Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(6):983-992. doi:10.7150/jca.18135 This issue Cite
Research Paper
Foretinib Enhances the Radiosensitivity in Esophageal Squamous Cell Carcinoma by Inhibiting Phosphorylation of c-Met
Guang-Zong Chen1*, Wang-Shu Dai1*, Hong-Cheng Zhu1*, Hong-Mei Song3, Xi Yang4, Yuan-Dong Wang1, Hua Min1, Qian Lu1, Shu Liu1, Xin-Chen Sun1 ![]() , Xiao-Ning Zeng2
, Xiao-Ning Zeng2 ![]()
1. Department of Radiation Oncology, the First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, China
2. Department of Respiratory & Critical Care Medicine, the First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, China
3. Department of Radiation Oncology, Lianyungang NO.2 People's Hospital Affiliated to Bengbu Medical College, Lianyungang 222000, China
4. Department of Radiation Oncology, the Cancer Hospital of Fudan University, Shanghai 200000, China
* These authors contributed equally to this work.
Received 2016-10-29; Accepted 2016-12-23; Published 2017-3-12
Citation:
Chen GZ, Dai WS, Zhu HC, Song HM, Yang X, Wang YD, Min H, Lu Q, Liu S, Sun XC, Zeng XN. Foretinib Enhances the Radiosensitivity in Esophageal Squamous Cell Carcinoma by Inhibiting Phosphorylation of c-Met. J Cancer 2017; 8(6):983-992. doi:10.7150/jca.18135. https://www.jcancer.org/v08p0983.htm
Other stylesAbstract
As a crucial event involved in the metastasis and relapse of esophageal cancer, c-Met overexpression has been considered as one of the culprits responsible for the failure in patients who received radiochemotherapy. Since c-Met has been confirmed to be pivotal for cell survival, proliferation and migration, little is known about its impact on the regulation of radiosensitivity in esophageal cancer. The present study investigated the radiosensitization effects of c-Met inhibitor foretinib in ECA-109 and TE-13 cell lines. Foretinib inhibited c-Met signaling in a dose-dependent manner resulting in decreases in the cell viability of ECA-109 and TE-13. Pretreatment with foretinib synergistically prompted cell apoptosis and G2/M arrest induced by irradiation. Moreover, decreases ability of DNA damage repair was also observed. In vivo studies confirmed that the combinatorial use of foretinib with irradiation significantly diminishes tumor burden compared to either treatment alone. The present findings implied a crucial role of c-Met in the modulation of radiosensitization in esophageal cancer, and foretinib increased the radiosensitivity in ECA-109 and TE-13 cells mainly via c-Met signaling, highlighting a novel profile of foretinib as a potential radiosensitizer for the treatment of esophageal cancer.
Keywords: Foretinib, Esophageal cancer, Radiosensitivity, c-Met
Introduction
Esophageal cancer is the eighth most common malignancy and the sixth most common cause of death worldwide (1). While surgery represents the primary modality of treatment, the prognosis and survival for this disease remains poor. Radiotherapy has been a critical treatment option for those unresectable esophageal cancer patients. However, insensitive to radiation contributes to the major restrictive factor for patients occurring treatment failure and reccurence (2, 3). As such, improved modalities of treatment are urgently needed to improve the prognosis and survival for radioresistant esophageal cancer patients.
c-Met, a receptor tyrosine kinase (RTK) encoded by MET gene, mediates cell proliferation, survival, invasion (4, 5). Previous studies by Stommel et al. found that silencing c-Met by using c-Met small interfering RNAs (siRNAs) can attenuate tumor growth (6). Hepatocyte growth factor (HGF) is widely expressed in mesenchymal cells as a ligand of c-Met and aberrant HGF/c-Met axis is known to correlate with tumor growth, metastasis and reccurrence in many malignancies (7). HGF can induce autophosphorylation of c-Met and activate downstream signaling pathways, such as PI3K/Akt and MAPK/Erk pathways (8). c-Met is commonly overexpressed in advanced and metastatic esophageal cancer and plays a critical role in the development and progression of malignances (9). Overexpression of c-Met has been reported to decrease apoptosis and enhance the repair of ionizing radiation induced DNA double-strand breaks (DSBs) (10). High levels of c-Met expression are closely related to poor prognosis and survival (11). As such, inhibition of c-Met expression coupled with radiotherapy may represent a promising option for the treatment of radioresistant esophageal cancer.
Foretinib, an oral receptor tyrosine kinase inhibitor, was demonstrated antitumor activity via inhibiting the phosphorylation of c-Met induced by HGF (12). Foretinib has been reported to block cancer cell proliferation, metastasis, angiogenesis and, most importantly, significantly improve survival of esophageal cancer patients (13). c-Met positive malignances have been reported to be more sensitive to c-Met inhibitor and present better prognosis in several clinical and preclinical trials (14, 15).
In this study we evaluated the effect of foretinib as a radiosensitizer on esophageal squamous carcinoma cells (ESCC) in vitro and in vivo. Furthermore, the potential mechanisms of radiosensitization effect were also studied.
Materials and methods
Cell culture and reagents
Esophageal squamous cell lines ECA-109 and TE-13 were purchased from KeyGEN Biotechnology (Nanjing, China) and maintained in Dulbecco's Modified Eagle Medium (Gibco) supplemented with 10% fetal bovine serum (Gibco). Cells were cultured at 37°C in an environment of 5% carbon dioxide. Foretinib was obtained from Selleck Chemicals. The antibodies were as follows: GAPDH (Cell Signaling Technology), Akt (Cell Signaling Technology), p-Akt (Cell Signaling Technology), c-Met (Bioworld Technology), p-Met (Tyr 1234/1235) (Cell Signaling Technology), γH2AX (Cell Signaling Technology).
Cell viability assay
Cell viability of ECA-109 and TE-13 were assessed using the Cell Counting Kit 8 (CCK8) assays. Cells were plated at a density of 5×103 cells/well in 96-well plate and cultured with different concentrations of foretinib for 24h and 48h. 10% CCK8 (Obio Technology) was added to each well and the absorbance was measured at 490 nm.
Clonogenic assay
Cells were seeded into 6-well plates at varying cell densities and cultured with different concentrations of foretinib (0.1 and 0.5 μM). Then cells were irradiated at 0, 2, 4, 6, 8 Gy at 4.5 Gy/min and cultured for another 14 days. Cells were fixed with paraformaldehyde and stained with crystal violet respectively for 1h. Colonies composed of greater than 50 cells were counted.
Apoptosis assay
Cells were plated at a density of 1×105 cells/well in 6-well plate and treated with foretinib or IR after 24h. Annexin V/PI Apoptosis Kit (KeyGENE Biotechnology) was used to detect the apoptosis cells by flow cytometry (BD FACS Calibur).
Cell cycle assay
Cells were divided into one of the following groups: control, foretinib-treatment, IR and combination treatment, and fixed overnight with 75% ice-cold ethanol. Cells were stained with propidium iodide and cell cycle distribution was assessed by flow cytometry (BD FACS Calibur).
Immunofluorescence assay
Cells were seeded into confocal laser small dishes and harvested at 2, 8, 24h postirradiation with or without foretinib. Cells were subsequently fixed in 4% paraformaldehyde at 4℃ overnight and permeabilized in 0.1% Triton X-100 (Sigma) for 10 min. Thereafter cells were blocked with 5% bovine serum albumin (Gibco) for 1h and incubated with γH2AX antibody overnight at 4℃. Cells were washed with PBS for three times before incubating with an Alexa Fluor 594-conjugated secondary antibody (Jackson ImmunoRearch) for 1h. Cells were treated with DAPI (Beyotime Biotechnology) and visualized using confocal fluorescence microscopy (Leica).
Western blotting analysis
Cells were seeded into 6-well plates and divided into the following groups: control, foretinib treatment, IR treatment and combination treatment. Cells were lysed using lysis buffer with phenylmethylsulfonyl fluoride (PMSF) and phosphatase inhibitor on ice. The cells were then centrifuged for 15min at 14000 rpm and the supernatant was collected to a new tube. Protein concentrations were identified by BCA Protein Quatification Kit (Vazyme Biotechnology). Primary antibodies were as follows: Bax (Cell Signaling Technology), Bcl-2 (Cell Signaling Technology), Bcl-xl (Bioworld Technology), Cdc2 (Santa Cruz Biotechnology), p-Cdc2 (Santa Cruz Biotechnology), Cyclin B1 (Cell Signaling Technology).
Animal experiments
Animal experiments were approved by Ethics Committee of Nanjing Medical University. Six to eight week-old female BALB/C nude mice were supplied by Nanjing Medical University Animal Center. A cell suspension of 1×106 ECA-109 cells was subcutaneously injected into the right leg of nude mice. Nude mice in the foretinib treatment group were administrated with 60 mg/kg/day foretinib orally for 14 days when tumors volume reached 150 mm3. Nude mice of IR treatment and combination treatment group received additional 8 Gy IR on Day 7 after foretinib administration compared with foretinib treatment alone. Body weight and tumor volume were evaluated every three days and mice were sacrificed on day 18 after foretinib treatment.
Statistical analysis
Numerous data were presented as mean ± standard deviation (SD). The t tests and One-way ANOVA were used to demonstrate the difference between different groups. SPSS 19.0 software was performed to statistical analysis and p value less than 0.05 was considered statistically significant.
Results
Foretinib inhibits the proliferation in ECA-109 and TE-13 cells
CCK8 assays demonstrated that foretinib could inhibit the proliferation of ECA-109 and TE-13 cells in a time and dose-dependent manner. As shown in Fig.1A, increasing concentrations of foretinib ranged from 0 to 4 μM surpressed cell viability gradually. The 50% inhibitory concentrations (IC50) of foretinib were 1.67 and 1.95 μM, for ECA-109 and TE-13 cells, respectively.
Foretinib radiosensitizes ECA-109 and TE-13 cells
Clonogenic assays were performed to determine the radiosensitization effect of foretinib on ECA-109 and TE-13 cells. As shown in Fig.1B, foretinib treatment (0.1 and 0.5 μM) combined ionizing radiation significantly shifted the dose-survival curves compared with the control group. The sensitization enhancement ratio (SER) of ECA-109 and TE-13 reached 1.73 and 1.63 respectively when treated with the concentration of 0.5 μM. D0, Dq, SF2 and SER were calculated in Table 1 and these data demonstrated that foretinib significantly resulted in a radiosensitizaiton of ECA-109 and TE-13 cells in vitro.
Table 1
Radiosensitization effects of Foretinib on ESCC cells in vitro
| ECA-109 | D0 | Dq | SF2 | SER |
| Control | 2.65 | 2.85 | 0.72 | |
| Foretinib 0.1 μM | 1.87 | 1.76 | 0.56 | 1.42 |
| Foretinib 0.5 μM | 1.53 | 0.23 | 0.31 | 1.73 |
| TE-13 | D0 | Dq | SF2 | SER |
| Control | 2.53 | 2.48 | 0.70 | |
| Foretinib 0.1 μM | 2.19 | 0.82 | 0.51 | 1.16 |
| Foretinib 0.5 μM | 1.55 | 0.66 | 0.37 | 1.63 |
D0, mean lethal dose; Dq, quasithre-should dose; SF2, survival fraction(2Gy);
SER, sensitization enhancement ratio.
Foretinib inhibits the phosphorylation of c-Met and the downstream signaling pathway
We detected the inhibitory effects of foretinib on c-Met phosphorylation and downstream signaling pathway in ECA-109 and TE-13 cells. As the PI3K/Akt pathway represents the major downstream mediator of c-Met signaling, resulting in changes to proliferation and survival, we evaluated this pathway. The status of c-Met, p-Met, Akt, p-Akt were determined by Western blotting. As shown in Fig 1C, foretinib significantly blocked c-Met phosphorylation and downstream PI3K/Akt signaling pathway in ECA-109 and TE-13 cells. These data demonstrated that foretinib was an efficient inhibitor of c-Met and downstream signaling pathways.
Foretinib induces the apoptosis in ECA-109 and TE-13 cells
ESCC cells were divided into the following groups: control, foretinib, IR and combination treatment. We evaluated apoptosis by flow cytometry and found that the degree of apoptosis as greatest in the combination treatment group versus the levels observed following foretinib treatment or IR treatment alone (Fig.2B). The results demonstrated that foretinib significantly enhances apoptosis induced by ionizing radiation. The Bcl-2 family consists of anti-apoptosis protein Bcl-2, Bcl-xl and pro-apoptosis protein Bax. Bax plays a crucial role in activating the caspase pathway to induce apoptosis via mitochondrial mechanisms. As shown in Fig.2C, foretinib combined with irradiation resulted in upregulation of Bax and concomitant downregulation of Bcl-2 and Bcl-xl.
Foretinib increases the G2/M arrest in ECA-109 and TE-13 cells
Flow cytometry was used to evaluate the cell cycle distribution in ECA-109 and TE-13 cells. Cells pretreated with foretinib alone induced G2/M arrest from 16.7% to 25% in ECA-109 and from 17.58% to 26.81% in TE-13 cells compared with controls. Irradiated cells pretreated with foretinib induced G2/M arrest from 48.81% to 69.10% and from 50.10% to 65.62% in ECA-109 and TE-13 cells respectively. Thus foretinib redistributed the cell cycle and enhanced the G2/M arrest in ECA-109 and TE-13 cells (Fig.3B). Cdc2/Cyclin B1 complex is a key component in mitosis process through regulating G2/M transition. As shown in Fig.3C, foretinib induced G2/M arrest via modulating phosphorylation of Cdc2 and decreasing the expression of Cyclin B1 while irradiation induced G2/M arrest by increasing the expression of Cyclin B1 and phosphorylation of Cdc2. These data is thus supportive of our flow cytometry findings.
Fig 1
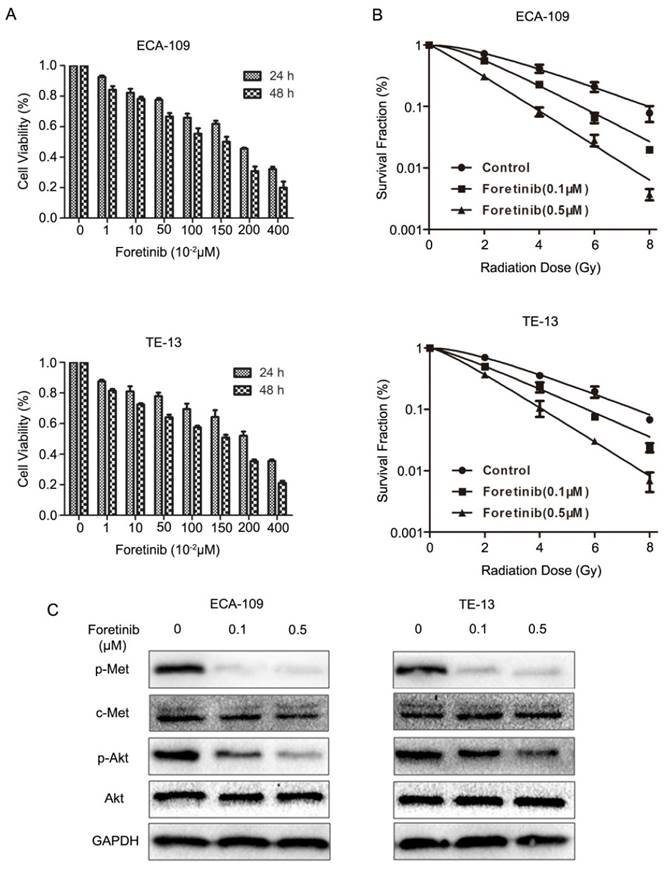
The effects of c-Met inhibitor foretinib on cell viability, survival, phosphorylation of c-Met and downstream PI3K/Akt pathway in ESCC cells. (A) Foretinib inhibited cell proliferation in a time and dose-dependent manner in ESCC cells. (B) The clonogenic survival of ESCC cells was suppressed significantly by foretinib at 0, 2, 4, 6, 8 Gy. (C) Western blotting results demonstrated that foretinib blocked the phosphorylation of c-Met and Akt obviously.
Fig 2
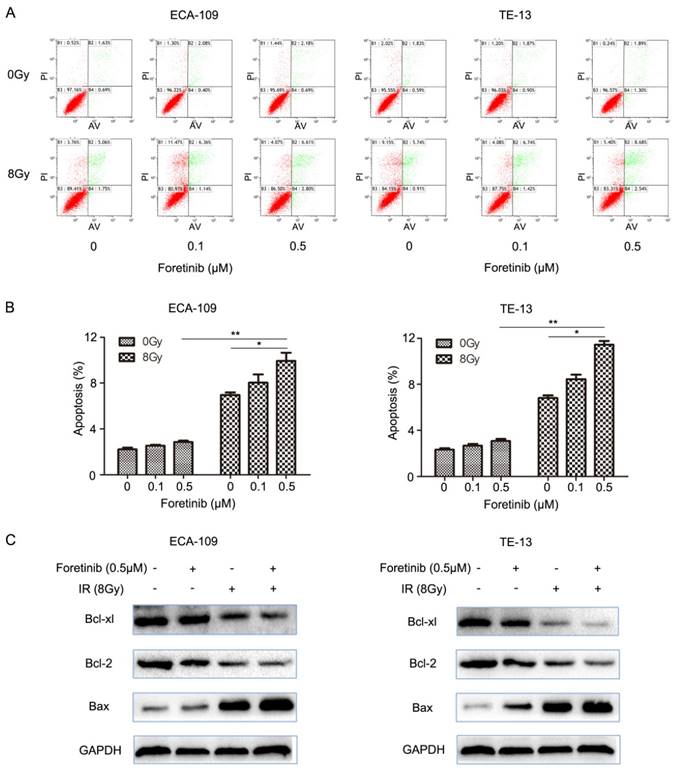
The effect of c-Met inhibitor foretinib on cell apoptosis in ESCC cells. (A) Flow cytometry was used to detect the apoptosis in ESCC cells treated with foretinib or irradiation. (B) Combination treatment increased the rate of apoptosis versus the use of foretinib or irradiation alone (*, P<0.05; **, P<0.01). (C) Western blotting results showed that foretinib resulted in improvement of Bax and degradation of Bcl-2 and Bcl-xl combined with irradiation.
Foretinib delays the repair of DNA double strand breaks following ionizing radiation
Immunofluorescence was performed to assess DNA double strand breaks repair in ECA-109 and TE-13 cells following irradiation. The average number of γH2AX foci per cell was calculated as a surrogate marker of DNA damage. We measured the number of γH2AX foci at 2, 8 and 24h postirradiation with or without foretinib pretreatment (0.5 μM). As shown in Fig.4, the average number of γH2AX foci in the foretinib pretreatment group was significantly higher than those observed in the control (DMSO) group. These data suggest that inhibition of c-Met phosphorylation results in a delay of DNA double strand breaks repair following irradiation.
Foretinib improves the radiosensitization in xenografts
As shown in Fig.5A, the combinatorial use of foretinib and IR significantly decreased tumor growth. Tumor volume increased at a much slower rate in mice who received combined treatment as compared with the control group (Fig.5B), while tumor weight was significantly lower in the combined group (Fig.5C). Relative tumor proliferation rate T/C (%) was measured to evaluate the anti-tumor activity. The T/C (%) of foretinib, IR and combination treatment respectively was 58.6%, 44.2% and 29% (Fig.5D), while tumor inhibition rates reached 38.5%, 53.4% and 67.2% in foretinib group, IR group and combination treatment group respectively. Furthermore, we calculated the doubling time and absolute growth delay in different treatment groups. As shown in Table 2, foretinib increased the doubling time from 5.7±0.8 days with IR alone to 13.7±1.4 days with combination group, resulting in an enhancement ratio of 3.8. These data demonstrate that foretinib enhanced the radioresponse in an ESCC xenograft model.
Table 2
Radiosensitization effects of Foretinib on ECA-109 xenograft model
| Treatment | Doubling time (Days) | Absolute Growth Delay (Days) | Normalized Growth Delay (Days) | Enhancement Factor |
|---|---|---|---|---|
| Control | 3.4±0.4 | |||
| Foretinib | 4.9±0.3 | 1.5±0.3 | ||
| IR | 5.7±0.8 | 2.3±0.8 | ||
| Foretinib+IR | 13.7±1.4 | 10.3±1.4 | 8.8±1.4 | 3.8(8.8/2.3) |
Absolute Growth Delay (Days), the doubling tumor time of treatment group minus that of control group;
Normalized Growth Delay (Days), the time of absolute growth delay of tumor in IR combined with foretinib group minus that of foretinib group.
Fig 3
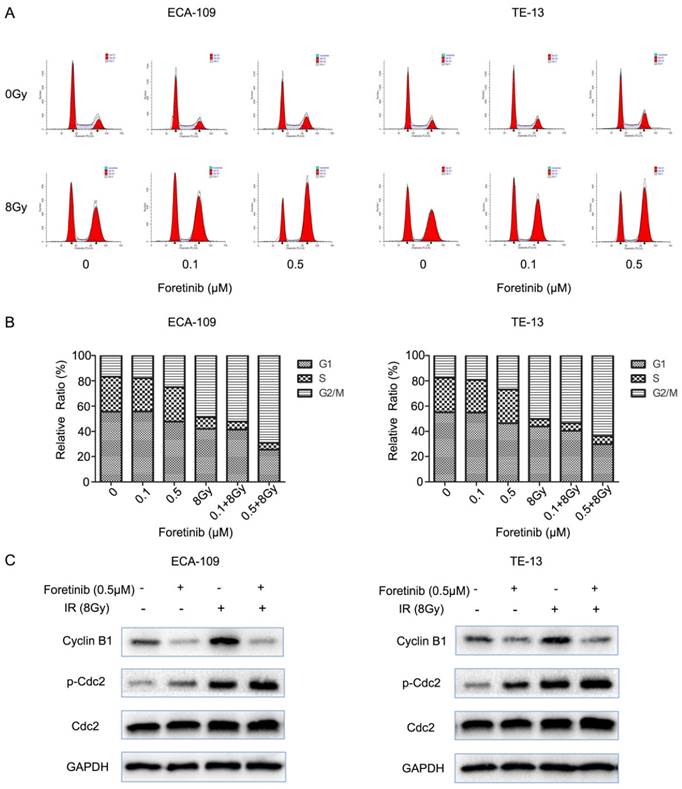
The effect of c-Met inhibitor foretinib on cell cycle in ESCC cells. (A) The cell cycle distribution of ESCC cells was examined by flow cytometry. (B) Foretinib increased the percentage of G2/M phase with increasing concentrations and enhanced G2/M arrest induced by irradiation. (C) Foretinib increased the expression of phosphorylated Cdc2 and decreased the expression of Cyclin B1. Irradiation improved the expression of phosphorylated Cdc2 and Cyclin B1.
Fig 4
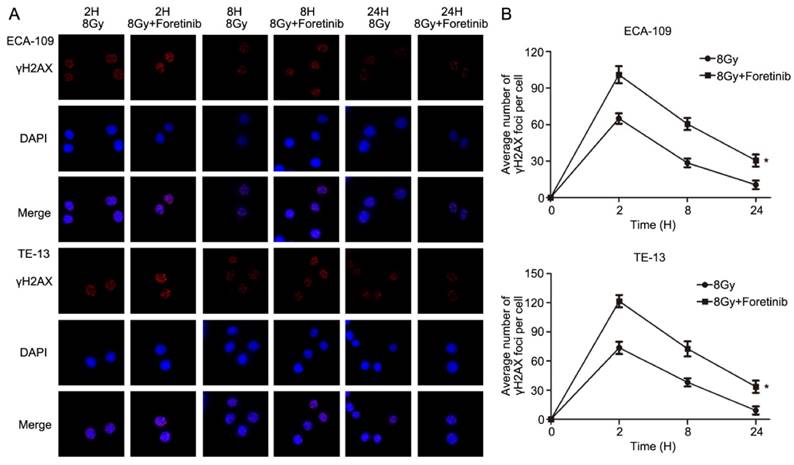
The effect of c-Met inhibitor foretinib on DNA damage repair in ESCC cells. (A) γH2AX foci was evaluated by immunofluorescence. (B) Foretinib treatment group significantly delayed the DNA damage repair compared with control group after irradiation at 2, 8, 24h (*, P<0.05).
Fig 5
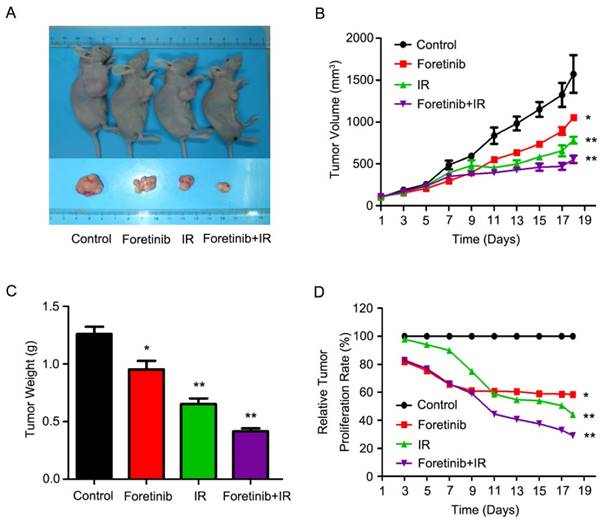
Foretinib enhances the radiosensitization of ESCC in vivo. ECA-109 xenograft-bearing female nude mice were divided into four groups: control, IR, foretinib, and a combination treatment group. Seven days following administration of foretinib, the mice were irradiated with a single fraction of 6 Gy X-rays. (A) Representative images of ECA-109 xenograft-bearing nude mice. (B) Measurements of tumor size. Data represent mean and SD (**, P<0.01). (C) Relative tumor proliferation rate T/C (%). T/C (%) was calculated according to the formula: TRTV/CRTV×100% (*, P<0.05; **, P<0.01). (D) Measurements of tumor weight. Data represent mean and SD (*, P<0.05; **, P<0.01).
Fig 6
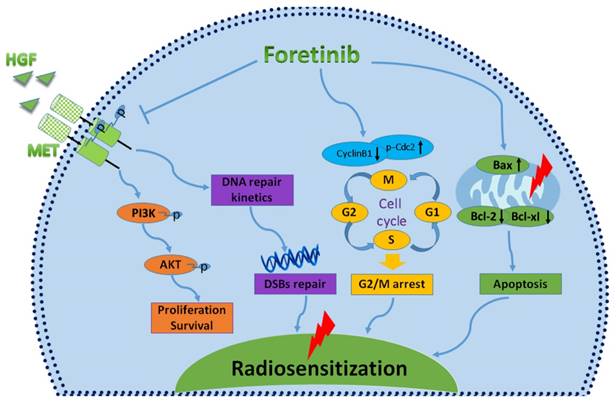
Characterizing the mechanisms of radiosensitization mediated by foretinib. Foretinib surpressed the proliferation and survival of ESCC through inhibiting the phosphorylation of c-Met. Foretinib enhanced the radioresponse via impairing the DNA repair ability of c-Met. Foretinib increased the radiosensitivity through inducing G2/M arrest and apoptosis postirradiation.
Discussion
Classical therapeutic strategies for the treatment of esophageal cancer include surgery coupled with radiotherapy and chemotherapy (16). However, chemo/radio-resistance often leads to treatment failure and poor prognosis in many patients (17). Fortunately, molecular therapies have rapidly developed in recent years, targeting aberrantly expressed receptor tyrosine kinases (18-20). HGF/c-Met signaling pathway plays a critical role in chemo- and radioresistance and c-Met has aroused considerable interest as a potential target due to its effects on tumor growth, invasion and metastasis (21, 22). Overexpression and dysregulation of c-Met has been verified tightly related to poor prognosis and survival in advanced esophageal cancer patients (23, 24). In addition, high levels of c-Met were reported to be associated with radiation resistance, activating downstream PI3K/Akt signaling pathway which was also linked to redioresistance (25, 26). Foretinib, a novel receptor tyrosine kinase inhibitor, was reported to significantly inhibit phosphorylation of c-Met and VEGFR-2 resulting in anti-tumor activity (27, 28). Notably, in recent Phase I/II clinical trials, studies have confirmed that foretinib improves the prognosis and the overall survival of patients with c-Met overexpression (29, 30). In the present study, we explored the radiosensitization effects of foretinib on ECA-109 and TE-13 cells and characterized molecular mechanisms of action. To our knowledge, this is the first time that foretinib has been coupled with radiotherapy and investigated as a possible therapeutic approach for the treatment of esophageal cancer.
As shown in Fig.1A, foretinib suppressed the cell viability of ECA-109 and TE-13 cells in a dose-dependent manner. As previously reported, phosphorylation of c-Met can activate PI3K/Akt pathway which has been shown to play a critical role in cell proliferation and survival. We observed that foretinib can inhibit the phosphorylation of c-Met and Akt in both ECA-109 and TE-13 cells (Fig.1C). Zillhardt et al. suggest that the inhibitory role of foretinib on cell growth is linked to changes in cell cycle distribution (31). They demonstrated that foretinib could suppress cell proliferation through G2/M arrest thereby inhibiting mitoses.
One mechanism by which foretinib enhanced radiosensitivity was related to changes in DNA damage repair following irradiation (32). DNA repair following double-stranded DNA breaks requires a variant histone protein called H2A.X. PI3K-like kinases like ATM, ATR and DNA-PK can induce phosphorylation of H2A.X at Ser139 site after DNA damage caused by ionizing radiation. As shown in Fig.4, foretinib treatment resulted in increased γH2AX foci compared with control group at 2, 8 and 24h following irradiation. These data demonstrated that foretinib efficiently delays the repair of DNA double strand breaks. Previous studies have suggested that c-Met may regulate DNA repair kinetics and attenuate DNA damage following irradiation. Li et al. demonstrated that high levels of c-Met were related to radioresistance, while small interfering RNA knockdown of c-MET enhanced the response to IR in non-small cell lung cell lines. We speculated that foretinib may exhibit its antitumor ability by inhibiting the repair of DNA double strand breaks regulated by c-Met (33). Apart from the role in regulating DNA repair, H2A.X is also required for checkpoint-mediated cell cycle arrest. Cells treated with foretinib exhibited more γH2AX foci and thus we hypothesized foretinib may induce cell cycle arrest.
As described previously, another mechanism by which foretinib enhanced radiosensitivity is associated with changes in cell cycle distribution. G2/M phase was reported to be most sensitive to radiation, and G2/M arrest is regulated by Cdc2/Cyclin B1 complex. Cdc2 regulates the entry of cells into mitosis, the phosphorylation of Cdc2 can contribute to inactivate Cdc2 and prevent cells mitosis. As such, dephosphorylation of Cdc2 at Thr14 and Tyr15 contributes to progression into mitosis. Cyclin B1 is a checkpoint regulatory protein and thought to modulate the translocation to nucleus at G2/M checkpoint and initiation of mitosis. The degradation of cyclin B1 is necessary for cells to progression out of mitosis (34). As shown in Fig.3C, foretinib could induce phosphorylation of Cdc2 and the degradation of Cyclin B1. Meanwhile, IR could result in the phosphorylation of Cdc2 and the accumulation of Cyclin B1 (35, 36). When combined foretinib with IR, more phosphorylation of Cdc2 and degradation of Cyclin B1 occured which ultimately led to a G2/M checkpoint arrest. To date, studies concerning cell cycle redistribution of c-Met inhibitors have demonstrated conflicting results. Huynh and his colleagues found that c-Met inhibitor foretinib blocked G2/M arrest via decreasing the expression of Cyclin B1 in hepatocellular carcinoma cells (37). Megiorni et al. found c-Met inhibitor crizotinib induced G2/M arrest in alveolar RMS cell lines via upregulation of Cyclin B1 and phosphorylation of Cdc2 (38). While Dai and his colleagues found that crizotinib caused G2/M arrest by increasing phosphorylation of Cdc2 and decreasing Cyclin B1 in primary effusion lymphoma cell lines (39). Yu represented c-Met inhibitor SU11274 radiosensitized prostate cell DU145 through abrogating G2/M arrest induced by irradiation (40).
The Bcl-2 family contains numerous evolutionarily conserved proteins that can be classified into three broad categories: pro-apoptotic effectors (Bax and Bak), pro-apoptotic BH3-only proteins; and anti-apoptotic proteins (Bcl-2, Bcl-xL, Bcl-w, and Mcl-1, A1/Bfl1). Bcl-2 homology (BH) domains play corresponding roles in modulating cell death and mitochondrial integrity. Bax, a pro-apoptotic protein, induces the initiation of caspase activated apoptosis pathway and mitochondrial cytochrome c release via changes in mitochondrial membrane permeability. Anti-apoptotic proteins Bcl-2 and Bcl-xl, suppress the pro-apoptosis activity and stabilize the mitochondria membrane integrity. Bax and Bcl-2 proteins are considered the potential mediators of cell death induced by ionizing radiation. Expression of Bax and degradation of Bcl-2 seem to be dependent on p53, whose consensus binding elements exist in the Bax promoter region (41). As shown in Fig.2C, foretinib combined with irradiation induced apoptosis by increasing expression of Bax and degrading Bcl-2 and Bcl-xl. Dai et al. demonstrated that c-Met inhibitor PF2341066 induced apoptosis through improving DNA damage and regulating DNA repair/damage-related proteins such as RRM2 and XRCC5.
Our studies using tumor-bearing xenografts demonstrated that the tumor volume and weight were decreased significantly in the combination treatment group as compared to use of foretinib or IR alone. These data suggest that foretinib could enhance the radiosensitivity of ESCC in vitro and in vivo.
Conclusions
In conclusion, foretinib enhanced the radioresponse of ECA-109 and TE-13 cells to IR through inhibiting phosphorylation of c-Met, inducing accumulation of G2/M phase and delaying DNA double strand breaks repair (Fig.6). This study highlights the promising therapeutic potential of targeting c-Met in patients with chemoradioresistant esophageal cancer patients. Further studies are needed to further characterize the radiosensitive mechanisms of foretinib in esophageal cancer.
Acknowledgements
This work was supported by the Natural Science Foundation of China (No. 81272504, 81472809, 81502653), Innovation Team [No. LJ201123 (EH11)], Jiangsu Provincial Science and Technology Projects [BK2011854 (DA11)], A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) (JX10231801), and grants from Key Academic Discipline of Jiangsu Province “Medical Aspects of Specific Environments”.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ajani JA, D'Amico TA, Almhanna K. et al. Esophageal and esophagogastric junction cancers, version 1.2015. J Natl Compr Canc Netw. 2015;13:194-227
2. Song M, He G, Wang Y. et al. Lentivirus-mediated Knockdown of HDAC1 Uncovers Its Role in Esophageal Cancer Metastasis and Chemosensitivity. J CANCER. 2016;7:1694-700
3. Yang H, Wu L, Ke S. et al. Downregulation of Ubiquitin-conjugating Enzyme UBE2D3 Promotes Telomere Maintenance and Radioresistance of Eca-109 Human Esophageal Carcinoma Cells. J CANCER. 2016;7:1152-62
4. Kentsis A, Reed C, Rice KL. et al. Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. NAT MED. 2012;18:1118-22
5. Xie Z, Lee YH, Boeke M. et al. MET Inhibition in Clear Cell Renal Cell Carcinoma. J CANCER. 2016;7:1205-14
6. Stommel JM, Kimmelman AC, Ying H. et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. SCIENCE. 2007;318:287-90
7. Posadas EM, Figlin RA. Understanding the role of MET kinase in cancer therapy. J CLIN ONCOL. 2013;31:169-70
8. Giordano S, Columbano A. Met as a therapeutic target in HCC: facts and hopes. J HEPATOL. 2014;60:442-52
9. Qian F, Engst S, Yamaguchi K. et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. CANCER RES. 2009;69:8009-16
10. Bhardwaj V, Zhan Y, Cortez MA. et al. C-Met inhibitor MK-8003 radiosensitizes c-Met-expressing non-small-cell lung cancer cells with radiation-induced c-Met-expression. J THORAC ONCOL. 2012;7:1211-7
11. Lennerz JK, Kwak EL, Ackerman A. et al. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J CLIN ONCOL. 2011;29:4803-10
12. Kataoka Y, Mukohara T, Tomioka H. et al. Foretinib (GSK1363089), a multi-kinase inhibitor of MET and VEGFRs, inhibits growth of gastric cancer cell lines by blocking inter-receptor tyrosine kinase networks. Invest New Drugs. 2012;30:1352-60
13. Eder JP, Shapiro GI, Appleman LJ. et al. A phase I study of foretinib, a multi-targeted inhibitor of c-Met and vascular endothelial growth factor receptor 2. CLIN CANCER RES. 2010;16:3507-16
14. Li B, Torossian A, Sun Y. et al. Higher levels of c-Met expression and phosphorylation identify cell lines with increased sensitivity to AMG-458, a novel selective c-Met inhibitor with radiosensitizing effects. Int J Radiat Oncol Biol Phys. 2012;84:e525-31
15. Toiyama Y, Yasuda H, Saigusa S. et al. Co-expression of hepatocyte growth factor and c-Met predicts peritoneal dissemination established by autocrine hepatocyte growth factor/c-Met signaling in gastric cancer. INT J CANCER. 2012;130:2912-21
16. Peter S. N. van Rossum1, Cai Xu1, David V. Fried, et al. The emerging field of radiomics in esophageal cancer: current evidence and future potential. Transl Cancer Res. 2016;5(4):410-423
17. Heynen GJ, Fonfara A, Bernards R. Resistance to targeted cancer drugs through hepatocyte growth factor signaling. CELL CYCLE. 2014;13:3808-17
18. Tumati V, Kumar S, Yu L. et al. Effect of PF-02341066 and radiation on non-small cell lung cancer cells. ONCOL REP. 2013;29:1094-100
19. Zhuang HQ, Bo QF, Yuan ZY. et al. The different radiosensitivity when combining erlotinib with radiation at different administration schedules might be related to activity variations in c-MET-PI3K-AKT signal transduction. Onco Targets Ther. 2013;6:603-8
20. Knubel KH, Pernu BM, Sufit A. et al. MerTK inhibition is a novel therapeutic approach for glioblastoma multiforme. ONCOTARGET. 2014;5:1338-51
21. Joffre C, Barrow R, Menard L. et al. A direct role for Met endocytosis in tumorigenesis. NAT CELL BIOL. 2011;13:827-37
22. You H, Ding W, Dang H. et al. c-Met represents a potential therapeutic target for personalized treatment in hepatocellular carcinoma. HEPATOLOGY. 2011;54:879-89
23. Ponzo MG, Lesurf R, Petkiewicz S. et al. Met induces mammary tumors with diverse histologies and is associated with poor outcome and human basal breast cancer. Proc Natl Acad Sci U S A. 2009;106:12903-8
24. Elaine Shum, Balazs Halmos. Skipping the line: bringing MET exon 14 skipping mutations to the forefront of targeted therapy. Transl Cancer Res. 2016;5(2):110-116
25. Kaldenbach M, Giebeler A, Tschaharganeh DF. et al. Hepatocyte growth factor/c-Met signalling is important for the selection of transplanted hepatocytes. GUT. 2012;61:1209-18
26. Ian Wong, Simon Law. The CROSS road in neoadjuvant therapy for esophageal cancer: long-term results of CROSS trial. Transl Cancer Res. 2016
27. Logan TF. Foretinib (XL880): c-MET inhibitor with activity in papillary renal cell cancer. CURR ONCOL REP. 2013;15:83-90
28. Faria CC, Golbourn BJ, Dubuc AM. et al. Foretinib is effective therapy for metastatic sonic hedgehog medulloblastoma. CANCER RES. 2015;75:134-46
29. Shah MA, Wainberg ZA, Catenacci DV. et al. Phase II study evaluating 2 dosing schedules of oral foretinib (GSK1363089), cMET/VEGFR2 inhibitor, in patients with metastatic gastric cancer. PLOS ONE. 2013;8:e54014
30. Choueiri TK, Vaishampayan U, Rosenberg JE. et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J CLIN ONCOL. 2013;31:181-6
31. Zillhardt M, Park SM, Romero IL. et al. Foretinib (GSK1363089), an orally available multikinase inhibitor of c-Met and VEGFR-2, blocks proliferation, induces anoikis, and impairs ovarian cancer metastasis. CLIN CANCER RES. 2011;17:4042-51
32. Yang H, Lee HW, Kim Y. et al. Radiosensitization of brain metastasis by targeting c-MET. LAB INVEST. 2013;93:344-53
33. Welsh JW, Mahadevan D, Ellsworth R. et al. The c-Met receptor tyrosine kinase inhibitor MP470 radiosensitizes glioblastoma cells. RADIAT ONCOL. 2009;4:69
34. Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys. 2004;59:928-42
35. Qin Q, Cheng H, Lu J. et al. Small-molecule survivin inhibitor YM155 enhances radiosensitization in esophageal squamous cell carcinoma by the abrogation of G2 checkpoint and suppression of homologous recombination repair. J HEMATOL ONCOL. 2014;7:62
36. Dufies M, Jacquel A, Robert G. et al. Mechanism of action of the multikinase inhibitor Foretinib. CELL CYCLE. 2011;10:4138-48
37. Huynh H, Ong R, Soo KC. Foretinib demonstrates anti-tumor activity and improves overall survival in preclinical models of hepatocellular carcinoma. ANGIOGENESIS. 2012;15:59-70
38. Megiorni F, McDowell HP, Camero S. et al. Crizotinib-induced antitumour activity in human alveolar rhabdomyosarcoma cells is not solely dependent on ALK and MET inhibition. J Exp Clin Cancer Res. 2015;34:112
39. Dai L, Trillo-Tinoco J, Cao Y. et al. Targeting HGF/c-MET induces cell cycle arrest, DNA damage, and apoptosis for primary effusion lymphoma. BLOOD. 2015;126:2821-31
40. Yu H, Li X, Sun S. et al. c-Met inhibitor SU11274 enhances the response of the prostate cancer cell line DU145 to ionizing radiation. Biochem Biophys Res Commun. 2012;427:659-65
41. Zhou L, Yuan R, Serggio L. Molecular mechanisms of irradiation-induced apoptosis. Front Biosci. 2003;8:d9-19
Author contact
![]() Corresponding authors: Xiaoning Zeng, Department of Respiratory & Critical Care Medicine, the First Affiliated Hospital of Nanjing Medical University, 300 Guanzhou Road, Nanjing 210029, China; zeng_xiao_ningcom. Xinchen Sun, Department of Radiation Oncology, the First Affiliated Hospital of Nanjing Medical University, 300 Guanzhou Road, Nanjing 210029, China; sunxinchen2012com.
Corresponding authors: Xiaoning Zeng, Department of Respiratory & Critical Care Medicine, the First Affiliated Hospital of Nanjing Medical University, 300 Guanzhou Road, Nanjing 210029, China; zeng_xiao_ningcom. Xinchen Sun, Department of Radiation Oncology, the First Affiliated Hospital of Nanjing Medical University, 300 Guanzhou Road, Nanjing 210029, China; sunxinchen2012com.
Citation styles
APA
Chen, G.Z., Dai, W.S., Zhu, H.C., Song, H.M., Yang, X., Wang, Y.D., Min, H., Lu, Q., Liu, S., Sun, X.C., Zeng, X.N. (2017). Foretinib Enhances the Radiosensitivity in Esophageal Squamous Cell Carcinoma by Inhibiting Phosphorylation of c-Met. Journal of Cancer, 8(6), 983-992. https://doi.org/10.7150/jca.18135.
ACS
Chen, G.Z.; Dai, W.S.; Zhu, H.C.; Song, H.M.; Yang, X.; Wang, Y.D.; Min, H.; Lu, Q.; Liu, S.; Sun, X.C.; Zeng, X.N. Foretinib Enhances the Radiosensitivity in Esophageal Squamous Cell Carcinoma by Inhibiting Phosphorylation of c-Met. J. Cancer 2017, 8 (6), 983-992. DOI: 10.7150/jca.18135.
NLM
Chen GZ, Dai WS, Zhu HC, Song HM, Yang X, Wang YD, Min H, Lu Q, Liu S, Sun XC, Zeng XN. Foretinib Enhances the Radiosensitivity in Esophageal Squamous Cell Carcinoma by Inhibiting Phosphorylation of c-Met. J Cancer 2017; 8(6):983-992. doi:10.7150/jca.18135. https://www.jcancer.org/v08p0983.htm
CSE
Chen GZ, Dai WS, Zhu HC, Song HM, Yang X, Wang YD, Min H, Lu Q, Liu S, Sun XC, Zeng XN. 2017. Foretinib Enhances the Radiosensitivity in Esophageal Squamous Cell Carcinoma by Inhibiting Phosphorylation of c-Met. J Cancer. 8(6):983-992.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.