pancreatic and colorectal cancer samples [68, 69]. miRNAs may also be an important component of this response as the upregulation of miRNAs, including miR-21, -20b, and 130b, in colorectal and esophageal cancers has been shown to suppress PTEN gene transcription [69, 70].
MAPK
In HNSCC, genetic alterations in MAPK family members are relatively rare. Only 5% of patients in the TCGA cohort displayed mutations or copy number changes in HRAS, with other alterations in this pathway (such as those affecting KRAS and MAPK1) occurring even less frequently [53]. In spite of this, signaling of the Ras-MEK-ERK pathway is often aberrant in HNSCC due to overexpression of EGFR or activation of other RTKs [71].
The signaling functions of the MAPK pathway, like those of the PI3K pathway, are important in interferon-induced changes in PD-L1 expression. MAPK acts in coordination with the JAK/STAT pathway to regulate gene transcription, and inhibitors of MAPK pathway members (including MEK, ERK, and JNK) block these effects [72]. Consistent with this, Liu et al. showed that PMA, a known MEK/ERK pathway activator, could increase PD-L1 expression in multiple myeloma; similarly, blocking MEK/ERK via pharmacological methods or siRNA knockdown resulted in decreased interferon-induced PD-L1 expression [73]. Melanoma cells resistant to BRAF inhibition also displayed MAPK activation and increased PD-L1 expression via c-Jun and STAT3, and this effect was reversed by MEK inhibitors [74]. Similar evidence for the role of the MAPK pathway in inducing PD-L1 expression was identified in bladder cancer, where inhibitors of ERK and JNK blocked the induction of PD-L1 expression by LPS treatment [75]. MAPK signaling also modulates PD-L1 expression in anaplastic large cell lymphoma (via ALK) and in Hodgkin's lymphoma (via p38 MAPK and MEK1/2) [76].
Recent studies of breast cancer cell lines and murine models, however, have indicated that MEK inhibitor trametinib might induce PD-L1 expression and therefore be useful in priming patients for immunotherapy treatment [77]. Further studies in BRAF-mutant and WT melanoma as well as KRAS-mutant NSCLC cells showed varying PD-L1 transcript levels following trametinib treatment [78]. Combination MAPK and PD-L1 inhibitor treatments have been proposed and tested in vitro and in vivo, and combination therapies are currently being evaluated in clinical trials (such as NCT02027961 and NCT01988896 for BRAF-mutant melanoma and NSCLC patients, respectively) [79].
While these dual therapies might be effective in HNSCC, further work regarding both the specific mechanism of MAPK pathway activation in PD-L1 expression and the modulation of PD-L1 levels by MAPK inhibitors are necessary. Initial studies by Ferris and colleagues suggest that MEK inhibitors do not significantly alter PD-L1 expression in HNSCC cell lines, but that JAK2 and STAT1 might be critical mediators of immunogenicity [34]. Additional research will aid in selecting patients who are the most likely to respond to combination MAPK-immunotherapy treatments, and one trial is currently underway in advanced solid malignancies including HNSCC (NCT02586987).
p53
The TP53 gene, encoding the p53 tumor suppressor, is the most commonly mutated gene in HNSCC [80]. Cortez et al recently reported that p53 can regulate PD-L1 production via miR-34. p53 deficient or mutant tumors express significantly higher PD-L1 levels than wild type p53 tumors [81]. In cell lines, p53 induced miR-34 repressed PD-L1 expression. Interestingly, injection of miR-34 mimics, such as the drug MRX34, reduces PDL1 expression and increases immune response to tumorous growth. When MRX34 was paired with radiation therapy (XRT), a common anti-cancer treatment that induces the adaptive immune response to promote tumor regression, exponential increases in CD8+ tumor-infiltrating cell expression were found [81]. Understanding the relationship between the p53 pathway and its effects on PDL1 expression may allow physicians to better tailor treatment of tumors with p53 mutations.
STAT3
The STAT signaling pathway is known to play an important role in many cellular processes, including division, apoptosis, and motility. Involvement of the transcriptional activator STAT3 has been reported in the development and growth of tumors [82], and recent studies aim to elucidate the role of STAT3 in PD-L1 expression on tumor cells [83]. STAT3 inhibition reduces PD-L1 expression in NSCLC [84]. Similarly, in multiple types of lymphoma, PD-L1 expression is enhanced by STAT3 and is decreased upon STAT3 inhibition [85, 86]. As activated STAT3 was elevated in HNSCC tumors compared to normal epithelium [87], elucidating a role for STAT3 in regulating PD-L1 in HNSCC may shed light on potential therapeutic targets or factors influencing the ability of these tumors to respond to PD-1 blockade. However, a recent study found no correlation between STAT3 and PD-L1 expression in HPV positive HNSCC, and only a weak correlation in HPV negative HNSCC [34].
HIF-1α
Hypoxia inducible factors (HIFs) are transcriptional regulators integral to the response to hypoxia in solid tumors, and are believed to be critical to metastasis [88]. Under hypoxic conditions, tumors utilize HIF-1α to upregulate glycolysis and to increase blood flow to the tumor by promoting angiogenesis. Recently, HIF-1α was suggested to play a role in PDL-1 expression [89]. Specifically, in myeloid derived stem cells (MDSCs), HIF-1α binds directly to the HRE-4 site in the proximal promoter of PD-L1 [89]. Another PD-L1 proximal promoter site, HRE-2, has also been shown to bind with HIF-1 in mammary cells [90]. Hypoxia-induced PD-L1 expression has been demonstrated in MDSCs, macrophages, dendritic cells, and tumor cells including breast and prostate cancer cell lines [89, 90], and this effect was HIF-1α dependent. While no direct link has been established between HIF-1 and PD-L1 in HNSCC, given the reported overexpression of HIF-1α in HNSCC tissue vs adjacent normal tissue [91] and the poor prognosis associated with low intratumoral oxygen levels in HNSCC [92], a more detailed understanding of the potential for interplay between immune evasion and hypoxia could reveal novel mechanisms of HNSCC pathogenesis.
Additional Targets
Finally, there are additional genomic mutations in head and neck squamous cell carcinomas noted in the data from The Cancer Genome Atlas HNSCC cohort that make attractive targets for investigation in the development of immunotherapies. In addition to those described above, defects of note include mutations affecting other components of the innate and adaptive immune response, including antigen processing machinery and HLA, as well as tumor necrosis factor (TNF) receptor-associated factor 3 (TRAF3). HLA molecules play a key role in tumor identification and antigen presentation in the functioning immune system. HLA associates with degraded tumor peptides and β2-microglobulins, and once properly folded, the HLA complex consisting of the HLA protein chains, β2-microglobulins, and tumor peptides is transported to the cell surface, where it is recognized by T cells [3]. However, in one study of head and neck cancers, approximately 40% of primary tumors had at least selective loss of HLA class I antigens, and 15% had complete loss [93]. Although no current therapies target this defect, it remains an attractive target for immunotherapies, as it could play an important role in activating T cells against tumor antigens.
TRAF3 plays an important role in regulating the crossroads between the anti-viral, anti-inflammatory, and cancer pathways by modulating toll-like receptors, TNF receptors, and producing interferons and anti-inflammatory cytokines [94]. They have been implicated in multiple myeloma in humans, and in one study of transgenic mice, overexpression was associated with autoimmunity and a predisposition to developing squamous cell carcinoma [95]. In the data from The Cancer Genome Atlas, TRAF3 mutation was a distinguishing mutation of HPV positive tumors [80]. No current therapies have been developed, but like HLA mutations, it remains as attractive area for investigation.
HPV and PD-L1
Human papillomavirus (HPV), a known risk factor for HNSCC, has been linked to PD-1 pathway activation in several settings. In a 2013 study, Yang et. al described a correlation between PD-1 on T cells, persistent high risk (HR)-HPV infection, and the development of cervical intraepithelial neoplasia (CIN) [96]. A positive correlation between PD-L1 expression and CIN grade was also observed in HR-HPV positive patients, suggesting that the PD-1:PD-L1 pathway may play a role in permitting HR-HPV-related CIN progression [96].
With rates of HPV associated oropharyngeal cancers rapidly rising, it has become clear that the distinct pathogenesis of these cancers may warrant the development of alternate treatment protocols [97]. HPV associated HNSCCs generally have favorable clinical outcomes independently predicted by HPV status [98, 99]. The oropharyngeal tumors that result from HPV infection grow in the tonsillar crypts and near the base of tongue, which are also the most common sites of HPV infection. PD-L1 expression is also found in deep tonsillar crypts in both normal and HNSCC patients [18]. It is therefore plausible that a dampened effector T cell response within these crypts may yield an environment especially permissive to both initial HPV infection and SCC tumorigenesis.
HPV positive HNSCCs are able to grow and evade anti-tumor immunity despite high levels of inflammation. In order to evaluate the relevance of PD-1:PD-L1 pathway in the development of HPV associated HNSCC, Lyford-Pike et. al analyzed PD-1 expression on TILs and peripheral blood mononuclear cells (PBMCs) isolated from patients with HPV positive HNSCC versus those from patients with a nonmalignant tonsillar pathology (such as hypertrophy or tonsillitis), and identified a population of PD-1 expressing CD8+ TILs in HPV positive HNSCC tumors. Such a population was not observed in T-cells infiltrating nonmalignant inflamed tonsils. Furthermore, the PD-1 expressing CD8+ TILs exhibited diminished ability to produce IFN-γ in response to stimulation with PMA/ionomycin compared to TILs not expressing PD-1, indicating the functional suppression of affected T-lymphocytes. Lyford-Pike also showed PD-L1 expression localized to the interface of the tumor and CD8+ TILs [18]. This study implies that the PD-1:PD-L1 pathway is likely important in both persistence of the initial HPV infection and suppression of anti-tumor immunity during tumorigenesis. Because high levels of membranous PD-L1 expression were observed within tumors, HPV positive HNSCC patients are logical candidates for PD-1:PD-L1-targeted therapy. Furthermore, a recent study in mice demonstrated improved anti-tumor activity in large HPV-induced tumors when immunization against HPV16 genes E6 and E7 was combined with an anti-PD-1 antibody [100].
Taken together, these data support the idea that blocking PD-1:PD-L1 interactions may be a potential therapeutic option for HPV-infected patients. Indeed, early reports from the CheckMate-141 trial indicate enhanced benefit from Nivolumab in HPV positive patients. Additionally, in the context of cervical lesions, HPV-induced malignant progression correlates with low or undetectable T cell response to HPV antigens [101]. Anti-tumor vaccines currently in clinical trials aim to stimulate an immune response against HPV specific antigens, such as viral oncoproteins E6 and E7, which function in malignant transformation to inactivate p53 and pRb, respectively (NCT02865135, NCT02864147, NCT02002182, NCT02596243, NCT02163057) [102, 103]. It is possible that future immunotherapeutic protocols for the management of HPV positive HNSCC may achieve maximal benefit by combining vaccines to promote generation of tumor-specific effector T cells with anti-PD-1/PD-L1 mAbs to relieve immunosuppression.
Predicting response to PD-1 blockade
PD-L1 expression was initially considered a logical potential biomarker predicting response to anti-PD-1 therapy, but the many factors complicating its detection have limited its current utility, and no correlation between PD-L1 expression and response to immunotherapy has been observed [29]. Thus, new methods for predicting potential responders are needed. Immunotherapy trials for other cancers may yield insight into potential biomarkers for response to PD-1 blockade in HNSCC. As in HNSCC, a main risk factor for non-small cell lung cancer (NSCLC) is smoking, which is associated with higher nonsynonymous mutation burden [104]. Recently, Rizvi et al reported improved objective response, durable clinical benefit, and progression free survival in Pembrolizumab-treated NSCLC patients with more non-synonymous mutations and a molecular smoking signature [105]. Not surprisingly, mutational load positively correlated with higher neoantigen levels, which were also associated with improved treatment efficacy. A similar trend of high mutational load was observed in bladder cancer patients responsive to PD-1 blockade [106]. It therefore seems likely that the neoantigens produced by highly mutated tumors could potentially improve the ability of T-cells to recognize tumor cells following release from PD-L1-mediated immunosuppression. Smoking status has not yet been reported for HNSCC patients treated with anti- PD-1 mAbs, and further investigation will be necessary to determine whether the correlations observed in NSCLC are recapitulated in the HNSCC setting.
In advanced melanoma patients who responded to pembrolizumab, tumors had less diverse T cell populations at baseline, but the number of expanded clones after treatment was 10 fold higher than in patients with disease progression [107]. These data suggest that a pre-existing T-cell repertoire poised to target tumor cells but negatively regulated by PD-1: PD-L1 may predict response to PD-1 blockade. Again, this mechanism has yet to be investigated in HNSCC patients.
Limitations
Given the recent approval of anti-PD-1 therapy in the treatment of recurrent HNSCC, immunotherapy represents an exciting new avenue in the management of a disease with a limited armamentarium of systemic treatment options. It is also important, however, to consider the current limitations of immune checkpoint blockade. Improvements to OS, ORR, and PFS have been reported in patients treated with nivolumab and pembrolizumab, but many patients do not respond to immunotherapy, and no criteria currently exist to aid in selection of patients likely benefit from PD-1 blockade. Additionally, many patients, such as those with preexisting autoimmune disease, are not considered candidates for immunotherapy due to concern for unacceptable toxicity.
If and when immunotherapy is extended beyond the metastatic/recurrent setting, important new considerations will arise. Anti-PD-1 is currently administered in the context of HNSCC recurrence to patients who have already received XRT, which may sensitize tumors to immune attack. In murine models, XRT increases type I IFN levels in tumors, thereby promoting T-cell effector function. XRT also induced IFN- dependent up-regulation of the CXCR3 chemokine, which enhanced the recruitment of T cells to tumors [108]. It is also speculated that the high mutational load in XRT treated tumors leads to higher neoantigen levels, and therefore improved detection by T cells. If these mechanisms are found to be at play, the utility of PD-1 blockade as a frontline monotherapy may be limited. However, further investigation of a role for RT in sensitizing cancers to anti-tumor immunity may provide rationale for protocols combining XRT with immune checkpoint blockade. Several studies in mice, as well as anecdotal evidence in humans, support the hypothesis that XRT and immunotherapy may synergize to promote tumor clearance by the immune system. Concurrent immune checkpoint therapy and XRT induced anti-tumor T-cell responses in mice, even outside the radiation field [109]. Instances of abscopal effects of XRT have been observed in melanoma patients also treated with immune checkpoint inhibitors [110, 111].
Conclusions
The exploitation of anti-tumor immunity in the treatment of cancer is a promising and rapidly expanding field, but more research is needed to understand mechanisms mediating response to immunotherapy and expand the population of patients who will benefit from this strategy. While anti-PD-1/PD-L1 therapy has improved outcomes in comparison to the standard of care for a subset of patients with HNSCC, the proportion of responding patients, as well as the degree of benefit, remains modest. The future of anti-PD-1/PD-L1 therapy in the treatment of HNSCC relies upon a better understanding of the dysregulated pathways in HNSCC that may alter its efficacy. With a vast and growing array of targeted therapies in development for the treatment of cancer, countless opportunities for modulation of PD-L1 can be envisioned. Trials capitalizing on mechanistic discoveries by combining anti-PD-1 therapy with other precision-medicine approaches may maximize the results seen with immunotherapy.
Table 2
List of current trials evaluating the use of targeted therapies towards the PD-1/PD-L1 pathway in combination with small molecule inhibitor treatments in HNSCC.
| PD-1/PD-L1 Inhibitors + Small molecule inhibitors | |||||
|---|---|---|---|---|---|
| Trial | Phase | Therapy | Description | Target | Condition |
| NCT02471846 | Phase Ib | Atezolizumab + GDC-0919 | Evaluate safety, tolerability, pharmokinetics, and preliminary efficacy of Atezolizumab + GDC-0919 (indoleamine 2,3-dioxygenase (IDO) pathway inhibitor) | PD-L1 Indoleamine 2,3-dioxygenase pathway | Locally advanced or metastatic solid tumors (including HNSCC) |
| NCT02264678 | Phase I | MEDI4736 + AZD6738 vs. AZD6738 + Carboplatin vs. AZD6738 + Olaparib | Evaluate the safety, tolerability, pharmacokinetics, and preliminary anti-tumor activity of MEDI4736 + AZD6738 (ATR kinase inhibitor) vs. AZD6738 + Carboplatin vs. AZD6738 + Olaparib (nuclear enzyme poly(ADP-ribose) polymerase (PARP) inhibitor) | PD-L1 ATR kinase Nuclear enzyme poly(ADP-ribose) polymerase (PARP) | Advanced solid malignancies (including HNSCC) |
| NCT02586987 | Phase I | MEDI4736 + Selumetinib | Evaluate the safety, tolerability, pharmacokinetics, and preliminary efficacy of MEDI4736 + Selumetinib (mitogen-activated protein kinase (MEK or MAPK/ERK kinase) 1 and 2 inhibitor) | PD-L1 MAP/ERK kinase | Advanced solid tumors (including HNSCC) |
| NCT02499328 | Phase 1b/2 | MEDI4736 + AZD5069 or AZD9150 | Evaluate safety, tolerability, and preliminary efficacy of AZD5069 (CXC2 inhibitor) versus AZD9150 (STAT3 inhibitor) alone and in combination with MEDI4736 (PD-L1 inhibitor) | PD-L1 CXC2 STAT3 | Recurrent or metastatic HNSCC |
| NCT02318277 | Phase I/II | MEDI4736 + Epacadostat | Evaluate the safety, tolerability, pharmacokinetics, and preliminary efficacy of MEDI4736 + Epacadostat (indoleamine 2,3-dioxygenase (IDO1) inhibitor) | PD-L1 Indoleamine 2,3-dioxygenase | Advanced solid tumors (including HNSCC) |
| NCT02526017 | Phase I | Nivolumab + FPA-008 | Evaluate safety and preliminary efficacy of Nivolumab + FPA-008 (colony stimulating factor-1 receptor (CSF1R) inhibitor) | PD-1 Colony stimulating factor-1 receptor | Advanced solid tumors (including HNSCC) |
| NCT02124850 | Phase Ib | Nivolumab + Cetuximab + Motolimod | Evaluate change in immune biomarkers and tumor markers after treatment with Cetuximab + Motolimod (small molecule agonist of toll-like receptor 8) versus Nivolumab + Cetuximab + Motolimod | PD-1 EGF receptor Toll-like receptor 8 | Stage II, III, or IVa HNSCC |
| NCT02834247 | Phase Ib | Nivolumab + TAK-659 | Evaluate maximum tolerated dose, safety, and preliminary efficacy of Nivolumab + TAK-659 (Spleen tyrosine kinase (SYK) inhibitor) | PD-1 Spleen tyrosine kinase | Advanced solid tumors (including HNSCC) |
| NCT02327078 | Phase I/II | Nivolumab + Epacadostat | Evaluate safety tolerability, and preliminary efficacy of Nivolumab + Epacadostat (indoleamine 2,3-dioxygenase (IDO1) inhibitor) | PD-1 Indoleamine 2,3 dioxygenase | Advanced malignancies (including HNSCC) |
| NCT02646748 | Phase I | Pembrolizumab + INCB039110 versus Pembrolizumab + INCB050465 | Evaluate the safety, tolerability, and preliminary efficacy of pembrolizumab + INCB039110 (Jak inhibitor specific for Jak1) versus INCB050465 (PI3K-delta inhibitor) | PD-1 JAK1 PI3K-delta | Advanced solid tumors (including HNSCC) |
| NCT02452424 | Phase 1/2a | Pembrolizumab + PLX3397 | Evaluate safety and preliminary efficacy of combined therapy of Pembrolizumab and PLX3397 (colony-stimulating factor-1 receptor (CSF1R) inhibitor) | PD-1 | Melanoma Solid tumors (including HNSCC) |
| NCT02501096 | Phase 1b/2 | Pembrolizumab + Lenvatinib | Evaluate safety and preliminary efficacy of Pembrolizumab + Lenvatinib (tyrosine kinase inhibitor) | PD-1 Tyrosine kinase | Solid malignancies (including HNSCC) |
| NCT02178722 | Phase I/II | Pembrolizumab + INCB024360 | Evaluate safety, toxicity, and preliminary efficacy of Pembrolizumab + Epacadostat (indoleamine 2,3-dioxygenase (IDO1) inhibitor) | PD-1 Indoleamine 2,3-dioxygenase | Selected cancers including HNSCC |
| NCT02538510 | Phase I/II | Pembrolizumab + Vorinostat | Evaluate safety and preliminary efficacy of Pembrolizumab + Vorinostat (Histone Deacetylase Inhibitor) | PD-1 Histone deacetylase | Recurrent or Metastatic HNSCC or salivary gland carcinoma |
| NCT02454179 | Phase II | Pembrolizumab + ACP-196 | Evaluate efficacy of Pembrolizumab + ACP-196 (Acalabrutinib, Bruton's tyrosine kinase inhibitor) | PD-1 Tyrosine kinase | Recurrent, metastatic, or unresectable HNSCC |
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bauernhofer T, Kuss I, Henderson B, Baum AS, Whiteside TL. Preferential apoptosis of CD56dim natural killer cell subset in patients with cancer. European journal of immunology. 2003;33:119-24
2. Dasgupta S, Bhattacharya-Chatterjee M, O'Malley BW, Chatterjee SK. Inhibition of NK Cell Activity through TGF-β1 by Down-Regulation of NKG2D in a Murine Model of Head and Neck Cancer. The Journal of Immunology. 2005;175:5541-50
3. Ferris RL, Whiteside TL, Ferrone S. Immune escape associated with functional defects in antigen-processing machinery in head and neck cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:3890-5
4. Almand B, Resser JR, Lindman B, Nadaf S, Clark JI, Kwon ED. et al. Clinical significance of defective dendritic cell differentiation in cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2000;6:1755-66
5. Kuss I, Hathaway B, Ferris RL, Gooding W, Whiteside TL. Decreased absolute counts of T lymphocyte subsets and their relation to disease in squamous cell carcinoma of the head and neck. Clinical cancer research: an official journal of the American Association for Cancer Research. 2004;10:3755-62
6. Chen Z, Malhotra PS, Thomas GR, Ondrey FG, Duffey DC, Smith CW. et al. Expression of proinflammatory and proangiogenic cytokines in patients with head and neck cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 1999;5:1369-79
7. Ferris RL. Immunology and Immunotherapy of Head and Neck Cancer. Journal of Clinical Oncology. 2015
8. Schaefer C, Kim GG, Albers A, Hoermann K, Myers EN, Whiteside TL. Characteristics of CD4+CD25+ regulatory T cells in the peripheral circulation of patients with head and neck cancer. British journal of cancer. 2005;92:913-20
9. Strauss L, Bergmann C, Szczepanski M, Gooding W, Johnson JT, Whiteside TL. A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-beta1 mediates suppression in the tumor microenvironment. Clinical cancer research: an official journal of the American Association for Cancer Research. 2007;13:4345-54
10. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-64
11. Ascierto PA, Marincola FM. 2015: The Year of Anti-PD-1/PD-L1s Against Melanoma and Beyond. EBioMedicine. 2015;2:92-3
12. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H. et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. The Journal of experimental medicine. 2000;192:1027-34
13. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. The EMBO journal. 1992;11:3887-95
14. Okiyama N, Katz SI. Programmed cell death 1 (PD-1) regulates the effector function of CD8 T cells via PD-L1 expressed on target keratinocytes. Journal of autoimmunity. 2014;53:1-9
15. Chen J, Feng Y, Lu L, Wang H, Dai L, Li Y. et al. Interferon-gamma-induced PD-L1 surface expression on human oral squamous carcinoma via PKD2 signal pathway. Immunobiology. 2012;217:385-93
16. He J, Hu Y, Hu M, Li B. Development of PD-1/PD-L1 Pathway in Tumor Immune Microenvironment and Treatment for Non-Small Cell Lung Cancer. Scientific reports. 2015;5:13110
17. Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL. et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Science translational medicine. 2012;4:127ra37
18. Lyford-Pike S, Peng S, Young GD, Taube JM, Westra WH, Akpeng B. et al. Evidence for a role of the PD-1:PD-L1 pathway in immune resistance of HPV-associated head and neck squamous cell carcinoma. Cancer Res. 2013;73:1733-41
19. Garcia-Pineres AJ, Hildesheim A, Herrero R, Trivett M, Williams M, Atmetlla I. et al. Persistent human papillomavirus infection is associated with a generalized decrease in immune responsiveness in older women. Cancer Res. 2006;66:11070-6
20. Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. International immunology. 2007;19:813-24
21. Lin Z, Chen X, Li Z, Luo Y, Fang Z, Xu B. et al. PD-1 Antibody Monotherapy for Malignant Melanoma: A Systematic Review and Meta-Analysis. PloS one. 2016;11:e0160485
22. Nivolumab Doubles Survival for Patients with HNSCC. Cancer Discov. 2016;6:Of3
23. Seiwert TY. et al. Antitumor activity and safety of pembrolizumab in patients (pts) with advanced squamous cell carcinoma of the head and neck (SCCHN): Preliminary results from KEYNOTE-012 expansion cohort. J Clin Oncol. 2015:abstrLBA6008. ASCO Annual Meeting. 2015; 33
24. Gillison M, Blumenschein G, Fayette J. et al. Nivolumab (nivo) vs investigator's choice (IC) for recurrent or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC): CheckMate-141. AACR Annual Meeting. 2016:AbstractCT099
25. Seiwert TY, Weiss J, Baxi SS, Ahn M, Fayette J, Gillison ML. et al. A phase 3, randomized, open-label study of first-line durvalumab (MEDI4736) ± tremelimumab versus standard of care (SoC; EXTREME regimen) in recurrent/metastatic (R/M) SCCHN: KESTREL. suppl; abstr TPS6101. J Clin Oncol, 2016 ASCO Annual Meeting. 2016:34
26. Lin Y-M, Sung W-W, Hsieh M-J, Tsai S-C, Lai H-W, Yang S-M. et al. High PD-L1 Expression Correlates with Metastasis and Poor Prognosis in Oral Squamous Cell Carcinoma. PloS one. 2015;10:e0142656
27. Strome SE, Dong H, Tamura H, Voss SG, Flies DB, Tamada K. et al. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res. 2003;63:6501-5
28. Ukpo OC, Thorstad WL, Lewis JS Jr. B7-H1 expression model for immune evasion in human papillomavirus-related oropharyngeal squamous cell carcinoma. Head and neck pathology. 2013;7:113-21
29. Badoual C, Hans S, Merillon N, Van Ryswick C, Ravel P, Benhamouda N. et al. PD-1-Expressing Tumor-Infiltrating T Cells Are a Favorable Prognostic Biomarker in HPV-Associated Head and Neck Cancer. Cancer Research. 2013;73:128-38
30. Cho Y-A, Yoon H-J, Lee J-I, Hong S-P, Hong S-D. Relationship between the expressions of PD-L1 and tumor-infiltrating lymphocytes in oral squamous cell carcinoma. Oral Oncology. 2011;47:1148-53
31. Hsu MC, Hsiao JR, Chang KC, Wu YH, Su IJ, Jin YT. et al. Increase of programmed death-1-expressing intratumoral CD8 T cells predicts a poor prognosis for nasopharyngeal carcinoma. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2010;23:1393-403
32. Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH. et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:5064-74
33. Wansom D, Light E, Thomas D, Worden F, Prince M, Urba S. et al. Infiltrating lymphocytes and human papillomavirus-16-associated oropharyngeal cancer. The Laryngoscope. 2012;122:121-7
34. Concha-Benavente F, Srivastava RM, Trivedi S, Lei Y, Chandran U, Seethala RR. et al. Identification of the Cell-Intrinsic and -Extrinsic Pathways Downstream of EGFR and IFNgamma That Induce PD-L1 Expression in Head and Neck Cancer. Cancer Res. 2016;76:1031-43
35. Birkeland AC, Ludwig ML, Meraj TS, Brenner JC, Prince ME. The Tip of the Iceberg: Clinical Implications of Genomic Sequencing Projects in Head and Neck Cancer. Cancers. 2015;7:2094-109
36. Birkeland AC, Uhlmann WR, Brenner JC, Shuman AG. Getting personal: Head and neck cancer management in the era of genomic medicine. Head & neck. 2015
37. Birkeland AC, Yanik M, Tillman BN, Scott MV, Foltin SK, Mann JE. et al. Identification of Targetable ERBB2 Aberrations in Head and Neck Squamous Cell Carcinoma. JAMA otolaryngology- head & neck surgery. 2016
38. Giefing M, Wierzbicka M, Szyfter K, Brenner JC, Braakhuis BJ, Brakenhoff RH. et al. Moving towards personalised therapy in head and neck squamous cell carcinoma through analysis of next generation sequencing data. European journal of cancer. 2016;55:147-57
39. Tillman BN, Yanik M, Birkeland AC, Liu CJ, Hovelson DH, Cani AK. et al. Fibroblast growth factor family aberrations as a putative driver of head and neck squamous cell carcinoma in an epidemiologically low-risk patient as defined by targeted sequencing. Head & neck. 2016;38(Suppl 1):E1646-52
40. Temam S, Kawaguchi H, El-Naggar AK, Jelinek J, Tang H, Liu DD. et al. Epidermal growth factor receptor copy number alterations correlate with poor clinical outcome in patients with head and neck squamous cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2007;25:2164-70
41. Grandis JR, Tweardy DJ. Elevated Levels of Transforming Growth Factor α and Epidermal Growth Factor Receptor Messenger RNA Are Early Markers of Carcinogenesis in Head and Neck Cancer. Cancer Research. 1993;53:3579-84
42. Birkeland AC, Brenner JC. Personalizing Medicine in Head and Neck Squamous Cell Carcinoma: The Rationale for Combination Therapies. Medical research archives. 2015:3
43. Birkeland AC, Ludwig ML, Spector ME, Brenner JC. The potential for tumor suppressor gene therapy in head and neck cancer. Discovery medicine. 2016;21:41-7
44. Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB. et al. Radiotherapy plus Cetuximab for Squamous-Cell Carcinoma of the Head and Neck. New England Journal of Medicine. 2006;354:567-78
45. Tang Y, Fang W, Zhang Y, Hong S, Kang S, Yan Y. et al. The association between PD-L1 and EGFR status and the prognostic value of PD-L1 in advanced non-small cell lung cancer patients treated with EGFR-TKIs. Oncotarget. 2015;6:14209-19
46. Lin K, Cheng J, Yang T, Li Y, Zhu B. EGFR-TKI down-regulates PD-L1 in EGFR mutant NSCLC through inhibiting NF-kappaB. Biochemical and biophysical research communications. 2015;463:95-101
47. Han JJ, Kim DW, Koh J, Keam B, Kim TM, Jeon YK. et al. Change in PD-L1 Expression After Acquiring Resistance to Gefitinib in EGFR-Mutant Non-Small-Cell Lung Cancer. Clinical lung cancer. 2016;17:263-70.e2
48. Kurai J, Chikumi H, Hashimoto K, Yamaguchi K, Yamasaki A, Sako T. et al. Antibody-Dependent Cellular Cytotoxicity Mediated by Cetuximab against Lung Cancer Cell Lines. Clinical Cancer Research. 2007;13:1552
49. Hara M, Nakanishi H, Tsujimura K, Matsui M, Yatabe Y, Manabe T. et al. Interleukin-2 potentiation of cetuximab antitumor activity for epidermal growth factor receptor-overexpressing gastric cancer xenografts through antibody-dependent cellular cytotoxicity. Cancer Science. 2008;99:1471-8
50. Kasper S, Breitenbuecher F, Reis H, Brandau S, Worm K, Kohler J. et al. Oncogenic RAS simultaneously protects against anti-EGFR antibody-dependent cellular cytotoxicity and EGFR signaling blockade. Oncogene. 2013;32:2873-81
51. Yang X, Zhang X, Mortenson ED, Radkevich-Brown O, Wang Y, Fu Y-X. Cetuximab-mediated Tumor Regression Depends on Innate and Adaptive Immune Responses. Mol Ther. 2013;21:91-100
52. Bertino EM, McMichael EL, Mo X, Trikha P, Davis M, Paul B. et al. A Phase I Trial to Evaluate Antibody-Dependent Cellular Cytotoxicity of Cetuximab and Lenalidomide in Advanced Colorectal and Head and Neck Cancer. Molecular Cancer Therapeutics. 2016;15:2244
53. Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576-82
54. Lui VW, Hedberg ML, Li H, Vangara BS, Pendleton K, Zeng Y. et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013;3:761-9
55. Michmerhuizen NL, Birkeland AC, Bradford CR, Brenner JC. Genetic determinants in head and neck squamous cell carcinoma and their influence on global personalized medicine. Genes & cancer. 2016;7:182-200
56. Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;9:667-76
57. Isaacsson Velho PH, Castro G Jr, Chung CH. Targeting the PI3K Pathway in Head and Neck Squamous Cell Carcinoma. Am Soc Clin Oncol Educ Book. 2015:123-8
58. Nguyen H, Ramana CV, Bayes J, Stark GR. Roles of phosphatidylinositol 3-kinase in interferon-gamma-dependent phosphorylation of STAT1 on serine 727 and activation of gene expression. J Biol Chem. 2001;276:33361-8
59. Kaur S, Uddin S, Platanias LC. The PI3' kinase pathway in interferon signaling. J Interferon Cytokine Res. 2005;25:780-7
60. Lee SK, Seo SH, Kim BS, Kim CD, Lee JH, Kang JS. et al. IFN-gamma regulates the expression of B7-H1 in dermal fibroblast cells. J Dermatol Sci. 2005;40:95-103
61. Le Good JA, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 1998;281:2042-5
62. Choudhury GG. A linear signal transduction pathway involving phosphatidylinositol 3-kinase, protein kinase Cepsilon, and MAPK in mesangial cells regulates interferon-gamma-induced STAT1alpha transcriptional activation. J Biol Chem. 2004;279:27399-409
63. DeVries TA, Kalkofen RL, Matassa AA, Reyland ME. Protein kinase Cdelta regulates apoptosis via activation of STAT1. J Biol Chem. 2004;279:45603-12
64. Kondo A, Yamashita T, Tamura H, Zhao W, Tsuji T, Shimizu M. et al. Interferon-gamma and tumor necrosis factor-alpha induce an immunoinhibitory molecule, B7-H1, via nuclear factor-kappaB activation in blasts in myelodysplastic syndromes. Blood. 2010;116:1124-31
65. Gowrishankar K, Gunatilake D, Gallagher SJ, Tiffen J, Rizos H, Hersey P. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-kappaB. PLoS One. 2015;10:e0123410
66. Xu C, Fillmore CM, Koyama S, Wu H, Zhao Y, Chen Z. et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell. 2014;25:590-604
67. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ. et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84-8
68. Zhang Y, Zhang J, Xu K, Xiao Z, Sun J, Xu J. et al. PTEN/PI3K/mTOR/B7-H1 signaling pathway regulates cell progression and immuno-resistance in pancreatic cancer. Hepatogastroenterology. 2013;60:1766-72
69. Zhu J, Chen L, Zou L, Yang P, Wu R, Mao Y. et al. MiR-20b, -21, and -130b inhibit PTEN expression resulting in B7-H1 over-expression in advanced colorectal cancer. Hum Immunol. 2014;75:348-53
70. Yu T, Cao R, Li S, Fu M, Ren L, Chen W. et al. MiR-130b plays an oncogenic role by repressing PTEN expression in esophageal squamous cell carcinoma cells. BMC Cancer. 2015;15:29
71. Ludwig ML, Birkeland AC, Hoesli R, Swiecicki P, Spector ME, Brenner JC. Changing the paradigm: the potential for targeted therapy in laryngeal squamous cell carcinoma. Cancer biology & medicine. 2016;13:87-100
72. Uddin S, Majchrzak B, Woodson J, Arunkumar P, Alsayed Y, Pine R. et al. Activation of the p38 mitogen-activated protein kinase by type I interferons. J Biol Chem. 1999;274:30127-31
73. Liu J, Hamrouni A, Wolowiec D, Coiteux V, Kuliczkowski K, Hetuin D. et al. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood. 2007;110:296-304
74. Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:598-609
75. Qian Y, Deng J, Geng L, Xie H, Jiang G, Zhou L. et al. TLR4 signaling induces B7-H1 expression through MAPK pathways in bladder cancer cells. Cancer Invest. 2008;26:816-21
76. Yamamoto R, Nishikori M, Tashima M, Sakai T, Ichinohe T, Takaori-Kondo A. et al. B7-H1 expression is regulated by MEK/ERK signaling pathway in anaplastic large cell lymphoma and Hodgkin lymphoma. Cancer Sci. 2009;100:2093-100
77. Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P. et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin Cancer Res. 2016;22:1499-509
78. Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang T. et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res. 2015;21:1639-51
79. Hughes PE, Caenepeel S, Wu LC. Targeted Therapy and Checkpoint Immunotherapy Combinations for the Treatment of Cancer. Trends Immunol. 2016;37:462-76
80. The Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576-82
81. Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y. et al. PDL1 Regulation by p53 via miR-34. JNCI Journal of the National Cancer Institute. 2016;108:djv303
82. Chan KS, Sano S, Kiguchi K, Anders J, Komazawa N, Takeda J. et al. Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. The Journal of clinical investigation. 2004;114:720-8
83. Wolfle SJ, Strebovsky J, Bartz H, Sahr A, Arnold C, Kaiser C. et al. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. European journal of immunology. 2011;41:413-24
84. Fujita Y, Yagishita S, Hagiwara K, Yoshioka Y, Kosaka N, Takeshita F. et al. The clinical relevance of the miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant non-small-cell lung cancer. Molecular therapy: the journal of the American Society of Gene Therapy. 2015;23:717-27
85. Hao Y, Chapuy B, Monti S, Sun HH, Rodig SJ, Shipp MA. Selective JAK2 Inhibition Specifically Decreases Hodgkin Lymphoma and Mediastinal Large B-cell Lymphoma Growth In Vitro and In Vivo. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:2674-83
86. Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O'Donnell E. et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116:3268-77
87. Grandis JR, Drenning SD, Zeng Q, Watkins SC, Melhem MF, Endo S. et al. Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:4227-32
88. Lu X, Kang Y. Hypoxia and hypoxia-inducible factors: master regulators of metastasis. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16:5928-35
89. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P. et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. The Journal of experimental medicine. 2014;211:781-90
90. Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014;74:665-74
91. Beasley NJ, Leek R, Alam M, Turley H, Cox GJ, Gatter K. et al. Hypoxia-inducible factors HIF-1alpha and HIF-2alpha in head and neck cancer: relationship to tumor biology and treatment outcome in surgically resected patients. Cancer Res. 2002;62:2493-7
92. Brizel DM, Dodge RK, Clough RW, Dewhirst MW. Oxygenation of head and neck cancer: changes during radiotherapy and impact on treatment outcome. Radiotherapy and oncology: journal of the European Society for Therapeutic Radiology and Oncology. 1999;53:113-7
93. Ferris RL, Hunt JL, Ferrone S. Human leukocyte antigen (HLA) class I defects in head and neck cancer: molecular mechanisms and clinical significance. Immunologic research. 2005;33:113-33
94. Guven-Maiorov E, Keskin O, Gursoy A, VanWaes C, Chen Z, Tsai CJ. et al. TRAF3 signaling: Competitive binding and evolvability of adaptive viral molecular mimicry. Biochimica et biophysica acta. 2016;1860:2646-55
95. Zapata JM, Llobet D, Krajewska M, Lefebvre S, Kress CL, Reed JC. Lymphocyte-specific TRAF3 transgenic mice have enhanced humoral responses and develop plasmacytosis, autoimmunity, inflammation, and cancer. Blood. 2009;113:4595-603
96. Yang W, Song Y, Lu YL, Sun JZ, Wang HW. Increased expression of programmed death (PD)-1 and its ligand PD-L1 correlates with impaired cell-mediated immunity in high-risk human papillomavirus-related cervical intraepithelial neoplasia. Immunology. 2013;139:513-22
97. Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tân PF. et al. Human Papillomavirus and Survival of Patients with Oropharyngeal Cancer. New England Journal of Medicine. 2010;363:24-35
98. Pignon JP, le Maitre A, Maillard E, Bourhis J. Meta-analysis of chemotherapy in head and neck cancer (MACH-NC): an update on 93 randomised trials and 17,346 patients. Radiotherapy and oncology: journal of the European Society for Therapeutic Radiology and Oncology. 2009;92:4-14
99. Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L. et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. Journal of the National Cancer Institute. 2000;92:709-20
100. Rice AE, Latchman YE, Balint JP, Lee JH, Gabitzsch ES, Jones FR. An HPV-E6/E7 immunotherapy plus PD-1 checkpoint inhibition results in tumor regression and reduction in PD-L1 expression. Cancer Gene Ther. 2015;22:454-62
101. de Jong A, van Poelgeest MI, van der Hulst JM, Drijfhout JW, Fleuren GJ, Melief CJ. et al. Human papillomavirus type 16-positive cervical cancer is associated with impaired CD4+ T-cell immunity against early antigens E2 and E6. Cancer Res. 2004;64:5449-55
102. Spanos WC, Hoover A, Harris GF, Wu S, Strand GL, Anderson ME. et al. The PDZ binding motif of human papillomavirus type 16 E6 induces PTPN13 loss, which allows anchorage-independent growth and synergizes with ras for invasive growth. Journal of virology. 2008;82:2493-500
103. Brehm A, Nielsen SJ, Miska EA, McCance DJ, Reid JL, Bannister AJ. et al. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. The EMBO journal. 1999;18:2449-58
104. Blackford A, Parmigiani G, Kensler TW, Wolfgang C, Jones S, Zhang X. et al. Genetic Mutations Associated With Cigarette Smoking in Pancreatic Cancer. Cancer research. 2009;69:3681-8
105. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ. et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124
106. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C. et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558-62
107. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L. et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568-71
108. Lim JY, Gerber SA, Murphy SP, Lord EM. Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8(+) T cells. Cancer immunology, immunotherapy: CII. 2014;63:259-71
109. Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC. et al. Fractionated but Not Single-Dose Radiotherapy Induces an Immune-Mediated Abscopal Effect when Combined with Anti-CTLA-4 Antibody. Clinical Cancer Research. 2009;15:5379-88
110. Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S. et al. Immunologic Correlates of the Abscopal Effect in a Patient with Melanoma. New England Journal of Medicine. 2012;366:925-31
111. Stamell EF, Wolchok JD, Gnjatic S, Lee NY, Brownell I. The Abscopal Effect Associated With a Systemic Anti-melanoma Immune Response. International Journal of Radiation Oncology*Biology*Physics. 2013;85:293-5
112. al Ce. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discovery. 2012:2
Author contact
![]() Corresponding author: J. Chad Brenner, Ph.D., 1150 E. Medical Center Dr., 9301B MSRB3, Ann Arbor, MI 48109-0602. Phone: (734)763-2761; Fax: (734)232-1007; Email: chadbrenedu.
Corresponding author: J. Chad Brenner, Ph.D., 1150 E. Medical Center Dr., 9301B MSRB3, Ann Arbor, MI 48109-0602. Phone: (734)763-2761; Fax: (734)232-1007; Email: chadbrenedu.
Citation styles
APA
Mann, JE., Hoesli, R., Michmerhuizen, NL., Devenport, SN., Ludwig, ML., Vandenberg, TR., Matovina, C., Jawad, N., Mierzwa, M., Shuman, AG., Spector, ME., Brenner, JC. (2017). Surveilling the Potential for Precision Medicine-driven PD-1/PD-L1-targeted Therapy in HNSCC. Journal of Cancer, 8(3), 332-344. https://doi.org/10.7150/jca.17547.
ACS
Mann, JE.; Hoesli, R.; Michmerhuizen, NL.; Devenport, SN.; Ludwig, ML.; Vandenberg, TR.; Matovina, C.; Jawad, N.; Mierzwa, M.; Shuman, AG.; Spector, ME.; Brenner, JC. Surveilling the Potential for Precision Medicine-driven PD-1/PD-L1-targeted Therapy in HNSCC. J. Cancer 2017, 8 (3), 332-344. DOI: 10.7150/jca.17547.
NLM
Mann JE, Hoesli R, Michmerhuizen NL, Devenport SN, Ludwig ML, Vandenberg TR, Matovina C, Jawad N, Mierzwa M, Shuman AG, Spector ME, Brenner JC. Surveilling the Potential for Precision Medicine-driven PD-1/PD-L1-targeted Therapy in HNSCC. J Cancer 2017; 8(3):332-344. doi:10.7150/jca.17547. https://www.jcancer.org/v08p0332.htm
CSE
Mann JE, Hoesli R, Michmerhuizen NL, Devenport SN, Ludwig ML, Vandenberg TR, Matovina C, Jawad N, Mierzwa M, Shuman AG, Spector ME, Brenner JC. 2017. Surveilling the Potential for Precision Medicine-driven PD-1/PD-L1-targeted Therapy in HNSCC. J Cancer. 8(3):332-344.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.