Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(9):1690-1703. doi:10.7150/jca.18681 This issue Cite
Review
The Multi-Purpose Tool of Tumor Immunotherapy: Gene-Engineered T Cells
Zeming Mo, Peixin Du, Guoping Wang, Yongsheng Wang ![]()
Department of Thoracic Oncology, State Key Laboratory of Biotherapy and Cancer center, West China Hospital, Sichuan University and Collaborative Innovation Center, Chengdu, Sichuan, China 610041.
Received 2016-12-9; Accepted 2017-2-27; Published 2017-6-23
Citation:
Mo Z, Du P, Wang G, Wang Y. The Multi-Purpose Tool of Tumor Immunotherapy: Gene-Engineered T Cells. J Cancer 2017; 8(9):1690-1703. doi:10.7150/jca.18681. https://www.jcancer.org/v08p1690.htm
Other stylesAbstract
A detailed summary of the published clinical trials of chimeric antigen receptor T cells (CAR-T) and TCR-transduced T cells (TCR-T) was constructed to understand the development trend of adoptive T cell therapy (ACT). In contrast to TCR-T, the number of CAR-T clinical trials has increased dramatically in China in the last three years. The ACT seems to be very prosperous. But, the multidimensional interaction of tumor, tumor associated antigen (TAA) and normal tissue exacerbates the uncontrolled outcome of T cells gene therapy. It reminds us the importance that optimizing treatment security to prevent the fatal serious adverse events. How to balance the safety and effectiveness of the ACT? At least six measures can potentially optimize the safety of ACT. At the same time, with the application of gene editing techniques, more endogenous receptors are disrupted while more exogenous receptors are expressed on T cells. As a multi-purpose tool of tumor immunotherapy, gene-engineered T cells (GE-T) have been given different functional weapons. A network which is likely to link radiation therapy, tumor vaccines, CAR-T and TCR-T is being built. Moreover, more and more evidences indicated that the combination of the ACT and other therapies would further enhance the anti-tumor capacity of the GE-T.
Keywords: Adoptive T cell therapy, Tumor immunotherapy, Gene-engineered T cell, Tumor associated antigen, Viral vectors and non-viral vectors.
Introduction
The adoptive T cell therapy (ACT) is that T cells are genetically modified to express a chimeric antigen receptor (CAR) or T-cell receptor (TCR) has obtained impressive results in treating multiple types of tumors, such as hematologic malignancies, sarcoma and melanoma, etc. The subjects are conducted a conditioning regimen of lymphodepletion, following the ACT [1-3]. The classical structure of CAR is built with a intracellular costimulatory domain, a intracellular CD3ζ and a chimera of single chain fragment (scFv) of antibodies that can directly target tumor associated antigens (TAA) in a major histocompatibility complex (MHC)-independent manner [4]. Analogously, the α chain and β chain of targeting TAA-specific peptide are concatenated as the activation domain of the TCR-transduced T cells (TCR-T) [5]. The activation of TCR requires the participation of MHC system-dependent peptides-presenting processes. In theory, around 6 × 206-7 and up to 12 × 2010 different peptides can be presented by MHC I and MHC II, respectively [6]. Therefore, based on the diversity of TAA-peptides, TCR α chain and TCR β chain are diverse in fine structure regulation when they identify and bind these peptides in a MHC-dependent manner.
CD19 is expressed on most B malignant cells malignancies, pro-B cells and mature B cells but not on other cell types, which restricts the “on target, off tumor” toxicity when efficient lymphodepletion chemotherapy is given [7]. The CAR-T immunotherapy has induced high remission rates of patients with refractory CD19+ B-cell hematologic malignancies. Excitingly, targeting NY-ESO-1, a cancer germline antigen (CGA) located on the X chromosome [8], has also showed encouraging results in some TCR-T clinical trials. Multiple successful results of CD19-CAR-T and NE-ESO-1-specific TCR-T clinical trials are summarized in Table 1. These results have greatly stimulated the enthusiasm for the exploration of adoptive cell therapy in tumors. Simultaneously, with the amazing achievements of immune checkpoint inhibitors in clinical trials including lung cancer, melanoma and renal cell carcinoma, the trend of tumor immunotherapy is almost inevitable [9]. Recently, the indispensable value of T cells in anti-tumor effect is further emphasized since FDA approved anti-PD-1 antibody pembrolizumab in metastatic NSCLC for first-line treatment of patients whose tumors have high PD-L1 expression [10]. But, immune checkpoint inhibitors are not perfect. Combination immune checkpoint blockade by ipilimumab and nivolumab, two patients with melanoma developed the fatal myocarditis [11]. A variety of factors will influence the resistance mechanisms to immune-checkpoint blockade, such as genetics, age, HLA type and background chronic infections, et al. [12]. As a multi-purpose tool of tumor immunotherapy, the CAR-T which can produce micro-pharmacies also offers the amazing expectations of anticancer application [13]. Thus, how to confer T cells with more powerful anti-tumor ability? It is becoming a hot research area. Especially, The GE-T plays one of the most important roles. As gene transfer tools, more accurate and safer viral vectors have been used for editing T cell functions, increasingly. Besides, these tools make it possible to finely edit the structure of TAA-special CAR or TCR. In particular, the exploration of GE-T immunotherapy is worth noting in both the United States and China.
The number of CAR-T clinical trials has exploded, particularly in China
Because of the breakthroughs in CAR-T and TCR-T tumor immunotherapy from some preclinical studies, multiple research centers in several countries have conducted hundreds related clinical trials. The development of new antibody of targeting TAA is rare in recent years. So, the types of scFv are relatively scarce, as well. In addition, some cDNAs of scFv have been published. Therefore, the CAR-T manufacturing technology enables rapid replication and propagation. It is easy to find that the number of CAR-T clinical trials is rapidly increasing, especially in China. In 2016, at least twenty-five CAR-T clinical trials have been initiated in China. Comparing in China, the number of newly initiated, published CAR-T clinical trials is maintained in a stable level in the United States. Of course, perhaps the difference in health care policy between China and the United States also was contributed to the divergence of the clinical trials trends. Meanwhile, Israel, Japan, Sweden, United Kingdom, Australia and other countries also have been carried out CAR-T clinical trials in recent years (Figure 1A). When accounted for 40% of the total numbers, CD19-targeted CAR-T is an undoubtedly star product in the history of CAR-T. In addition, as a TAA derived from solid tumors, mesothelin-targeted CAR-T had obtained some encouraging results in pre-clinical studies, especially in regional delivery therapy [14]. At least seven clinical trials of mesothelin-special CAR-T have been or are being carried out at present. Amazingly, a phase I clinical results illustrated that utilizing mesothelin-targeted mRNA-CAR-T cells gene therapy was safe and effective in patients with advanced solid malignancies [15].
The CAR-T targeted antigen spectrum is constantly enriched, including hematologic malignancies TAA, such as CD30, CD20, CD22, ROR1, CD138, BCMA, CD70, LeY [16-23], and also including solid malignancies TAA, such as GPC3, HER2, GD2, EGFR variant III (EGFR vIII), EGFR, CEA, PSMA, FRα, EPCAM, MUC1, ROR1, MUCI16eto, VEGFR2, CD171, PSCA and EphA2, etc (Figure 1B) [24-38].
The clinical trials of TCR-T, the United States outshines others
In contrast to CAR-T, The HLA restriction is one of the most marked characteristics for TCR-T gene therapy. The characteristic increased the difficulty of patients enrolled in the TCR-T clinical trials. For example, because the number of enrollment patients did not meet the expected requirement, a clinical trial was terminated (ClinicalTrials.gov Identifier: NCT00706992). HLA-A*02:01 is a dominant HLA-matched subtype, more than 77% (37/48) of TCR-T clinical trial's inclusion criteria contained HLA-A*02:01 positive. In addition, some TCR-T clinical trials of targeting NY-ESO-1 and melanoma differentiation antigens MART-1 were terminated, according to low accrual (ClinicalTrials.gov Identifier: NCT00509496, NCT00610311, NCT00612222 and NCT02062359). These terminated trials indicated that it is difficult to find and construct enough effective TCR-T. Peptides derived from the same antigen are very diverse in amino acids and structures. Even from targeting the same TAA, the different peptide/MHC-redirected TCR-T may exhibit different treatment outcomes and adverse effects. For example, two different MART-1-special TCR-Ts, DMF5 TCR-T(against gp100:154-162 epitope) and DMF4 TCR-T(against the gp100:209-217 epitope), which had a significant difference in the outcome of treatment [39, 40]. Therefore, the threshold of TCR-T tumor immunotherapy is higher than CAR-T tumor immunotherapy. The United States has occupied an absolute advantage in the number and the quality of TCR-T clinical trials (Figure 2 A-C).
Table 1
Some successful clinical trials of CD19-special CAR-T and NE-ESO-1-special TCR-T cancer immunotherapy
| Institution | CT.GI | TAA | transfer tool | scFv or TCR | Patient populations | Lymphodepleting regimens | Infused cells dose | Responses of treatment |
|---|---|---|---|---|---|---|---|---|
| NCI | NCT01593696 | CD19 | Retrovirus | FMC63 | child and young adult: ALL; n=21 | Cy 900 mg/ m2 ×1+ Flu 25 mg/ m2×3d | 1 ×106 (n=15) vs 3 × 106 (n=4) CAR-T cells/kg | CR: 14/20 (MRD-in 12/14); LFS: 79% at 4.8 mo (MRD- CR patients); OS: 52% at 7.8 mo (all) |
| FHCRC | NCT01865617 | CD19 | Lentivirus | FMC63 | adult: NHL; n=32 | Cy 2-4 g/m2×1 vs Cy 2-4 g/m2 ×1+etoposide 100 to 200 mg/m2×3d vs Cy 60 mg/m2 ×1+flu 25 mg/m2×3 or ×5d | 1 × 105(n=5) vs 1 × 106(n=18) vs 1 ×107(n=7) CAR-T cells/kg | Cy/Flu:50% CR (9/18), 72% ORR (13/18) VS Cy or Cy/E:8% CR (1/12), 50% ORR (6/12) |
| CHP HTUP | NCT01626495 NCT01029366 | CD19 | Lentivirus | FMC63 | child and young adult: ALL; n=30 | Cy/VP: Eto100mg/ m2 x2+Cy 440 mg/ m2 x2d Cy/Flu: Cy 50 mg/m2×2+flu 30 mg/ m2×4d or CVAD-B or CVAD-A | 0.76×106 to 20.6×106 CAR-T cells/kg | CR:90% (27/30); event-free survival rate:67% at 6 mo; OS:78% at 6 mo |
| HFCRC | NCT01865617 | CD19 | Lentivirus | FMC63 | adult: ALL; n=30 | Cy 2-4 g/ m2×1 (n=11) vs Cy 2-3 g/ m2 ×1 +etoposide 100 mg/ m2×3d (n=2)vs Cy 60 mg/ m2 ×1+flu 25 mg/ m2×3 or×5d (Cy/Flu)(n=17) | 1 × 105(n=13) vs 1 ×106(n=15) vs 1 ×107(n=2) CAR-T cells/kg | 27 of 29 patients (93%) achieved BM remission |
| NCI | [NCI] 08-C-0121 | NY-ESO-1 | Retrovirus | 1G4-α95:LY | adult: Mela; n=11 adult: SCS; n=6 | Cy 60 mg/ m2 ×2+ Flu 25 mg/ m2×5 d | a median of 5.5×1010 TCR-T cells/patient(range, 16 to 130×109) | CR: 2/11 in Mela(persisted over 1 year in two patients); ORR:5/11 in Mela; 4/5 in SCS( over 18 months in one patient) |
| NCI | NCT00670748 | NY-ESO-1 | Retrovirus | 1G4-α95:LY | adult: Mela; n=20 adult: SCS; n=18 | Cy 60 mg/ m2 ×2+ Flu 25 mg/ m2×5 d | a median of 5×1010 TCR-T cells/patient (range, 0.9 to 13×1010) | CR:1/18 in SCS,4/20 in Mela; OR:11/20 in Mela,11/18 in SCS; OS:38% and 14% at 3 and 5 year in SCS, respectively; both 33% at 3 and 5 year in Mela |
CT.GI: ClinicalTrials.gov Identifier; FHCRC: Fred Hutchinson Cancer Research Center; FHCRC: Fred Hutchinson Cancer Research Center; CHP: Children's Hospital of Philadelphia; HTUP: Hospital of the University of Pennsylvania; NCI: National Cancer Institute; NHL: non-Hodgkin's lymphoma; ALL: acute lymphoblastic leukemia; Mela: melanoma; SCS: synovial cell sarcoma; Flu: fludarabine; Cy: cyclophosphamide; mo: month; Vp: Etoposide; CVAD-B: Methotrexate 1000mg/m2 day1,Cytarabine 1000 mg/m2 every 12hours days 2,3,5; CVAD-A: Cyclophosphamide 300 mg/m2 every 12 hours days 1-3, Vincristine 2 mg day 3, Adriamycin 50 mg/m2 day 3.
Figure 1
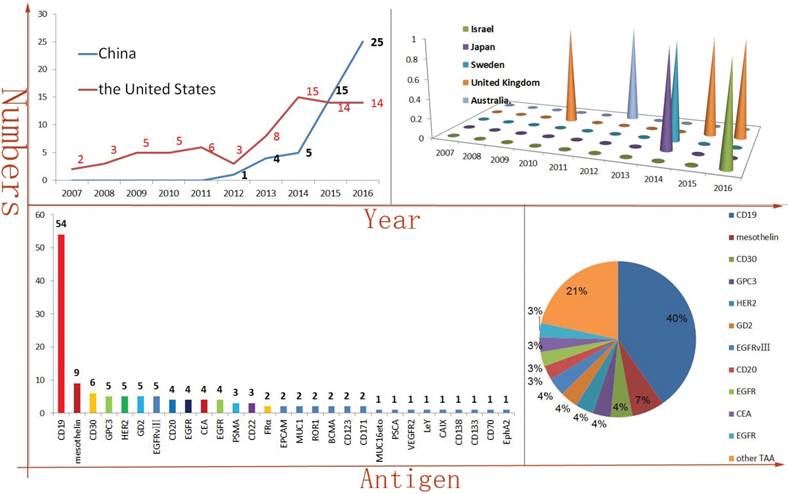
CAR-T immunotherapy is rapidly developing in China and CD19 is the dominant TAA (The vast majority of the data comes from: clinicaltrials.gov.) Figure 1A is indicated the numbers of new initiated CAR-T clinical trials each year and the United States and China account for most of them. But, Israel, Japan, Sweden, United Kingdom and Australia have also carried out similar clinical trials, as well. Figure 1B is indicated the antigen distribution of CAR-T immunotherapy targeting (in total). It is very clearly that CD19 is the dominant TAA. At the same time, a wide variety of TAAs are selected.
Figure 2
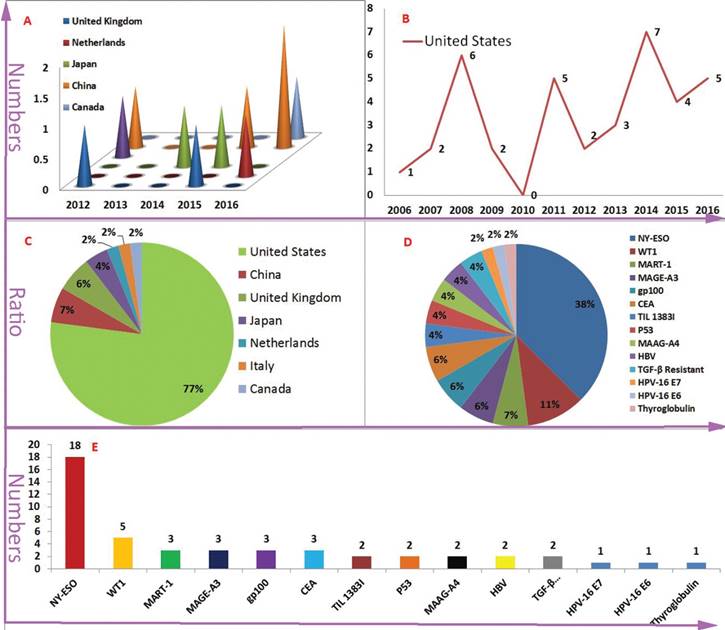
The speed of TCR-T therapy development is in a stable level and NY-ESO is the dominant TAA (The vast majority of the data comes from: clinicaltrials.gov.) Figure 2A-B is indicated the numbers of new initiated TCR-T clinical trials each year and the United States is the dominant leader. But, United Kingdom, China, Japan, Netherlands and Canada have also carried out similar clinical trials in recent year. Figure 2C-E is indicated antigen distribution of TCR-T immunotherapy targeting (in total). It is very clearly that NY-ESO-1 is the dominant TAA. At the same time, a wide variety of TAAs are selected.
As mentioned above, the NY-ESO-1-activated TCR-T clinical trials accounted for more than 30% of all TCR-T clinical trials. Meanwhile, as a TAA, the wilms' tumor antigen 1 (WT1) is highly expressed in a majority of hematologic malignancies, such as acute myeloid leukaemia (AML), acute lymphoid leukaemia (ALL), myelodysplastic syndrome (MDS). WT1 was founded in the solid tumors of various tissue origins, such as lung, breast, digestive organs, brain, head and neck, thyroid, and female genital tract, etc [41]. For this reason, it has been developing rapidly that HLA-A*0201-restricted/WT1-specific TCR-T gene therapy for chronic myeloid leukaemia (CML), AML, MDS, NSCLC and mesothelioma. Of course, other tumor associated antigens (TAAs) including MART-1, MAGE-A3, MAGE-A4, gp100, CEA, TIL 1383I, P53, HPV-16 E6, HPV-16 E7 and HBV were also selected as the TCR-T targets (Figure 2 D-F) [42-48].
Although there are a series of the targets of TCR-T and CAR-T, the action of TAA exploration will not stop. On the one hand the heterogeneity of cancer cells may contribute to the difference of antigen distribution, on the other hand the heterogeneous cancer cells still can't get rid of the majority inherent nature of normal cell. It is very difficult to find antigens or antigenic peptides whose expression level is high enough to activate the signal of specific recognition receptors in cancer tissues while low enough to inactivate the signal of specific recognition receptors in normal tissues. Cancer represents a paradox in which the generality and the heterogeneity coexist. The paradox forms the multidimensional interaction in tumor, TAA and normal tissue.
The multidimensional interaction: tumor, TAA and normal tissue
The complexity and diversity of TAAs make us puzzled, as shown in Figure 1B and Figure 2D. For example, there are many alternative TAAs to target hematological malignancies. When adoptive CAR-T gene therapy for hematological malignancies, there are lots of TAAs can be selected, such as CD19, CD20, CD30, CD22, CD138, CD70, BMCA, etc. It's amazing that WT1 and NY-ESO-1 can be selected as targets for TCR-T cancer immunotherapy in hematological malignancies, as well. For solid tumors, it is a more complicated situation. For example, for lung cancer, the candidate TAAs may contain mesothelin, EGFR, MUC1, RORI, CEA, WT1, NY-ESO-1, MAGE-A3/4, etc [49]. A further increase in TAA species is no doubt in the future. Therefore, the concept of tandem CAR has been gradually popular. One encouraging result showed that an adoptive tandem-CAR-T therapy in glioblastomas by targeting heterogeneous expression antigens human epidermal growth factor receptor 2 (HER2) and IL13Rα2 on glioblastomas. The tandem-CAR-T got a better control of cancer progression, comparing single CAR that specially targets HER2 or IL13Rα2, respectively. By inducing HER2-IL13Rα2 heterodimers, the tandem-CAR engaged the both antigens to promote superadditive T cell activation and mitigate tumor antigen escape [50]. Moreover, if antigen-activated CAR and antigenic peptide/MHC complex-activated TCR co-exist, what effect will T cells produce? A preclinical study indicated that HER2-special CAR expression in CD8+T cells does not affect stimulation through peptide (SIINFEKL)-activated TCR [51]. Besides, The GE-T cells demonstrated a similar cytotoxic effect through CAR and TCR stimulation in the short term. But, CAR expression was downregulated, and TCR expression was reversely unchanged over the first 20 hours of coincubation. Thus, from 20 to 50 hours, the cytotoxic effect of TCR mediated was better than CAR mediated [51]. Alternatively, the sequential ACT with different targets becomes popular. A clinical trial of the sequential ACT that CD19-targeted CAR-T and CD20-targeted CAR-T for Diffuse Large B Cell Lymphoma (DLBCL) has been executed (ClinicalTrials.gov Identifier: NCT02737085).
It is worth noting that not only diversified TAAs are expressed in one kind of cancer, but also one TAA is expressed in multiple kinds of cancers. For example the NY-ESO-1 is highly expressed in melanoma, multiple myeloma, NSCLC, synovial sarcoma, breast cancer, renal cell cancer, hepatocellular cancer, esophageal cancer, ovarian cancer, bladder cancer, etc [52-54]. Similarly, the mesothelin is highly expressed in mesothelioma and breast cancer, cervical cancer, pancreatic cancer, ovarian cancer, lung cancer and endometrial cancer [55]. Table 2 and Figure 3 provide a good illustration of the multi-dimension interaction. Particularly, when GE-T specifically binds and kills tumors, we cannot ignore that these targeted TAAs may be normally or restrictedly expressed in multiple normal tissues. The multidimensional interactions make the potential toxicity of "on target, off tumor" unavoidable. For instance, the prostate-specific membrane antigen (PSMA) is expressed in prostate cancer, bladder carcinoma, schwannoma, tumor neovasculature of many solid tumors. But it was expressed in not only prostatic acinarcells but also many other normal tissues, including the kidney, small intestine, the central and peripheral nervous system, as well [56].
A wide variety of cytokines are released from the T cells cytotoxic effect and the tumor lysis syndrome, such as TNF-α, IFN-γ, IL-2, IL-6, etc. On the one hand these cytokines enhance anticancer capacity of the GE-T, on the other hand the excessive release of cytokines may induce the T cells exhaustion and the severe cytokine release syndrome (CRS). As serious adverse event, CRS is frequently occurred and potentially caused the fatal complication in the GE-T tumor immunotherapy [57-59]. The GE-T seems as if a double-edged sword. Thus, how we weaken the potential serious adverse effects of GE-T therapy?
Table 2
The multidimensional interaction of TAAs and tumors in CAR-T gene therapy
| Tumor Associated Antigen | Targeted Tumor | Tumor Associated Antigen | Targeted Tumor |
|---|---|---|---|
| CD19/CD20/CD30/CD22/ CD138/CD70/BMCA | Lymphoma, Leukemia | EGFR | Cholangiocarcinoma/Glioma/ Cancer of Lung, Colorectal, Ovary, Pancreatic/Renal, et al |
| MUC16ecto | Ovarian Cancer/ Primary Peritoneal Cancer/ Fallopian Tube Cancer | EPCAM | Liver Neoplasms/ Stomach Neoplasms |
| Mesothelin | Mesothelioma / Caner of Breast, Cervical, Pancreatic, Ovarian, Lung, Endometrial, et al. | MUC1 | NSCLC/TNBC/Cancer of Hepatocellular, Pancreatic |
| GPC3 | Hepatocellular Carcinoma | ROR1 | CLL/MLL/ALL/NSCLC/TNBC |
| HER2 | Glioma / Sarcoma / Cancer of Breast, Ovarian, Lung, Gastric, Colorectal, Pancreatic, et al | CEA | Cancer of Lung, Colorectal, Gastric, Breast, Pancreatic, et al |
| GD2 | Sarcoma, Osteosarcoma, Neuroblastoma, Melanoma | EGFRvIII | Glioblastoma |
| CD171 | Neuroblastoma |
Footers: CLL: chronic lymphocytic leukemia; MLL: myeloid/lymphoid leukemia; ALL: acute lymphoblastic leukemia; NSCLC: non-small cell lung cancer; TNBC: three negative breast cancer.
Figure 3
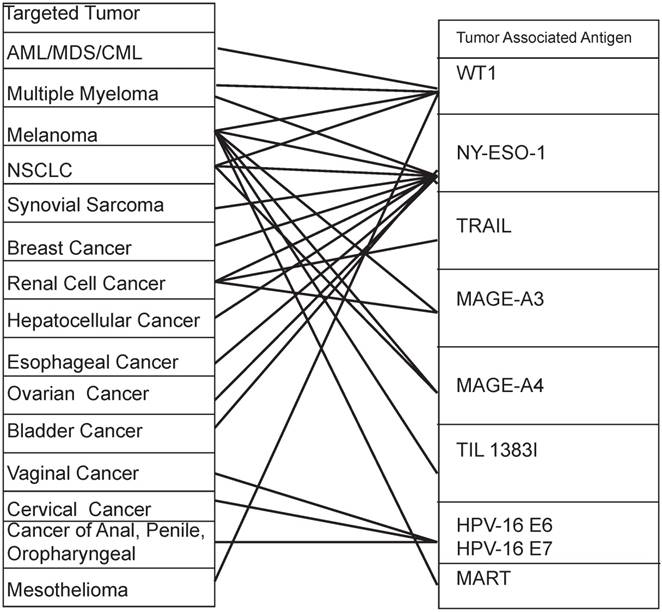
The multidimensional interaction of TAAs and tumors in TCR-T gene therapy A tumor can be killed by a variety of antigenic peptide-specific TCR-T. At the same time, an antigen peptide-specific TCR-T can kill a variety of different organ-derived tumors. Footers: AML: acute myelogenous leukemia; MDS: myelodysplastic syndrome; CML: chronic myelogenous leukemia; NSCLC: non-small cell lung cancer
The GE-T cancer immunotherapy was followed by fatal serious adverse events
Animal disease models cannot fully reveal the potential risk of fatal serious adverse effect from new drug research and the GE-T is no exception [60]. The antigen co-expression on tumor tissue and normal tissue can easily induce the “on target, off tumor” toxicity [61]. For instance, a carboxy-anhydrase-IX (CAIX)-special CAR-T was administered to 12 patients with CAIX-expressing metastatic renal cell carcinoma (RCC). Unfortunately, these patients who were infused as low as 0.2×109 CAR-T cells would occur the CTC grade 2-4 liver enzyme disturbance and develop necessitated cessation of treatment. The examination of liver biopsies revealed CAIX was expressed on bile duct epithelium in liver [62]. A more common event is B cell aplasia resulting from CD19-redirected CAR-T therapy for CD19+ hematological malignancies. The conditioning regimen of lymphodepletion and chemotherapy may contribute B cell aplasia, as well [63, 64]. Even more, some regrettable fatal serious adverse events occurred in the development history of the GE-T tumor immunotherapy. As the events are listed in Table 3, we cannot ignore that the double-edged sword of the GE-T could kill ourselves in some particular conditions, including the clinical trials of CAR-T and TCR-T.
The star drug of Juno Therapeutics, JCAR015, a second generation of CD19-redirected CAR-T, has caused three patients who received the cyclophosphamide + fludarabine (Cy/Flu)-based lymphodepletion to die. The cause of death may be neurologic toxicity, originating from the Cy/Flu regimen to enhance CAR-T cells expansion, persistence and higher response rates. The Cy/Flu regimen also increased the incidence of severe CRS and grade ≥3 neurotoxicity. The speculation was verified in a certain degree in another clinical trial, a phase I/II study of CD19-redirected CAR-T therapy for advanced CD19+ CLL/ALL/Lymphoma with defined subsets of CAR-T (NCT01865617). Comparing patients who received Cy-based lymphodepletion without Flu, patients who received Cy/Flu lymphodepletion had higher response rates. Table 1 is a good illustration of the clinical trial results [3]. Maybe the cerebral CRS contributes to the grade ≥3 neurotoxicity. A Chinese clinical trial results demonstrated that cerebral CRS can be triggered by the blood-brain barrier (BBB)-penetrating CD19-directed CAR-T. The cerebral CRS could induce an extremely higher level of IFN-γ and IL-6 in cerebrospinal fluid than in serum [65]. In particular, when we reviewed a clinical trial of the HLA-A*0201/MAGE-A3-positive TCR-T gene therapy for metastatic saynovial sarcoma, melanoma and esophageal cancer, one melanoma patient and one esophageal cancer patient died of neurologic toxicity. The cause of death is the HLA-A*0201/MAGE-A3-positive TCR-T cross-presentation epitopes from MAGE-A12 which was expressed in human brain cells (and possibly MAGE-A1, MAGE-A8 and MAGE-A9) [66]. Those patients who died of neurologic toxicity were likely to experience a long and painful process of death in the ACT. In this clinical trial, the spans of time from first infusion of the GE-T to dead were 167 days and 94 days, respectively [66]. Even if identify and combine same TAA, the different HLA/peptide-positive TCRs may induce totally different fatal serious adverse event. For example, another clinical trial of HLA-A*01/MAGEA3 (EVDPIGHLY)-special TCR-T gene therapy for melanoma and myeloma was transduced. Two patients who were infused the HLA-A*01/MAGEA3 (EVDPIGHLY)-special TCR-T died within 5 days, due to cardiovascular toxicity. An unrelated peptide derived from the striated muscle-specific protein titin was targeted by the TCR-T, inducing powerful “on-target off-tumor” toxicity [60]. The total infusion doses of GE-T were from 2.4 ×109 to 7.9 ×1010 cells. Thus, it's difficult to find an exact death dose. How can we weaken the serious adverse effects of the adoptive GE-T gene therapy?
Table 3
The clinical trials of adoptive gene-engineered T cell immunotherapy were followed by fatal serious adverse events
| Institution | Registered of number | TAA | Gene transfer tool | Positive-receptor (scFv or TCR) | Patient populations | Lymphodepleting regimens | Infused cell doses (total) | Time | Cause of death (number of deaths) |
|---|---|---|---|---|---|---|---|---|---|
| JunoTherapeutics, Inc. | NCT02535364 | CD19 | Retrovirus | scFv: 19z1-28ζ(2nd) | Relapsed or refractory B-ALL | Cy + Flu | Unknown | Unknown | Neurologic toxicity (3) |
| Chinese PLA General Hospital | NCT01735604 | CD20 | Lentivirus | scFv: CAR.20-CD137ζ* (2nd) | Diffuse large B-cell lymphoma | COD | 107/kg | 3 weeks | Massive hemorrhage of alimentary tract (1) |
| National Cancer Institute | NCT00924287 | HER2 | Retrovirus | scFv: 4D5-CD8-28BBζ (3rd) | Colon cancer metastatic to the lungs and liver | Cy 60 mg/kg for 2 days followed by Flu 25 mg/m2 for 5 days | 1010 cells | 5 days | CRS; Speculate that off tumor, targeting lung epithelial Cells (1) |
| National Cancer Institute | NCT01273181 | MAGE-A3/A12 | Retrovirus | TCR: HLA-A*0201-restricted MAGE-A3 peptide: KMAELVHFL | Patient 5: Melanoma 8: esophageal cancer | Cy 60 mg/kg for 2 days followed by Flu 25 mg/m2 for 5 days | Patient 5: 7.9 ×1010 cells Patient 8: 6.1×1010 cells | Patient 5: 167 days; Patient 8: 94 days | Neurologic toxicity (2) |
| Washington University; the University of Pennsylvania | NCT01350401 NCT01352286 | MAGE-A3 | Retrovirus | TCR: HLA-A*01-restricted MAGE-A3 peptide: EVDPIGHLY | Patient 1: Melanoma Patient 2: Myeloma | Patient 1 :Cy 60 mg/kg for 2 days; Patient 2: Melphalan at 200 mg/m2 followed HSCT | 5.3 ×109 cells 2.4 ×109 cells. | Patient 1: 4 days; Patient 2: 5 days | Cardiovascular toxicity (2) |
| The Netherlands Cancer Institute | NL.37327.000.11 | MART-1 | Retrovirus | HLA A*0201 -restricted MART-1 epitope: EAAGIGILTV | melanoma | cy: 60 mg/kg for 2 days followed by Flu: 25 mg/m2/day for 5 days | 5×109 cells | 9 days | Multiple organ failure and irreversible neurologic damage (1) |
Time: the span of time from first infused GE-T to death; *: scFv domain targeting the CD20 was derived from AY160760.1; 2nd: the second generation of CAR-T; 3rd: the third generation of CAR-T; CRS: cytokine release syndrome; Cy: Cyclophosphamide; Flu: fludarabine; COD: day1, Cyclophosphamide, 500 mg/m2, day1, Vincristine, 2 mg, day1-3, Dexamethasone.
The road of optimizing security
Change HLA-restricted type of TCR and insert suicide genes
The disappointing outcomes of the two MAGE-A3-targeted clinical trials did not stop the team of surgery branch, national cancer institute from exploring new MAGE-A3 TCR-T for cancer immunotherapy. An HLA-DPB1*04: 01-restricted MAGE-A3 TCR, 6F9 TCR, was isolated from a regulatory T-cell clone which derived from the peripheral blood of melanoma patient after MAGE-A3 vaccination. The MHC class II-restricted TCR-T could specifically target the MAGE-A3 (+) /DP4 (+) cell lines Mel 526-CIITA and H1299-CIITA. Because the HLA-DPB1*04:01 is present in ~ 60% of the Caucasian population, the 6F9-specific TCR-T may have a strong application prospects [67]. In addition, HLA-DP4/NY-ESO-1-specific TCR-T gene therapy for NY-ESO-1/HLA-DP4-expressing melanoma cells had been constructed at early stage [68, 69]. But it's difficult to find some significant breakthroughs in the area of HLA-DP4 restriction. Maybe another alternative strategy of optimizing security is the insertion of the suicide genes. Either the herpes simplex virus thymidine kinase (HSV-TK), or inducible caspase 9 safety switch (iCasp9), or a truncated human epidermal growth factor receptor (tEGFR), which confer lethal sensitivity of GE-T according to the anti-herpes drug-ganciclovir, or the small molecule dimerizer, or triggering antibody-dependent cellular cytotoxicity (ADCC) by the infusion of the EGFR-specific monoclonal antibody (mAb) cetuximab, respectively [70-72]. Interestingly, an oligopeptide, Strep-tag II, was introduced into in the CD19-redirected GE-T. The strep-tag II conferred GE-T many functions, such as marker identification and purification, functional optimization, cell sorting and large-scale cell expansion [73]. All these functions will depend on specific recognition of the antibody to Strep-tag II. In accordance with the principles of ADCC, Strep-tag II is also acted as suicide gene [73].
Reduce affinity of scFv or TCR
The serious adverse effects of multiple clinical trials were due to the high affinity of receptor-antigens. The security may improve when we reduce receptor recognition affinity. For example, a second-generation HER2-specific CAR that scFv was derived from the HER2-specific MAb FRP5 (FRP5-CAR-T), was transduced into T cells. The results of a phase I/II clinical study in recurrent/refractory HER2-positive sarcoma with FRP5-CAR-T demonstrated that the FRP5-CAR-T could persist for 6 weeks without evident toxicities and had a good efficacy of anti-tumor [74]. Similarly, two types of EGFR-special CAR-T were constructed from monoclonal antibodies which differ in affinity. Engineered T cells with low affinity EGFR-CAR not only selectively targeted cells overexpressing EGFR, but also exhibited attenuate effector function as the density of EGFR declined [75].
Non-viral vectors and regional delivery of GE-T
The non-durable target genes expression may be a good choice to improve the security of GE-T. Comparing lentiviral and retroviral vectors technology, the CARs expression using a RNA platform may bypass “on-target, off-tumor” toxic effects according to the property that RNA CAR-T could be completely and rapidly removed as the metabolism of RNA, without depending on suicide genes. For example, C4-27z-redirected and C4opt-27z-redirected CAR-T, two CAR-Ts targeting folate receptor-α(FRα) expression cancers, exhibited significant proliferation and anti-tumor activity in animal models of human ovarian cancer [76]. Another preclinical data of CD19-directed RNA CAR T cells also illustrated that when multiple reinfusions of RNA CAR T cells were administered, the improved survival and sustained antitumor responses were observed in a robust leukemia xenograft model [77]. Another good example comes from CD20-specific RNA CAR-T in dogs with spontaneous B cell lymphoma [78]. These preclinical outcomes undoubtedly enhance the application potential of RNA CAR-T.
The incidence of CRS and “on-target, off-tumor” toxic effects may be enhanced, when GE-T is infused by intravenous, because the incidence that GE-T encounters and binds the normal tissue of co-expression TAAs is increased. It can be very effective in controlling the intensity of CRS that the application of tocilizumab, a monoclonal antibody against interleukin-6 (IL-6), and corticosteroids [79]. But, CRS still cannot be completely eliminated. It may be able to enhance security through the physical and chemical barrier in pleural cavity, abdominal cavity and cranial cavity which limit GE-T excessive systemic diffusion via regional delivery [80]. Therefore, the regional delivery of GE-T is more and more striking. Further, the high concentration of GE-T delivered by local injection may further enhance the therapeutic value of solid tumors. Whether intra-pleurally administered CAR-T targeting mesothelin, regional intraperitoneal (IP) infusion of CAR-T targeting MUC-16ecto and CEA, or intracranial injection of ErbB and IL13R -targeted CAR-T, which were repeatedly proved that regional CAR-T cell infusions for solid tumors of multiple organs may be superior than systemic delivery [14, 34, 81-83].
Humanized scFv
What we cannot ignore is that multiple scFvs are derived from murine mAb, such as FMC63 (CD19 targeted), 14G2a (GD2 targeted), Mov19 (FRα targeted), CE7 (CD171 targeted), etc. If we use these scFv to construct CAR-T, there are always risks of graft-versus-host diseases (GVHD) in different species. The innovation for humanized scFv has been going. A humanized CD19-targeted CAR-T showed the same anti-tumor efficacy and prolonged survivability as those were expressed murine scFv [84]. Other humanized scFvs contain chA21 (HER2 targeted), 2173 (EGFRvIII targeted) had been constructed and applied in models of human cancer or clinical studies [84-86].
Spatiotemporal control of receptor activation
At the cellular level, other strategies to enhance safety are more vividly described by Christopher A Klebanoff and his colleagues [87]. They make a good summary about the spatiotemporal control of TAA-redirected receptor activation in CAR-T [88-90].
Stringent examination the potential co-expression of peptides and antigens
In the clinical treatment level, the inclusion criteria and exclusion criteria of clinical trial need to be strictly implemented. Before the GE-T immunotherapy administration, the co-expression of antigenic peptides or TAAs that are present in normal tissues and organs need to be examined and confirmed, as far as possible. How to recognize and manage the toxicities of CAR-T? Jennifer N. Brudno and James N. Kochenderfer presented some good guidelines for before, during and after tumor immunotherapy of gene-modified T cells [91].
Are all of the above strategies sufficient to drive GE-T into a safe road? It is obviously not enough. When we apply viral vectors of constant insertion, both retroviral vectors and lentiviral vectors, the risk of insertional mutation is the sword of Damocles. Retroviral vectors as gene transfer tool had temporarily cured some inherited disorders by gene-engineered hematopoietic stem cells transplantation, while following the genotoxicity mediated by insertional mutagenesis, including leukemogenesis [92]. Luckily, we still haven't found that genetically modified T cells can be caused leukemogenesis via viral vectors, so far. On the one hand the survival time of genetically modified T cells in clinical subject bodies may be too short to induce tumorigenesis, on the other hand the subjects with tumor may have a greater ability to recognize and kill viral vectors-induced mutant antigens than the ability of patients with inherited disorders. For CAR-T or for TCR-T, maybe the emerge of the sleeping beauty transposon/transposase system (SB) or multiplex artificial nucleases gene editing technology is a reliable strategy for eliminating this potential insertional mutation. But the toxicity of off-target cleavage by artificial nucleases cannot be ignored, as well [93-96].
The road of optimizing security seems to be endless. T cells have been modifying to stronger viability, stronger cytotoxicity capacity, more precise targets identification, and broader applicability. Maybe these modifications are accompanied with new risks. In addition, perhaps tumors are also evolving more intelligent. The GE-T needs to be equipped with enough weapons to fight malignancies (Figure 4).
Figure 4
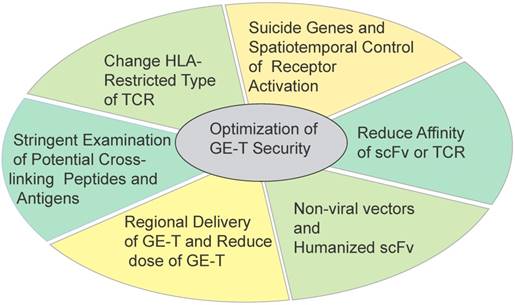
Some strategies to optimize the security and efficacy of genetically T cells immunotherapy Six strategies in different directions were applied for optimizing the safety and efficacy of ACT.
The weapons of gene-modified T cells
Although some encouraging clinical trial results were demonstrated from the CAR-T immunotherapy for haematological malignances and TCR-T immunotherapy for melanoma, the GE-T gene therapy is still young, comparing the history of cancers. As mentioned above, the GE-T has to be given some weapons on the road of optimization security and efficacy (Figure 5).
Figure 5
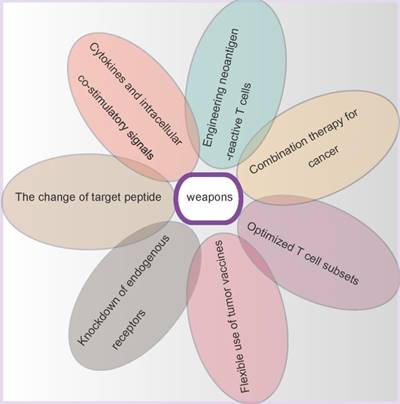
The weapons of gene-modified T cells. In order to confer T cells the stronger anti-tumor ability, seven different weapons are equipped on the gene-modified T cells bodies.
Cytokines and intracellular co-stimulatory signals
Common cytokine-receptor γ-chain (γc) family cytokines, which contains interleukin-2 (IL-2), IL-4, IL-7, IL-9, IL-15 and IL-21, have critical roles in the development, proliferation, survival and differentiation of T cells [97]. The addition of cytokines during the culture of genetically T cells in vitro has a significant influence on the subsequent immunotherapeutic effect in vivo. Facilitating the formation of memory T cells, IL-7, IL-15 and IL-21 can promote the survival time and long-term anti-tumor effect of CD19-redirected CAR-T and GD2-redirected CAR-T in vivo [98, 99]. A gp100/HLA-A2-directed TCR-T gene therapy was also showed similar results in vitro [100]. One potential risk is IL-7-mediated and IL-15-mediated TCR sensitization enables T cell responses to autoantigens associated with autoimmune disease, such as rheumatoid arthritis (RA) and autoimmune diabetes [101, 102]. Other cytokines, including IL-12 which mediated a second activation mechanism of TCR and led to a 10 to 100 folds increase in persistence and anti-tumor efficacy of ACT in vivo, may be further developed and utilized in the future [103, 104]. At the same time, the CAR that was incorporated IL15 gene also showed a similar therapy effect [105].
Additionally, with the evolution of the costimulatory endodomain in CAR-T that is consisted of the first generation, the second generation and the third generation, it may be confirmed that CD137 (also named 4-1BB) is superior to CD28 for application value. CD137 (also named 4-1BB) was able to attenuate the exhaustion induced by persistent CAR signaling, however, CD28 may accelerate the exhaustion [106]. CD137 confers CAR-T a mild and less terminal differentiation state, which allows CAR-T to survive longer in vivo and may attenuate severe adverse effects while enhancing antitumor effects. It is consistent with the theory that hierarchical T cell exhaustion during chronic infection [58]. The third generation of CAR may confer T cells a higher level of activation and earlier exhaustion tendency. For instance, when we review an attention death event of the HER2-special CAR-T that immunotherapy for metastatic colon cancer, the HER2-special CAR consisted 4D5-CD8-28BBζ, a third generation of CAR-T [25]. Another study was also indicated that CD28-ζ CAR (second generation) was more efficient than the CD28-ζ-OX40 CAR (third generation) [107]. Interestingly, Zhao and colleagues considered a combined CAR that named 1928z-41BBL, including CD19-special scFv, the CD28CD3ζ intracellular domain and 4-1BB ligand (41BBL), could balance effector functions and persistence functions of CAR-T [108]. An interesting thing to note is that these evaluations on the modification of costimulatory signal and the choices for second generation CARs or third-generation CARs were based on viral vectors. Do the same conclusions apply to non-viral vectors? Perhaps the characteristics of the third generation of CAR are more suitable for multiple reinfusions of GE-T immunotherapy.
The change of target peptide
In contrast to scFv species which is relatively simple, the species of TCR is more diversified. It creates a variety of antigenic peptides and activated TCRs that the complicated repertoire of presented peptides and the subsequent T cell response by MHC system [6]. Petra Simon and colleagues found 16 functional antigen-specific TCRs that specially target the well-known TAA NY-ESO-1 via an integrated approach [109]. However, these TCRs exhibit diverse therapeutic capability, according to the affinity to recognize MHC-multimers loaded with NY-ESO-1 peptide and the capacity of recognizing endogenously processed antigen [110]. For example, the study of NY-ESO-1/HLA-A*0201-specific peptide 157-165 (SLLMWITQC)-redirected TCR-T immunotherapy for advanced myeloma got an amazing result that clinical responses were observed in 16 of 20 patients (80%), with a median progression-free survival of 19.1 months. In contrast, the anti-tumor ability of NY-ESO-1/ HLA-DP4-specific peptide p161-180-redirected TCR-T was much weaker.[61] Another typical case comes from WT1-special TCR-T. Three different TCR-T targeting WT1 peptide, HLA-DRB1*04:05-restricted WT1332, HLA-A*02:01-restricted 9-amino acid peptide (RMFPNAPYL) and HLA-A*24:02-restricted WT1235-243, which were also revealed significant immunotherapy capacity [111-113].
Knockdown of endogenous receptors
The exogenous and endogenous TCR chains which are co-expressed in genetically engineered T cells lead to competition for surface expression and inappropriate pairing, with suboptimal vitality and potentially harmful unpredicted specificities. The knockdown of endogenous TCRs will confer a higher level of expression and more efficient antigen recognition of exogenous TCRs. Even more, the knockout of endogenous TCRs will reduced the probability of TCR gene transfer-induced graft-versus-host-disease [113]. The knockdown can be achieved by nucleases genome editing, contained zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALEN), CRISPR/Cas9 and megaTAL nucleases, or by RNA interfere (RNAi). The megaTAL nucleases and CRISPR/Cas9 seem to have better disruption efficiency and low levels of toxicity and off target cleavage [96, 114]. In order to confer more capacity on genetically modified T cells, increasingly, more and more endogenous receptors have been knocked down. A "Off-the-Shelf" CD19-targeted CAR-T that was deficient in expression of both their αβ T-cell receptor (TCR) and CD52, a protein targeted by alemtuzumab, a chemotherapeutic agent, was allogeneic transplanted to CD19+ tumor patient. Even in the presence of the chemotherapeutic agent, the "Off-the-Shelf" CAR-T was still able to effectively destroy CD19+ tumors. It is an idealized third-party CAR T immunotherapy product [115]. Similarly, a triple genome editing that simultaneously disrupts the genes of TCR, beta-2 microglobulin (B2M) and programmed cell death gene-1(PD-1) in CAR-T by using CRISPR/Cas9 system. The deficient of CAR-T provide an alternative as a universal donor to adoptive T cells therapy for cancers [116]. The small hairpin RNA (shRNA) could also blockade cell-intrinsic PD-1 in CAR-T [117]. Another alternative is that viral vectors encode anti-immunosuppressive factors antibodies while expressing a specific receptor. For example, a human CAIX-targeted CAR-T was engineered to secrete human anti-programmed death ligand 1 (PD-L1) antibodies [118]. As more and more GE-T is demanded for diversification functions, more and more endogenous receptors will be abolished.
The most valuable thing is that the knockdown of endogenous receptors is equally applicable to both CAR-T and TCR-T, in most cases.
Flexible use of tumor vaccines
The early injection of tumor vaccines is an effective and relatively safe way to isolate antigenic peptide-positive TCRs. It is widely used in TCR-T cancer immunotherapy [2, 61, 119]. For a long time, independent of MHC system has been considered the unique advantage of CAR-T immunotherapy. However, with the difficult progress in the solid tumor treatments, we need to find a reliable path to break through solid tumor additional obstacles that contain immunosuppressive microenvironment, membrane antigen mutagenesis, tumor cell heterogeneity, et al. A fully humanized CAR which selectively and especially targeted the HLA-A*02:01/alpha-fetoprotein (AFP) 158-166, ET1402L1-CAR, was transduced into T cells via a pCDH lentiviral vector. ET1402L1-special CAR-T was shown robust anti-tumor activity in vivo by intravenous administration or intraperitoneal injection [120]. This result is interesting, because it can be served as a bridge between cancer vaccines, CAR-T and TCR-T.
Optimized T cell subsets
T cells are divided into many subsets, such as CD4+ T cells, CD8+ T cells, central memory TCM, effector memory TEM cells, or regulatory T cells. In tumor immunotherapy, the single subset of GE-T may not be the best choice. A clinical trial which CD19 CAR-T cells were manufactured from defined T cell subsets in a 1:1 CD4+/CD8+ ratio to 32 adults with relapsed and/or refractory B cell non-Hodgkin's lymphoma was verified the important of optimized T cell subsets [3]. At the same time, along with the technology of T cells amplification and sorting in vitro to further improve, more functional ratio of T cell subset will be further exploited.
Combination therapy for cancer
In the course of cancer treatment, the ACT can be existed alone, or acted as a part of the combination therapy. For example, a human IgG1 T-cell receptor mimic monoclonal antibody directed to a peptide (RMFPNAPYL) of WT1 in HLA-A*02 dependent was therapeutically effective, alone and in combination with tyrosine kinase inhibitors (TKIs), against a leukemia with the most common, pan-TKI, gatekeeper resistance mutation, T315I [111]. Whether WT1-targeted TCR and the mimic monoclonal antibody combination therapy will produce a similar effect? If so, this strategy will become an option for TKIs resistance in cancer therapy. As noted above, tumor vaccines have a unique advantage in inducing TAA-specific TCR. The tumor radiotherapy can produce a similar effect. The vaccine-similar effect was contributed non-redundant immune mechanisms in cancer by a combination of radiotherapy and dual checkpoint blockade [121]. Maybe it is a very imaginative treatment combination that ACT is combined with radiation therapy.
Engineering neoantigen-reactive T cells
Perhaps in the future, it can be implemented that relying on high-throughput sequencing technology to find tumor specific antigens (TSA) which are caused by mutations. Neoantigen is one of the TSA. Using the technology of high-throughput TCR gene-capture to search neoantigen-reactive TCR and constructing neoantigen-targeted TCR-T are no longer distant [87, 122]. Here, the high-throughput sequencing technologies not only become a link between TSA and activated receptors, but also act as a booster for customizing GE-T. It is possible to develop more precise ACT, depending on the TAA expression assay of individual tumor cells. But, the sad fact gene therapy is the expensive treatment. The overwhelming majority of patients cannot afford the cost of gene therapy, including GE-T tumor immunotherapy [123].
In summary, the functions of T cell are continuously developed. More and more weapons will be added to the menu of personalized treatment. However, with the abundance weapons being applied, tumors may undergo modulation and deletion of TAA. Tumors may resistant to chemotherapy, radiotherapy, targeted therapy, and immunotherapy in combination therapy. The arms race is far from over.
Abbreviations
PD-1: programmed death-1 receptor; NSCLC: non-small cell lung cancer; HLA: histocompatibility leukocyte antigen; ROR1: tyrosine kinase-like orphan receptor 1; BCMA: B-cell maturation antigen; LeY: Lewis (Le)-Y; GPC3: Glypican-3; HER2: human epidermal growth factor receptor-2; GD2: The tumor-associated ganglioside GD2; EGFR: epidermal growth factor receptor; CEA: carcinoembryonic antigen; PSMA: prostate-specific membrane antigen; FRα: folate receptor-alpha; EPCAM: epithelial cell adhesion molecule; MUC1: mucin 1; VEGFR2: vascular Endothelial Growth Factor Receptor-2; PSCA: prostate stem cell antigen; EphA2: erythropoietin-producing hepatocellular carcinoma A2; HPV: human papillomavirus; MAGE: melanoma-associated antigen-encoding gene; TNF-α: tumor necrosis factor-α.
Authorship
Conception and design: Zeming Mo, Peixin Du, Guoping Wang, Yongsheng Wang
Administrative support: Yongsheng Wang; Collection and assembly of data: Zeming Mo
Data analysis and interpretation: Zeming Mo; Manuscript writing: All authors
Final approval of manuscript: All authors
Competing Interests
The authors have declared that no competing interest exists.
References
1. Maude SL, Frey N, Shaw PA. et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507-17
2. Robbins PF, Kassim SH, Tran TL. et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:1019-27
3. Cameron J TL, Carolina B, Michael H. et al. Immunotherapy of non-Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016;8:355ra116
4. Sommermeyer D, Hudecek M, Kosasih PL. et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30:492-500
5. Cyrille J CZ, Regina B, Yangbing Z. et al. Recognition of Fresh Human Tumor by Human Peripheral Blood Lymphocytes Transduced with a Bicistronic Retroviral Vector Encoding a Murine Anti-p53 TCR. Journal of immunology. 2005;175:5799-808
6. Rock KL, Reits E, Neefjes J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends in immunology. 2016;37:724-37
7. Ghorashian S, Pule M, Amrolia P. CD19 chimeric antigen receptor T cell therapy for haematological malignancies. British journal of haematology. 2015;169:463-78
8. Caballero OL, Chen YT. Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci. 2009;100:2014-21
9. Wolchok JD. PD-1 Blockers. Cell. 2015;162:937
10. 2016. FDA Accepts Supplemental Biologics License Application, Assigns Priority Review and Grants Breakthrough Therapy Designation to Merck's KEYTRUDA® (pembrolizumab) for First-Line Treatment of Patients with Advanced Non-Small Cell Lung Cancer. http://wwwmercknewsroomcom
11. Johnson DB, Balko JM, Compton ML. et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. New England Journal of Medicine. 2016;375:1749-55
12. Pitt JM, Vetizou M, Daillere R. et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity. 2016;44:1255-69
13. Boice M, Salloum D, Mourcin F. et al. Loss of the HVEM Tumor Suppressor in Lymphoma and Restoration by Modified CAR-T Cells. Cell. 2016;167:405-18 e13
14. Adusumilli PS, Cherkassky L, Villena-Vargas J. et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014;6:261ra151
15. Beatty GL, Haas AR, Maus MV. et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2:112-20
16. Wang C WZ, Wang Y, Guo Y. et al. Autologous T cells expressing CD30 chimeric antigen receptors for relapsed or refractory Hodgkin's lymphoma: an open-label phase I trial. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016 pii: clincanres.1365.2016
17. Brian G. TM, Wang JJ, Qian XJ, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012;119:3940-50
18. Haso W, Lee DW, Shah NN. et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165-74
19. Hudecek M, Lupo-Stanghellini MT, Kosasih PL. et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:3153-64
20. Wang L ZS, Ou J, Bai H. Cytotoxity of pomalidomide combined CAR-T cell for multiple myeloma cell RPMI8226 and U266. Zhonghua Xue Ye Xue Za Zhi. 2015;36:497-500
21. P S. Haematological cancer: Anti-BCMA CAR T cells show promise in MM. Nat Rev Clin Oncol. 2016;13:530
22. Shaffer DR SB, Yi Z, Chow KK. et al. T cells redirected against CD70 for the immunotherapy of CD70-positive malignancies. Blood. 2011;117:4304-14
23. Ritchie DS, Neeson PJ, Khot A. et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther. 2013;21:2122-9
24. Li W, Guo L, Rathi P. et al. Redirecting T Cells to Glypican-3 with 4-1BB Zeta Chimeric Antigen Receptors Results in Th1 Polarization and Potent Antitumor Activity. Hum Gene Ther. 2016 [Epub ahead of print]
25. Morgan RA, Yang JC, Kitano M. et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843-51
26. Louis CU SB, Dotti G, Pule M. et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050-6
27. Liu KY, Liu XJ, Peng ZP. et al. Retargeted human avidin-CAR T cells for adoptive immunotherapy of EGFRvIII expressing gliomas and their evaluation via optical imaging. Oncotarget. 2015;6:23735-47
28. Feng K, Guo Y, Dai H. et al. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci. 2016;59:468-79
29. Katz SC, Burga RA, McCormack E. et al. Phase I Hepatic Immunotherapy for Metastases Study of Intra-Arterial Chimeric Antigen Receptor-Modified T-cell Therapy for CEA+ Liver Metastases. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:3149-59
30. Santoro SP, Kim S, Motz GT. et al. T cells bearing a chimeric antigen receptor against prostate-specific membrane antigen mediate vascular disruption and result in tumor regression. Cancer Immunol Res. 2015;3:68-84
31. Kandalaft LE, Powell DJ Jr. et al. A phase I clinical trial of adoptive transfer of folate receptor-alpha redirected autologous T cells for recurrent ovarian cancer. Journal of translational medicine. 2012;10:157
32. Deng Z, Wu Y, Ma W. et al. Adoptive T-cell therapy of prostate cancer targeting the cancer stem cell antigen EpCAM. BMC Immunol. 2015;16:1
33. Posey AD Jr, Schwab RD, Boesteanu AC. et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity. 2016;44:1444-54
34. Koneru M, O'Cearbhaill R, Pendharkar S. et al. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. Journal of translational medicine. 2015;13:102
35. Hillerdal V RM, Leja J, Essand M. Systemic treatment with CAR-engineered T cells against PSCA delays subcutaneous tumor growth and prolongs survival of mice. BMC Cancer. 2014;14:1-9
36. Huang J, Liang J, Tang Q. et al. An active murine-human chimeric Fab antibody derived from Escherichia coli, potential therapy against over-expressing VEGFR2 solid tumors. Appl Microbiol Biotechnol. 2011;91:1341-51
37. Künkele A, Taraseviciute A, Finn LS. et al. Preclinical Assessment of CD171-Directed CAR T Cell Adoptive Therapy For Childhood Neuroblastoma: CE7 Epitope Target Safety and Product Manufacturing Feasibility. Clinical cancer research. 2017;23:466-77
38. Chow KK, Naik S, Kakarla S. et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther. 2013;21:629-37
39. Johnson LA, Morgan RA, Dudley ME. et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535-46
40. Marybeth SY, Mark ED, Zheng ZL. et al. Transfer of a TCR Gene Derived from a Patient with a Marked Antitumor Response Conveys Highly Active T-Cell Effector Functions. Hum Gene Ther. 2005;16:457-72
41. Sugiyama H. WT1 (Wilms' tumor gene 1): biology and cancer immunotherapy. Jpn J Clin Oncol. 2010;40:377-87
42. Johnson LA, Morgan RA, Dudley ME. et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535-46
43. Gerald PL, Marcela VM, Aaron PR. et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Clinical Trials And Observations. 2013;122:863-71
44. Kageyama S, Ikeda H, Miyahara Y. et al. Adoptive Transfer of MAGE-A4 T-cell Receptor Gene-Transduced Lymphocytes in Patients with Recurrent Esophageal Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:2268-77
45. Parkhurst MR, Yang JC, Langan RC. et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19:620-6
46. Mehrotra S, Al-Khami AA, Klarquist J. et al. A Coreceptor-Independent Transgenic Human TCR Mediates Anti-Tumor and Anti-Self Immunity in Mice. The Journal of Immunology. 2012;189:1627-38
47. Scholten KB, Turksma AW, Ruizendaal JJ. et al. Generating HPV specific T helper cells for the treatment of HPV induced malignancies using TCR gene transfer. Journal of translational medicine. 2011;9:147
48. Gehring AJ, Xue SA, Ho ZZ. et al. Engineering virus-specific T cells that target HBV infected hepatocytes and hepatocellular carcinoma cell lines. J Hepatol. 2011;55:103-10
49. Robbins PF, Morgan RA, Feldman SA. et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:917-24
50. Hegde M, Mukherjee M, Grada Z. et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. The Journal of clinical investigation. 2016;126:3036-52
51. Davenport AJ, Jenkins MR, Cross RS. et al. CAR-T Cells Inflict Sequential Killing of Multiple Tumor Target Cells. Cancer Immunol Res. 2015;3:483-94
52. Chen YT SM, Sahin U, Türeci O. et al. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:1914-8
53. Gure AO CR, Williamson B, Gonen M. et al. Cancer-testis genes are coordinately expressed and are markers of poor outcome in non-small cell lung cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2005;11:8055-62
54. Singh N, Kulikovskaya I, Barrett DM. et al. T cells targeting NY-ESO-1 demonstrate efficacy against disseminated neuroblastoma. Oncoimmunology. 2016;5:e1040216
55. Feng Y, Xiao X, Zhu Z. et al. A novel human monoclonal antibody that binds with high affinity to mesothelin-expressing cells and kills them by antibody-dependent cell-mediated cytotoxicity. Molecular cancer therapeutics. 2009;8:1113-8
56. Kinoshita Y, Kuratsukuri K, Landas S. et al. Expression of prostate-specific membrane antigen in normal and malignant human tissues. World J Surg. 2006;30:628-36
57. Farber DL, Yudanin NA, Restifo NP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol. 2014;14:24-35
58. Wherry EJ. T cell exhaustion. Nature immunology. 2011;12:492-9
59. Xu XJ, Tang YM. Cytokine release syndrome in cancer immunotherapy with chimeric antigen receptor engineered T cells. Cancer Lett. 2014;343:172-8
60. Linette GP SE, Maus MV, Rapoport AP. et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863-71
61. Rapoport AP, Stadtmauer EA, Binder-Scholl GK. et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nature medicine. 2015;21:914-21
62. Lamers CH, Sleijfer S, van Steenbergen S. et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21:904-12
63. Brentjens RJ, Riviere I, Park JH. et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817-28
64. Davila ML, Kloss CC, Gunset G. et al. CD19 CAR-targeted T cells induce long-term remission and B Cell Aplasia in an immunocompetent mouse model of B cell acute lymphoblastic leukemia. PloS one. 2013;8:e61338
65. Hu Y, Sun J, Wu Z. et al. Predominant cerebral cytokine release syndrome in CD19-directed chimeric antigen receptor-modified T cell therapy. J Hematol Oncol. 2016;9:70
66. Morgan RA, Chinnasamy N, Abate-Daga D. et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36:133-51
67. Yao X LY, Parker LL, Li YF. et al. Isolation and Characterization of an HLA-DPB1*04: 01-restricted MAGE-A3 T-Cell Receptor for Cancer Immunotherapy. J Immunother. 2016;39:191-201
68. Mandic M, Castelli F, Janjic B. et al. One NY-ESO-1-Derived Epitope That Promiscuously Binds to Multiple HLA-DR and HLA-DP4 Molecules and Stimulates Autologous CD4+ T Cells from Patients with NY-ESO-1-Expressing Melanoma. The Journal of Immunology. 2005;174:1751-9
69. Zhao Y ZZ, Khong HT, Rosenberg SA. et al. Transduction of an HLA-DP4-restricted NY-ESO-1-specific TCR into primary human CD4+ lymphocytes. J Immunother. 2006;29:398-406
70. Kobold S, Steffen J, Chaloupka M. et al. Selective bispecific T cell recruiting antibody and antitumor activity of adoptive T cell transfer. J Natl Cancer Inst. 2015;107:364
71. Greco R, Oliveira G, Stanghellini MT. et al. Improving the safety of cell therapy with the TK-suicide gene. Front Pharmacol. 2015;6:95
72. Straathof KC, Pule MA, Yotnda P. et al. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105:4247-54
73. Liu L, Sommermeyer D, Cabanov A. et al. Inclusion of Strep-tag II in design of antigen receptors for T-cell immunotherapy. Nat Biotechnol. 2016;34:430-4
74. Ahmed N, Brawley VS, Hegde M. et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2015;33:1688-96
75. Caruso HG, Hurton LV, Najjar A. et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer research. 2015;75:3505-18
76. Schutsky K SD, Lynn R, Smith JB. et al. Rigorous optimization and validation of potent RNA CAR T cell therapy for the treatment of common epithelial cancers expressing folate receptor. Oncotarget. 2015;6:28911-28
77. Barrett DM, Liu X, Jiang S. et al. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum Gene Ther. 2013;24:717-27
78. Panjwani MK SJ, Schutsky K, Gnanandarajah J. et al. Feasibility and Safety of RNA-transfected CD20-specific Chimeric Antigen Receptor T Cells in Dogs with Spontaneous B Cell Lymphoma. Mol Ther. 2016;24:1602-14
79. Abboud R, Keller J, Slade M. et al. Severe Cytokine-Release Syndrome after T Cell-Replete Peripheral Blood Haploidentical Donor Transplantation Is Associated with Poor Survival and Anti-IL-6 Therapy Is Safe and Well Tolerated. Biol Blood Marrow Transplant. 2016;22:1851-60
80. Presland RB DB. Epithelial structural proteins of the skin and oral cavity: function in health and disease. Crit Rev Oral Biol Med. 2000;11:383-408
81. Katz SC, Point GR, Cunetta M. et al. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther. 2016;23:142-8
82. Brown CE, Badie B, Barish ME. et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:4062-72
83. Papa S vSM, Maher J. Clinical Evaluation of ErbB-Targeted CAR T-Cells, Following Intracavity Delivery in Patients with ErbB-Expressing Solid Tumors. Methods Mol Biol. 2015;1317:365-82
84. Qian L, Li D, Ma L. et al. The novel anti-CD19 chimeric antigen receptors with humanized scFv (single-chain variable fragment) trigger leukemia cell killing. Cell Immunol. 2016;304-305:49-54
85. Sun M SH, Liu C, Liu J. et al. Construction and evaluation of a novel humanized HER2-specific chimeric receptor. Breast Cancer Res. 2014;16:R61
86. Johnson LA, Scholler J, Ohkuri T. et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. 2015;7:275ra22
87. Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nature medicine. 2016;22:26-36
88. Kloss CC, Condomines M, Cartellieri M. et al. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71-5
89. Fedorov VD TM, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5:215ra172
90. Wu CY, Roybal KT, Puchner EM. et al. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. science. 2015;350:aab4077
91. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127:3321-30
92. Hacein-Bey-Abina S, Von KC, Schmidt M. et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415-9
93. Singh H, Moyes JS, Huls MH. et al. Manufacture of T cells using the Sleeping Beauty system to enforce expression of a CD19-specific chimeric antigen receptor. Cancer Gene Ther. 2015;22:95-100
94. Deniger DC, Pasetto A, Tran E. et al. Stable, Nonviral Expression of Mutated Tumor Neoantigen-specific T-cell Receptors Using the Sleeping Beauty Transposon/Transposase System. Mol Ther. 2016;24:1078-89
95. Cox DB, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nature medicine. 2015;21:121-31
96. Osborn MJ, Webber BR, Knipping F. et al. Evaluation of TCR Gene Editing Achieved by TALENs, CRISPR/Cas9, and megaTAL Nucleases. Mol Ther. 2016;24:570-81
97. Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol. 2009;9:480-90
98. Markley JC, Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood. 2010;115:3508-19
99. Gargett T, Brown MP. Different cytokine and stimulation conditions influence the expansion and immune phenotype of third-generation chimeric antigen receptor T cells specific for tumor antigen GD2. Cytotherapy. 2015;17:487-95
100. Pouw N, Treffers-Westerlaken E, Kraan J. et al. Combination of IL-21 and IL-15 enhances tumour-specific cytotoxicity and cytokine production of TCR-transduced primary T cells. Cancer Immunol Immunother. 2010;59:921-31
101. Deshpande P, Cavanagh MM, Le Saux S. et al. IL-7- and IL-15-mediated TCR sensitization enables T cell responses to self-antigens. Journal of immunology. 2013;190:1416-23
102. Chen XL, Bobbala D, Rodriguez GM. et al. Induction of autoimmune diabetes in non-obese diabetic mice requires interleukin-21-dependent activation of autoreactive CD8(+) T cells. Clin Exp Immunol. 2013;173:184-94
103. Braun M RM, Yoo YE, Scholz CJ. et al. IL12-mediated sensitizing of T-cell receptor-dependent and -independent tumor cell killing. Oncoimmunology. 2016;5:e1188245
104. Rubinstein MP, Su EW, Suriano S. et al. Interleukin-12 enhances the function and anti-tumor activity in murine and human CD8(+) T cells. Cancer Immunol Immunother. 2015;64:539-49
105. Hoyos V, Savoldo B, Quintarelli C. et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24:1160-70
106. Long AH, Haso WM, Shern JF. et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nature medicine. 2015;21:581-90
107. Hombach AA, Rappl G, Abken H. Arming cytokine-induced killer cells with chimeric antigen receptors: CD28 outperforms combined CD28-OX40 "super-stimulation". Mol Ther. 2013;21:2268-77
108. Zhao Z, Condomines M, van der Stegen SJ. et al. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer cell. 2015;28:415-28
109. Simon P, Omokoko TA, Breitkreuz A. et al. Functional TCR retrieval from single antigen-specific human T cells reveals multiple novel epitopes. Cancer Immunol Res. 2014;2:1230-44
110. Sommermeyer D, Conrad H, Kronig H. et al. NY-ESO-1 antigen-reactive T cell receptors exhibit diverse therapeutic capability. Int J Cancer. 2013;132:1360-7
111. Dubrovsky L, Pankov D, Brea EJ. et al. A TCR-mimic antibody to WT1 bypasses tyrosine kinase inhibitor resistance in human BCR-ABL+ leukemias. Blood. 2014;123:3296-304
112. Ochi T, Fujiwara H, Okamoto S. et al. Novel adoptive T-cell immunotherapy using a WT1-specific TCR vector encoding silencers for endogenous TCRs shows marked antileukemia reactivity and safety. Blood. 2011;118:1495-503
113. Katsuhara A, Aoyama F, Tanii N. et al. Transduction of a novel HLA-DRB1*04:05-restricted, WT1-specific TCR gene into human CD4+ T cells confers killing activity against human leukemia cells. Anticancer research. 2015;35:1251-61
114. Bunse M, Bendle GM, Linnemann C. et al. RNAi-mediated TCR knockdown prevents autoimmunity in mice caused by mixed TCR dimers following TCR gene transfer. Mol Ther. 2014;22:1983-91
115. Poirot L, Philip B, Schiffer-Mannioui C. et al. Multiplex Genome-Edited T-cell Manufacturing Platform for "Off-the-Shelf" Adoptive T-cell Immunotherapies. Cancer research. 2015;75:3853-64
116. Ren J LX, Fang C, Jiang S. et al. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clinical cancer research. 2016 [Epub ahead of print]
117. Cherkassky L, Morello A, Villena-Vargas J. et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. Journal of Clinical Investigation. 2016;126:3130-44
118. Suarez ER CdK, Sun J, Sui J. et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. 2016;7:34341-55
119. Chapuis AG, Ragnarsson GB, Nguyen HN. et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Transl Med. 2013;5:174ra27
120. Liu H, Xiang JY, Long L. et al. Targeting alpha-fetoprotein (AFP)-MHC complex with CAR T cell therapy for liver cancer. Clinical cancer research. 2017;23:478-88
121. Twyman-Saint Victor C, Rech AJ, Maity A. et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373-7
122. Linnemann C HB, Kvistborg P, Kluin RJ. et al. High-throughput identification of antigen-specific TCRs by TCR gene capture. Nature medicine. 2013;19:1534-41
123. Orkin SH, Reilly P. Paying for future success in gene therapy. Science. 2016;352:1059-61
Author contact
![]() Corresponding author: Yongsheng Wang Telephone Number: 18980602258 Email: wangys75com
Corresponding author: Yongsheng Wang Telephone Number: 18980602258 Email: wangys75com
Citation styles
APA
Mo, Z., Du, P., Wang, G., Wang, Y. (2017). The Multi-Purpose Tool of Tumor Immunotherapy: Gene-Engineered T Cells. Journal of Cancer, 8(9), 1690-1703. https://doi.org/10.7150/jca.18681.
ACS
Mo, Z.; Du, P.; Wang, G.; Wang, Y. The Multi-Purpose Tool of Tumor Immunotherapy: Gene-Engineered T Cells. J. Cancer 2017, 8 (9), 1690-1703. DOI: 10.7150/jca.18681.
NLM
Mo Z, Du P, Wang G, Wang Y. The Multi-Purpose Tool of Tumor Immunotherapy: Gene-Engineered T Cells. J Cancer 2017; 8(9):1690-1703. doi:10.7150/jca.18681. https://www.jcancer.org/v08p1690.htm
CSE
Mo Z, Du P, Wang G, Wang Y. 2017. The Multi-Purpose Tool of Tumor Immunotherapy: Gene-Engineered T Cells. J Cancer. 8(9):1690-1703.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.