Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(4):555-562. doi:10.7150/jca.17205 This issue Cite
Research Paper
Combined Targeting of mTOR and Akt Using Rapamycin and MK-2206 in The Treatment of Tuberous Sclerosis Complex
Shuang Ji1,2*, Wei Lin3,4*, Li Wang2, Zhaofei Ni2, Fuquan Jin2, Xiaojun Zha2 ![]() , Guanghe Fei1
, Guanghe Fei1 ![]()
1. Pulmonary Department, First Affiliated Hospital of Anhui Medical University, Hefei, Anhui, China;
2. Department of Biochemistry & Molecular Biology, School of Basic Medical Sciences, Anhui Medical University, Hefei, Anhui, China;
3. Department of Stomatology, First Affiliated Hospital of Anhui Medical University, Hefei, Anhui, China;
4. Department of Prosthodontics, Hospital of Stomatology, Wuhan University, Wuhan, Hubei, China.
* Contributed equally to this article.
Received 2016-8-15; Accepted 2016-10-29; Published 2017-2-11
Citation:
Ji S, Lin W, Wang L, Ni Z, Jin F, Zha X, Fei G. Combined Targeting of mTOR and Akt Using Rapamycin and MK-2206 in The Treatment of Tuberous Sclerosis Complex. J Cancer 2017; 8(4):555-562. doi:10.7150/jca.17205. https://www.jcancer.org/v08p0555.htm
Other stylesAbstract
Tuberous sclerosis complex (TSC), caused by loss-of-function mutations in the TSC1 or TSC2 genes, is an autosomal dominant disease characterized by benign tumor formation in multiple organs. Hyperactivation of mammalian target of rapamycin (mTOR) is the primary alteration underlying TSC tumor. Thus, rapamycin, as an mTOR specific inhibitor, has been assumed as a potential drug for the treatment of TSC. However, its application in TSC patients has been limited due to side effects. By analyzing Tsc1- or Tsc2-null mouse embryonic fibroblasts (MEFs), we found that loss of TSC1 or TSC2 led to a decreased sensitivity to MK-2206, a novel allosteric Akt inhibitor. Ectopic expression of a constitutively activated Akt (myristoylated Akt-1, myrAkt-1) sensitized Tsc2-null and Tsc1-null MEFs to MK-2206. Furthermore, MK-2206 increased the cytotoxicity of rapamycin in Tsc1-/- or Tsc2-/- MEFs. Moreover, the benefit of the combinatorial treatment was also demonstrated in a TSC xenograft mouse model. We conclude that the combination of rapamycin and MK-2206 may be utilized as a new therapeutic regimen for TSC.
Keywords: TSC, mTOR, Akt, rapamycin, MK-2206
Introduction
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder characterized by benign tumor formation in multiple organs such as kidney, liver, brain and skin; there is no treatment available to date beyond the alleviation of its symptoms [1-3]. Genetically, TSC is caused by loss-of-function mutations of one of the two tumor suppressor genes: TSC1 and TSC2 [4-6]. TSC1 located at 9q34 and TSC2 located at 16p13 encode protein hamartin and tuberin, respectively [7, 8]. The two proteins function in an interdependent manner in a stable complex [9]. The TSC1/2 (TSC1/TSC2) heterodimer acts as a brake bridging upstream Akt and downstream mammalian target of rapamycin (mTOR). Akt activation phosphorylates TSC2 which disrupts the association of the TSC1/TSC2 protein complex within the membrane; The TSC1/TSC2 complex is required for GAP function of the RAS homolog enriched in brain (Rheb)-GTP [10, 11]. Loss-of-function mutation in either TSC1 or TSC2 results in the accumulation of Rheb-GTP, which in turn activates mTOR [12]. Aberrant mTOR-activated signaling leads to uncontrolled cell growth and tumorigenesis in TSC.
The mTOR protein is a serine/threonine protein kinase consisting of rapamycin-sensitive complex (mTORC1) and rapamycin-insensitive multimeric complex (mTORC2) [13-15]. The mTORC1 complex is composed of mTOR, Raptor and PRAS40; mTORC1 phosphorylates and activates its downstream targets S6 kinase1 (S6K1) and eukaryotic initiation factor 4E-binding protein 1(eIF4E-BP1) [3, 16, 17]. The activated S6K1 phosphorylates ribosomal protein S6 (RpS6), and promotes protein translation. Activated eIF4E-BP1 can no longer bind and inhibit eIF4E. These molecules play distinctly to promote translation initiation [18]. The mTORC2 complex is comprised of mTOR, Rictor, Sen1, and Raptor; mTORC2 phosphorylates Akt at Ser473 [15]. Loss of the TSC1/TSC2 complex leads to Akt suppression and its activity is rapidly reversed by rapamycin, an inhibitor of mTORC1. Therefore, rapamycin had been anticipated to be effective in the treatment of TSC patients in recent years, but it has only demonstrated modest clinical efficiency [19]. Moreover, rapamycin treatment can induce significant side effects, including chronic immunosuppression and associated opportunistic infections, which makes the long-term treatment less feasibility. Thus, development of a novel and effective therapeutic approaches are urgently needed.
MK-2206 is a selective Akt inhibitor that binds to the Akt protein at a site in the pleckstrin homology domain, leading to a Akt conformational change that prevents its localization to the plasma membrane and its subsequent activation [20]. MK-2206 has been examined in both preclinical settings and clinical trials as an anticancer agent that can synergistically enhance the antitumor efficacy of certain molecular targeted agents in preclinical animal models of lung cancer, ovarian cancer and breast cancer [21-23].
In this study, we investigated whether Akt inhibition increased the sensitivity of Tsc-deficient MEFs to rapamycin. A synergistic effect was detected using a combination of MK-2206 in vitro and rapamycin through MTT analysis and apoptosis. A significantly increased antitumor growth effect was observed in vivo when treating a TSC xenograft mouse model with the combination of MK-2206 and rapamycin. We provided evidence that a combination of drugs that target both Akt and mTOR sheds light on a new therapeutic tactic warranting clinical investigation for treating patients with TSC or other related cancers.
Materials and Methods
Reagents and antibodies
Rapamycin and MK-2206 were obtained from Selleck Chemicals (Houston, TX, USA). DMEM (Dulbecco's Modified Eagle Medium), FBS (Fetal Bovine Serum), lipofectamine 2000 and 4%-12%Bis-Tris Nu-PAGE gels were from Life Technologies (Carlsbad, CA, USA). MTT (3-[4,5-dimehyl-2-thiazolyl]-2,5-diphenyl-2H-tetrazolium bromide) kits were from Beyotime Biotechnology (Shanghai, China). All HRP-labeled secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-phospho-Akt (ser473) (#4060), anti-Akt-1 (#2967), anti-phospho-S6 (#2217), anti-S6 (#4857) and anti-Caspase-3 (#9665) were from Cell Signaling Technology (Danvers, MA, USA).
Cell culture
Immortalized Tsc1+/+, Tsc1-/-, Tsc2+/+ and Tsc2-/- MEFs (mouse embryonic fibroblasts) used in this study were described previously [6, 24]. All cells were cultured in DMEM containing 10% FBS in a humidified atmosphere of 5% CO2 at 37℃. MyrAkt-1 (myristoylated Akt-1) expression plasmids were contransfected with lipofectamine 2000 into wildtype MEF lines. Cells were harvested and proteins were isolated two days later for western blot analysis.
Western blot analysis
After being washed in PBS, cells were harvested using RIPA buffer (Beyotime Biotechnology, Haimen, China), boiled for 5 minutes. Total protein were separated on SDS-PAGE and transferred to a PVDF membrane. After blockade with 5% non-fat dry milk in TBST (50 nM Tris-HCl, 150 mM NaCl, 0.05% Tween 20, pH 7.5) for 45 min at room temperature, membrane was incubated with the primary antibodies specific to p-Akt (1:1000), Akt-1 (1:1000), p-S6 (1:1000), Caspase-3 (1:1000) or S6 (1:1000) at 4℃ for overnight, and subsequent incubation with a secondary HRP-conjugated antibody (Santa Cruz Biotechnology) at 1:5000 dilution for 4 h at room temperature. An enhanced chemo-luminescence detection kit (ECL Advance, Amersham, UK) was employed and blots were exposed to X-ray film.
Cell proliferation assays
To assess the effects of rapamycin and MK-2206, MEFs were seeded in triplicate in 96-well plates at 4000 cells/well and incubated for 8 h. The medium was removed and replaced with fresh medium containing rapamycin, MK-2206 or both at various concentrations. At different time points, 10 μl MTT (5 mg/ml in PBS) was added to each well, followed by incubation at 37℃. The culture medium was removed and 100 μl dimethylsulfoxide (DMSO) was added to each well followed by shaking at room temperature for 10 min. The spectrometric absorbance at 490 nm was determined with a microplate reader (Thermo Scientific, Waltham, MA, USA). Data were presented as mean ± standard deviation (SD) of separate experiments (n≥3). The results of MTT assays represented the average of each individual experiment.
Induction of subcutaneous tumors in nude mice
BALB/c nude male mice (18-20 g, 6 weeks) were purchased from Vital River Laboratories Animal Technology (Beijing, China). All experiments involving animals follow the guidelines of the Animal Center of Anhui Medical University, and all animal experimental protocols were approved by the experimental Animal Ethical Committee of Anhui Medical University. For tumorigenicity assays, 4.0 × 106 Tsc1-null MEFs in 0.2 ml solvent control were injected subcutaneously near the axillary fossa of the nude mice. Three weeks later, when the tumor diameters reached 4.0 mm × 5.0 mm, the tumor cell-inoculated mice were randomly divided into four treatment groups with five mice per group. Group A were treated with solvent control; group B with 0.2mg/kg rapamycin (20 mg/ml stock in ethanol) diluted in 0.25% PEG plus 0.25% Tween-20 were given by intraperitoneal injection; Group C with 100mg/kg MK-2206 (14 mg/ml stock in DMSO) diluted in 0.25% PEG plus 0.25% Tween-20 were administered by oral gavage; group D with 0.2mg/kg rapamycin by intraperitoneal and 100mg/kg MK-2206 by intraperitoneal injection every day. Tumors were measured with a caliper every 2 days and the tumor volume were calculated using the formula V=1/2 (width2×length); body weights were also recorded. On day 40 after tumor cell inoculation, all experimental mice were euthanized and the total weight of the tumors in each mouse was measured.
Statistical analysis
Data were expressed as mean ± SD. Results were analyzed by a one- or two-way ANOVA. If significance was found, post hoc Bonferroni multiple comparison was used. All statistical tests were conducted using SPSS 19.0 software package (IBM, Armonk, NY, USA) with p < 0.05 considered statistically significant.
Results
Tsc1/2-null MEFs present a low sensitivity to MK-2206
The MEFs with and without Tsc1/2 deficiency were treated with MK-2206 (a selective Akt inhibitor) for 36 h. The primary MEF cells and Tsc1/2-deficient MEF cells did not present morphological differences before treatment. However, 36 h after MK-2206 treatment, Tsc2-/-- (Fig. 1A) and Tsc1-/-- (Fig. 1B) deficient MEF cells presented a higher cellular viability as compared to primary cell lines.
The cell viability was quantified with an MTT cell survival assay. MK-2206 treatment decreased the cell viability in Tsc2+/+ MEFs. A two-way ANOVA test revealed a significant effect of MK-2206 treatment (F (1, 16) = 255.86, P < 0.01), time course (F (3, 16) = 3.40, P < 0.05) and an interaction of treatment × time course (F (3, 16) = 52.65, P < 0.01). MK-2206 decreased the cell viability in Tsc2-/- MEF cells. A two-way ANOVA test revealed a significant main effect of MK-2206 treatment (F (1, 16) = 18.85, P < 0.01), time course (F (3, 16) = 30.99, P < 0.01) and the interaction of treatment × time course (F (3, 16) = 3.38, P < 0.05). MK-2206 treatment presented a low effect on cell proliferation of Tsc2-/- MEFs. A two-way ANOVA test revealed a significant main effect of cell type (F (1, 16) = 367.37, P < 0.01) and the interaction of cell type × time course (F (3, 16) = 54.42, P < 0.01). There was no difference in time course (F (3, 16) = 2.78, P = 0.08) (Fig. 1C).
MK-2206 decreased the cell viability in Tsc1+/+ MEFs. A two-way ANOVA test revealed a significant main effect of MK-2206 treatment (F (1, 16) = 571.54, P < 0.01), time course (F (3, 16) = 130.54, P < 0.01) and an interaction of treatment × time course (F (3, 16) = 102.95, P < 0.01). MK-2206 treatment decreased the cell viability in Tsc1-/- MEFs. A two-way ANOVA test revealed a significant effect of MK-2206 treatment (F (1, 16) = 175.65, P < 0.01), time course (F (3, 16) = 277.90, P < 0.01) and an interaction of treatment × time course (F (3, 16) = 37.42, P < 0.01). MK-2206 presented a low effect on Tsc1-/- MEFs. A two-way ANOVA test revealed a significant effect of cell type (F (1, 16) = 127.17, P < 0.01), time course (F (3, 16) = 47.61, P < 0.01) and an interaction of cell type × time course (F (3, 16) = 23.04, P < 0.01) (Fig. 1D). Western Blot assay showed that Tsc2-/- (Fig. 1E) and Tsc1-/- (Fig. 1F) MEF cells presented a lower p-Akt expression comparted to primary MEF cells.
Overexpressing a constitutively active Akt-1 sensitizes Tsc2-null or Tsc1-null MEF cells to MK-2206
Because loss of TSC1 or TSC2 genes suppresses Akt activity (Fig. 1E and 1F) and MK-2206 is a specific inhibitor of Akt, we speculated that deficiency of TSC1 or TSC2 could decrease MEFs sensitivity to MK-2206 due to the inhibition of Akt activity. Tsc1/2-null MEF cells were transfected with retroviral vectors to overexpress (myrAkt-1); Akt overexpression was confirmed by western blot analysis (Figs. 2A and 2B). Consistently, ectopic myrAkt-1 expression in Tsc2-/- MEF cells (Fig. 2C) and Tsc1-/- MEF cells (Fig. 2D) increased cell sensitivity to MK-2206. Increasing Akt-1 expression enhanced the sensitivity of Tsc2-/- MEFs to MK-2206. A two-way ANOVA test revealed a significant effect of Akt-1 transfection (F (1, 16) = 75.92, P < 0.01), time course (F (3, 16) = 110.63, P < 0.01) and an interaction of Akt-1 transfection × time course (F (3, 16) = 22.29, P < 0.01) (Fig. 2E). Consistently, an increase in Akt-1 expression enhanced the sensitivity of Tsc1-/- MEFs to MK-2206. A two-way ANOVA test revealed a significant effect of Akt-1 transfection (F (1, 16) = 152.63, P < 0.01), time course (F (3, 16) = 29.54, P < 0.01) and an interaction of Akt-1 transfection × time course (F (3, 16) = 33.66, P < 0.01) (Fig. 2F).
Combining rapamycin with MK-2206 significantly inhibits proliferation and induces apoptosis in Tsc2-null or Tsc1-null MEF cells
We examined whether the combination of rapamycin and MK-2206 would achieve a better inhibitory effect on the growth of Tsc2-/- MEF cells. The combination of 2.0 nM rapamycin and 100 nM MK-2206 exerted a stronger inhibitory effect on the growth of Tsc2-/- MEF cells than either compound alone (Fig. 3A). Western blot analysis showed that MK-2206 treatment abolished the up-regulated Akt activity triggered by rapamycin (Fig. 3B). Rapamycin and MK-2206 administered in combination similarly potentiated the cytotoxicity in Tsc1-/- MEF cells (Fig. 3C). The increased Akt activity induced by rapamycin was also attenuated by MK-2206 in Tsc1-/- MEFs (Fig. 3D). To assess the cell apoptosis of diminished signaling to Akt in MEFs lacking TSC2 and TSC1, we examined the expression of Caspase3, an indicator of cell apoptosis. In contrast to untreated cells, both Tsc2-null and Tsc1-null MEF lines demonstrated an increased expression of Caspase3 when given the two drugs in combination (Fig. 3B and 3D). Combining rapamycin and MK-2206 significantly decreased the cell proliferation in Tsc2-/- MEF cells (Fig. 3E). A one-way ANOVA test revealed a significant difference (F (3.12) = 9.16, P < 0.01). Post hoc Bonferroni's multiple comparison analysis showed a significant difference between the control group and rapamycin and MK-2206 combination group (P < 0.01). Combination of rapamycin and MK-2206 significantly decreased the cell proliferation in Tsc1-/- MEF cells (Fig. 3F). A one-way ANOVA test revealed a significant difference (F (3.12) = 23.11, P < 0.01). Post hoc Bonferroni's multiple comparison showed a significant difference between the control group and rapamycin and MK-2206 combination group (P < 0.01).
Figure 1
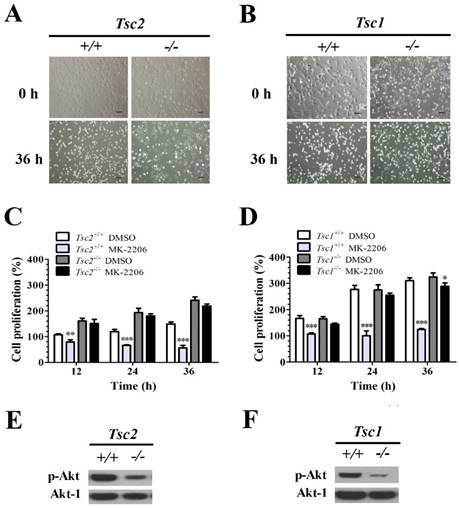
Tsc1/2-null MEF cells present a low sensitivity to MK-2206. (A, B) Microscopically, MK-2206 had apparently different effects on Tsc1 and Tsc2 deficient MEF cells (original magnification, ×100). (C, D) Tsc2 and Tsc1 deficient MEF cells were treated with or without 100 nM MK-2206 for up to 36 h and cell survival was tested by MTT assay. Cells treated with DMSO only acted as controls with cell viability set at 100%. The percent cell viability in each treatment group was calculated relative to cells treated with DMSO vehicle control. (E, F) Western blot assay showed that Tsc2-/- and Tsc1-/- MEF cells presented lower p-Akt and Akt-1 expression. (*denotes P<0.05, **denotes P<0.01, ***denotes P<0.001 or significant differences between treatment groups and DMSO control groups).
Figure 2
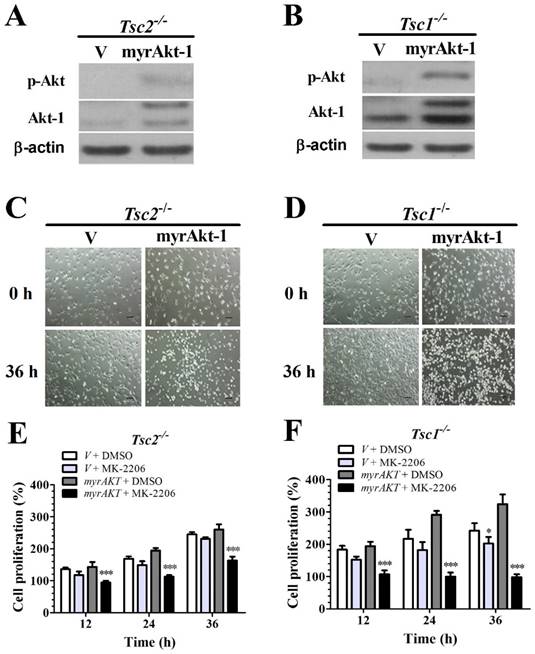
Overexpressing a constitutively active Akt-1 sensitizes Tsc2-null or Tsc1-null MEF cells to MK-2206. (A, B) Cell lysates were extracted from TSC2- and TSC1-null MEF cells transduced with the retroviruses for myristoylated Akt-1 (myrAkt-1) in pLXIN-hyg or its control vector pLXIN-hyg (v) and subjected to immunoblotting for p-Akt and Akt-1. (C, D) The changes in cell morphology in MEF cells without TSC2 or TSC1 expressing v and myrAkt-1 treated with 100 nM MK-2206 for 36 h were observed, and cells were photographed using a microscope fitted with digital camera (original magnification, ×100). (E, F) Cell proliferation of Tsc2-null and Tsc1-null MEF cells expressing v or myrAkt-1 and treated with 100 nM MK-2206 was examined by MTT assays. (*denotes P<0.05, ***denotes P<0.001 or significant differences between treatment groups and DMSO control groups).
MK-2206 enhances the rapamycin-mediated inhibition of Tsc1-null MEF cells in a xenograft tumor model
We evaluated the efficacy of combining rapamycin and MK-2206 in a subcutaneous TSC xenograft mouse model. 4.0×106 Tsc1-/- MEF cells were injected subcutaneously into the axillary fossa of BALB/c-mice (n = 5 per group). After establishment of palpable tumors, the mice were randomly assigned to four groups with rapamycin, MK-2206 alone or together for 17 days. Rapamycin and MK-2206 alone or in combination did not alter the body weight. A two-way repeated ANOVA test did not reveal a difference in treatment (F (3, 16) = 0.918) and treatment × time interaction (F (24, 128) = 1.42, P = 0.11) (Fig. 4A). Rapamycin and MK-2206 treatment significantly altered the tumor weight (F (3.16) = 151, P < 0.01). Post hoc Bonferroni's multiple comparison showed that combination of rapamycin and MK-2206 significantly decreased tumor weight compared to control, rapamycin only and MK-2206 only groups (P < 0.01) (Fig. 4C).
Both rapamycin and MK-2206 treatments significantly altered tumor size. A two-way ANOVA test revealed a significant main effect of treatment (F (3,144) = 72.90, P < 0.01), time (F (8,144) = 101.38, P < 0.01) and treatment × time (F (24, 144) = 9.26, P < 0.01). Post hoc Bonferroni's multiple comparison showed that combination of rapamycin and MK-2206 significantly decreased tumor size compared to controls, rapamycin only and MK-2206 only groups (P < 0.01) (Fig. 4D).
Figure 3
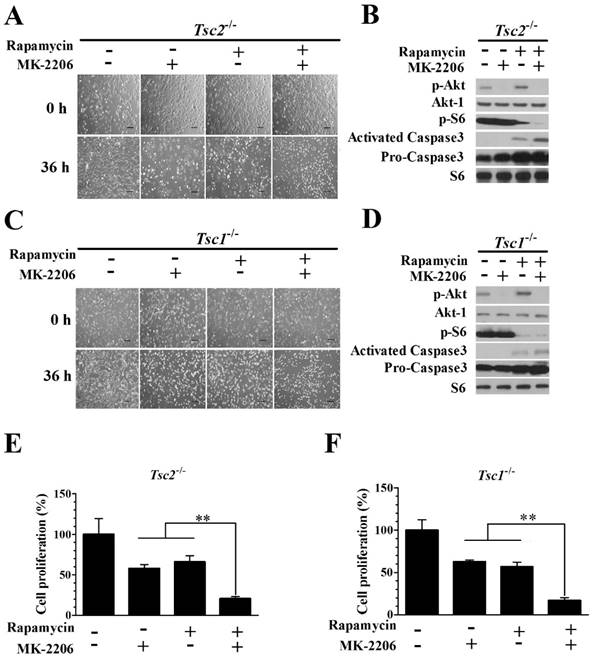
Combining rapamycin with MK-2206 strongly inhibits proliferation and induces apoptosis in Tsc2-null or Tsc1-null MEF cells. (A, C) Images of Tsc2-null and Tsc1-null MEF cells were taken after treatment with or without rapamycin (2.0 nM) or MK-2206 (100 nM) for 36 h. (B, D) Tsc2- and Tsc1-null MEF cells were incubated with or without MK-2206 (100 nM) for 36 h after pre-treatment with 2.0 nM rapamycin for 6 h. Cell lysates were subjected to immunoblotting with the indicated antibodies. Phospho-S6 (p-S6) is a downstream indicator of mTORC1 activity. S6 was used as a loading control. (E, F) The proliferation of Tsc2-and Tsc1-null MEF cells treated with or without 2.0 nM rapamycin or 100 nM MK-2206 was examined by MTT assay. A schematic of MK-2206 and rapamycin-treated cell proliferation compared with untreated cells after 36 h (**denotes P<0.01 or significant differences between treatment groups and DMSO control groups).
Discussion
Loss-of-function mutations of the TSC1 or TSC2 genes cause the development of TSC and hyperactivation of mTORC1 is the primary change of TSC, Tsc1- or Tsc2-null MEF cells and control cells are widely used as cell models in the research of TSC and mTORC1 signaling [25-27]. Analyzed Tsc1- and Tsc2-null MEF cells, we found that cells lacking Tsc1 or Tsc2 showed a decreased sensitivity to MK-2206, while ectopic expression of myrAkt-1 restored MK-2206 sensitivity in Tsc1 or Tsc2-deficient MEF cells. Furthermore, an enhanced anti-cell proliferation effect was found in cells treated with a combination of MK-2206 and rapamycin via apoptosis in vitro. The in vivo antitumor growth effect was also increased in MEFs treated with a combination of MK-2206 with rapamycin. Therefore, the results of our study suggest that co-administration of MK-2206 and rapamycin may represent a novel strategy for the treatment of TSC and other related tumors.
MK-2206 showed great inhibited effect on tumor cell growth in many types of cancers [23, 28, 29]. In a phase I study of solid tumors, MK-2206 have demonstrated anti-proliferative activity as a single agent or in combination with other agents [30]. However, to our knowledge, there is no report about whether MK-2206 could be used for the treatment of TSC. In the current study, we evaluated the inhibitory effects of MK-2206 on the proliferation of Tsc1- or Tsc2-null MEF cells. Unexpectedly, Tsc1- or Tsc2-null MEF cells showed greater tolerance than the control cells in response to MK-2206 treatment. This result indicated that MK-2206 alone is not an ideal drug for TSC treatment. Since mTORC1 activation (caused by the loss of the TSC1/TSC2 complex) leads to a dramatic downregulation of Akt activity and MK-2206 acts as a specific Akt inhibitor, we speculated that downregulated Akt activity was responsible for the loss of MK-2206 sensitivity in Tsc1- or Tsc2-null MEF cells, as compared to control cells. Subsequent experiments of restoring Akt activity by overexpression of a constitutively activated Akt-1 in Tsc1- or Tsc2-deficient MEF cells confirmed this hypothesis. It is likely that, besides TSC, MK-2206 is not suitable for the treatment of cancers in which Akt activity is inhibited.
Aberrant activation of mTORC1 is considered to be the main reason of TSC tumor formation. Thus, rapamycin and its analogues have been suggested to be potential drugs for the treatment of TSC and other related cancers [31, 32]. For example, everolimus, a rapamycin analog, has recently been approved for treatment of subependymal giant cell astrocytomas (SEGA) and angiomyolipomas (AML) growth in TSC patients [33]. There has been similar approval for treatment of other TSC-related tumors with rapamycin analogs, such as LAM [34] and angiofibromas [35]. Although everolimus have proven efficacy in decreasing tumor growth for SEGA and AML in TSC patients, a potential limitation is the ill-defined duration of therapy. Cessation of treatment seems to result in tumor regrowth indicating that mTOR inhibitors alone are not ideal cures for TSC tumors. One explanation of this limitation is that rapamycin activates Akt. On the other hand, as an immunosuppressor, significant side effects may occur with rapamycin, including chronic immunosuppression and associated opportunistic infections [36-38]. Reducing cytotoxicity and increasing treatment efficacy by rational combination of different agents is a commonly used strategy for tumor treatment. Given that rapamycin treatment leads to activation of Akt, it is reasonable to assume that combined administration of rapamycin with Akt inhibitors could help to eliminate the unwanted effects caused by up-regulated Akt activity and improve the antitumor effect of rapamycin in TSC treatment. Strikingly, we observed a strong inhibitory effect on the proliferation of Tsc1- or Tsc2-null MEF cells in vitro and in vivo after combined treatment with rapamycin and MK-2206. As a mechanism, we found that MK-2206 eliminated rapamycin-induced Akt up-regulation and enhanced rapamycin-triggered apoptosis.
In conclusion, our data provide in vitro and preclinical evidence for clinical trials of rapamycin in combination with MK-2206 to improve treatment efficacy in TSC.
Figure 4
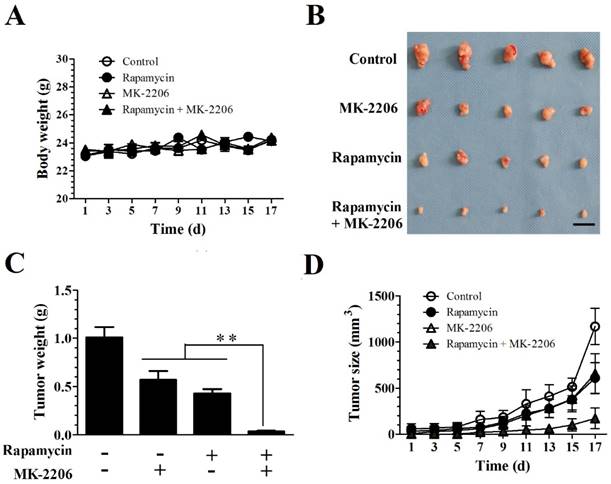
MK-2206 enhances the rapamycin-mediated inhibition of Tsc1-null MEF cells in a xenograft tumor model. Tsc1-null MEF cells were injected subcutaneously into nude mice to evaluate the effects of rapamycin and MK-2206 on: (A) body weight (B) tumor pictures (C) tumor weight (D) tumor volumes at different times (**denotes P<0.01 or significant differences between treatment groups and solvent control groups; N = 5 mice for each group).
Acknowledgements
This work was supported by the Natural Science Foundation of China (81372475, 81570034 and 81270081), the Outstanding Young Scholars Plan of Anhui Medical University (for X Zha) and National Undergraduate Training Programs for Innovation and Entrepreneurship (201410366020).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cheadle JP, Reeve MP, Sampson JR, Kwiatkowski DJ. Molecular genetic advances in tuberous sclerosis. Human genetics. 2000;107:97-114
2. Dabora SL, Jozwiak S, Franz DN, Roberts PS, Nieto A, Chung J. et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. American journal of human genetics. 2001;68:64-80
3. Sabatini DM. mTOR and cancer: insights into a complex relationship. Nature reviews Cancer. 2006;6:729-34
4. Orlova KA, Crino PB. The tuberous sclerosis complex. Annals of the New York Academy of Sciences. 2010;1184:87-105
5. Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. The New England journal of medicine. 2006;355:1345-56
6. Zha X, Hu Z, Ji S, Jin F, Jiang K, Li C. et al. NFkappaB up-regulation of glucose transporter 3 is essential for hyperactive mammalian target of rapamycin-induced aerobic glycolysis and tumor growth. Cancer letters. 2015;359:97-106
7. van Slegtenhorst M, de Hoogt R, Hermans C, Nellist M, Janssen B, Verhoef S. et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277:805-8
8. European Chromosome 16 Tuberous Sclerosis C. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75:1305-15
9. Plank TL, Yeung RS, Henske EP. Hamartin, the product of the tuberous sclerosis 1 (TSC1) gene, interacts with tuberin and appears to be localized to cytoplasmic vesicles. Cancer research. 1998;58:4766-70
10. Li Y, Corradetti MN, Inoki K, Guan KL. TSC2: filling the GAP in the mTOR signaling pathway. Trends in biochemical sciences. 2004;29:32-8
11. Cai SL, Tee AR, Short JD, Bergeron JM, Kim J, Shen J. et al. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. The Journal of cell biology. 2006;173:279-89
12. Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes & development. 2003;17:1829-34
13. Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H. et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163-75
14. Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S. et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177-89
15. Zha X, Wang F, Wang Y, He S, Jing Y, Wu X. et al. Lactate dehydrogenase B is critical for hyperactive mTOR-mediated tumorigenesis. Cancer research. 2011;71:13-8
16. Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer cell. 2007;12:9-22
17. Zhang Z, Miao L, Wu X, Liu G, Peng Y, Xin X. et al. Carnosine Inhibits the Proliferation of Human Gastric Carcinoma Cells by Retarding Akt/mTOR/p70S6K Signaling. Journal of Cancer. 2014;5:382-9
18. Beugnet A, Wang X, Proud CG. Target of rapamycin (TOR)-signaling and RAIP motifs play distinct roles in the mammalian TOR-dependent phosphorylation of initiation factor 4E-binding protein 1. The Journal of biological chemistry. 2003;278:40717-22
19. Chan JA, Zhang H, Roberts PS, Jozwiak S, Wieslawa G, Lewin-Kowalik J. et al. Pathogenesis of tuberous sclerosis subependymal giant cell astrocytomas: biallelic inactivation of TSC1 or TSC2 leads to mTOR activation. Journal of neuropathology and experimental neurology. 2004;63:1236-42
20. Lindsley CW, Barnett SF, Yaroschak M, Bilodeau MT, Layton ME. Recent progress in the development of ATP-competitive and allosteric Akt kinase inhibitors. Current topics in medicinal chemistry. 2007;7:1349-63
21. Balasis ME, Forinash KD, Chen YA, Fulp WJ, Coppola D, Hamilton AD. et al. Combination of farnesyltransferase and Akt inhibitors is synergistic in breast cancer cells and causes significant breast tumor regression in ErbB2 transgenic mice. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:2852-62
22. Meng J, Dai B, Fang B, Bekele BN, Bornmann WG, Sun D. et al. Combination treatment with MEK and AKT inhibitors is more effective than each drug alone in human non-small cell lung cancer in vitro and in vivo. PloS one. 2010;5:e14124
23. Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K. et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Molecular cancer therapeutics. 2010;9:1956-67
24. Zhang H, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N. et al. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. The Journal of clinical investigation. 2003;112:1223-33
25. Zhang H, Bajraszewski N, Wu E, Wang H, Moseman AP, Dabora SL. et al. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. The Journal of clinical investigation. 2007;117:730-8
26. Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X. et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955-68
27. Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL. et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Molecular cell. 2010;39:171-83
28. Cheng Y, Zhang Y, Zhang L, Ren X, Huber-Keener KJ, Liu X. et al. MK-2206, a novel allosteric inhibitor of Akt, synergizes with gefitinib against malignant glioma via modulating both autophagy and apoptosis. Molecular cancer therapeutics. 2012;11:154-64
29. Liu R, Liu D, Trink E, Bojdani E, Ning G, Xing M. The Akt-specific inhibitor MK2206 selectively inhibits thyroid cancer cells harboring mutations that can activate the PI3K/Akt pathway. The Journal of clinical endocrinology and metabolism. 2011;96:E577-85
30. Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K. et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:4688-95
31. Franz DN. Everolimus in the treatment of subependymal giant cell astrocytomas, angiomyolipomas, and pulmonary and skin lesions associated with tuberous sclerosis complex. Biologics: targets & therapy. 2013;7:211-21
32. Yang G, Yang L, Yang X, Shi X, Wang J, Liu Y. et al. Efficacy and safety of a mammalian target of rapamycin inhibitor in pediatric patients with tuberous sclerosis complex: A systematic review and meta-analysis. Experimental and therapeutic medicine. 2015;9:626-30
33. Zemke D, Azhar S, Majid A. The mTOR pathway as a potential target for the development of therapies against neurological disease. Drug news & perspectives. 2007;20:495-9
34. Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM. et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. The New England journal of medicine. 2008;358:140-51
35. Haemel AK, O'Brian AL, Teng JM. Topical rapamycin: a novel approach to facial angiofibromas in tuberous sclerosis. Archives of dermatology. 2010;146:715-8
36. Boulay A, Zumstein-Mecker S, Stephan C, Beuvink I, Zilbermann F, Haller R. et al. Antitumor efficacy of intermittent treatment schedules with the rapamycin derivative RAD001 correlates with prolonged inactivation of ribosomal protein S6 kinase 1 in peripheral blood mononuclear cells. Cancer research. 2004;64:252-61
37. Woodrum C, Nobil A, Dabora SL. Comparison of three rapamycin dosing schedules in A/J Tsc2+/- mice and improved survival with angiogenesis inhibitor or asparaginase treatment in mice with subcutaneous tuberous sclerosis related tumors. Journal of translational medicine. 2010;8:14
38. Rivera VM, Squillace RM, Miller D, Berk L, Wardwell SD, Ning Y. et al. Ridaforolimus (AP23573; MK-8669), a potent mTOR inhibitor, has broad antitumor activity and can be optimally administered using intermittent dosing regimens. Molecular cancer therapeutics. 2011;10:1059-71
Author contact
![]() Corresponding authors: Prof. Guanghe Fei, Pulmonary Department, First Affiliated Hospital of Anhui Medical University, Hefei 230022, Anhui, China. Tel:+86-51-6292-2013, Fax: +86-551- 6363-5578. E-mail: guanghefeicom. Associate Prof. Xiaojun Zha, Department of Biochemistry & Molecular Biology, School of Basic Medicine, Anhui Medical University, Hefei 230032, Anhui, China. E-mail: zhaxiaojunedu.cn.
Corresponding authors: Prof. Guanghe Fei, Pulmonary Department, First Affiliated Hospital of Anhui Medical University, Hefei 230022, Anhui, China. Tel:+86-51-6292-2013, Fax: +86-551- 6363-5578. E-mail: guanghefeicom. Associate Prof. Xiaojun Zha, Department of Biochemistry & Molecular Biology, School of Basic Medicine, Anhui Medical University, Hefei 230032, Anhui, China. E-mail: zhaxiaojunedu.cn.
Citation styles
APA
Ji, S., Lin, W., Wang, L., Ni, Z., Jin, F., Zha, X., Fei, G. (2017). Combined Targeting of mTOR and Akt Using Rapamycin and MK-2206 in The Treatment of Tuberous Sclerosis Complex. Journal of Cancer, 8(4), 555-562. https://doi.org/10.7150/jca.17205.
ACS
Ji, S.; Lin, W.; Wang, L.; Ni, Z.; Jin, F.; Zha, X.; Fei, G. Combined Targeting of mTOR and Akt Using Rapamycin and MK-2206 in The Treatment of Tuberous Sclerosis Complex. J. Cancer 2017, 8 (4), 555-562. DOI: 10.7150/jca.17205.
NLM
Ji S, Lin W, Wang L, Ni Z, Jin F, Zha X, Fei G. Combined Targeting of mTOR and Akt Using Rapamycin and MK-2206 in The Treatment of Tuberous Sclerosis Complex. J Cancer 2017; 8(4):555-562. doi:10.7150/jca.17205. https://www.jcancer.org/v08p0555.htm
CSE
Ji S, Lin W, Wang L, Ni Z, Jin F, Zha X, Fei G. 2017. Combined Targeting of mTOR and Akt Using Rapamycin and MK-2206 in The Treatment of Tuberous Sclerosis Complex. J Cancer. 8(4):555-562.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.