Journal of Cancer 3.2
Impact Factor
ISSN: 1837-9664
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(10):1884-1891. doi:10.7150/jca.17595 This issue Cite
Review
The tumor microenvironment and inflammatory breast cancer
Aji Huang1, Shousong Cao2, Lili Tang1 ![]()
1. Department of Breast Surgery, Xiangya Hospital, Central South University, Changsha, Hunan 410008, China;
2. Laboratory of Cancer Pharmacology, Department of Pharmacology, School of Pharmacy, Southwest Medical University, Luzhou, Sichuan 646000, China.
Received 2016-9-15; Accepted 2017-4-1; Published 2017-7-3
Citation:
Huang A, Cao S, Tang L. The tumor microenvironment and inflammatory breast cancer. J Cancer 2017; 8(10):1884-1891. doi:10.7150/jca.17595. https://www.jcancer.org/v08p1884.htm
Other stylesAbstract
Inflammatory breast cancer (IBC) is a rare and very aggressive subtype of breast cancer with clinical manifestations similar to acute inflammation. The prognosis of IBC is still poor even though combination therapy with surgery, chemotherapy, and target therapy, mainly due to a lack of fully understanding of the cellular and molecular mechanisms of IBC pathogenesis and progression. In the present article, we have comprehensively reviewed the connection of the pathogenesis of IBC and inflammation, immune reaction and cancer, particularly focused on the role and mechanism of tumor microenvironment related to IBC formation, tumor cell proliferation, migration, invasion and metastasis as well as the clinical manifestations of IBC. As the diverse cells including inflammatory cells, immune cells, and tumor cells and the soluble molecules produced by these cells in the microenvironment play an essential role in IBC development and progression. Therefore, anti-inflammatory therapy and immunotherapy with available agents warrant further investigation in the treatment of IBC.
Keywords: tumor microenvironment, inflammatory breast cancer
Background
It is well known that the cancerogenesis, cancer cell proliferation, survival, invasion and metastasis are closely associated to the tumor microenvironment. This tumor environment is composed of tumor surrounding blood vessels, inflammatory cells, immune cells, lymphocytes, fibroblasts, extracellular matrix and a variety of biological active molecules derived from tumor ccells and the surrounding non-tumor cells in the microenvironment. These factors, together with their associated signal pathways, interact with each other in the tumor microenvironment, forming an extremely complicated signal network. Any change in the network may affect the metabolism and behavior of tumor cells, and consequently alter the tumor progression, clinical manifestations and outcomes in a direct or indirect manner.
The existence of an intrinsic link between inflammation and cancer has been noticed more than a century ago. In 1863, Rudolf Virchow and his colleagues found that white blood cells were present in tumor tissues, hypothesized that inflammation was associated with the tumor, and suggested that the "lymphoreticular infiltrate” reflected the origin of cancer at the site of chronic inflammation [1]. Over the last decade, the role of chronic inflammation in the development of cancer has been explored more intensively and growing evidence has supported Virchow's hypothesis. Epidemiological studies have showed that approximately 25% of tumors are involved in chronic infection or inflammation and the cancer risk in subjects with chronic inflammation or oxygen free radicals overload is much higher than the general population, suggesting an association between inflammation and cancer [2]. Inflammatory mediators such as cytokines, chemokines, free radicals, prostaglandins, and growth hormones can cause genetic and epigenetic alterations, including mutations of tumor suppressor genes, DNA methylation and post-translational modifications, resulting in genomic instability and changes in the critical pathways for the maintenance of normal cell functions, and consequently leading to cell transformation and carcinogenesis [2]. It is now recognized that chronic inflammatory lesions may lead to cancer, and cytokines produced by the inflammatory cells in the tumor microenvironment are the necessary factors for cancer cell survival, proliferation, differentiation, and progression. Meanwhile, the inflammatory cells and released cytokines may interact with other cells, cytokines and external harmful physical and chemical substances in the microenvironment, and together, alter cancer cell behavior. In addition, inflammation may be either a cause of cancer or a consequence induced by cancer. These data collectively indicate the complexity of inflammation and cancer. Providentially, the recent discovery of the interaction between microRNAs and the immune system has shed some light into the understanding of the connection between inflammation and cancer [2]. Furthermore, as tumor microenvironment plays such critical role in tumor initiation, development and progression, interventions targeted to this network may become an effective approach for fighting with cancer.
Inflammatory breast cancer (IBC) is a rare and very aggressive subtype of breast cancer and needs early diagnosis and multimodal treatment. IBC was first described in 1814, and subsequently named by Lee and Tannenbaum in 1924 due to its fast progress and similar appearance as acute inflammation [3]. The incidence of IBC is quite low but it is very aggressive and possesses, rapid progression, and poor prognosis. Since the initial clinical manifestations of IBC are similar to breast infection or inflammation, it is easily misdiagnosed as acute mastitis and other infectious diseases, treated with antibiotics consequently, and delay the anticancer treatment [4]. The strategy for the management of IBC is a multimodal approach with an individualized combination of chemotherapy, surgery, radiation and targeted therapy due to a lack of specific therapy [4].
Currently, the pathogenesis and molecular mechanisms of IBC development are poorly understood. With the advancement of modern technologies, more and more studies have been conducted to elucidate the molecular mechanisms, and determine the differences between IBC and other breast cancer. Studies over the years have suggested that the etiology and pathogenesis of IBC may be closely related to inflammation due to the apparent common inflammatory manifestations and changes in microenvironment. In the present article, we have reviewed the association among inflammation, immunity and cancer, especially focused on the relationship between inflammatory mediators and IBC, and on the mechanism related to the clinical manifestations of IBC.
Clinical characteristics and epidemiological profile of IBC
The clinical features of IBC
According to the American Joint Committee on Cancer (AJCC) Tumor-Node-Metastasis (TNM) staging system of breast cancer, IBC is a rare subtype of locally advanced breast cancer, and classified as T4d [5, 6]. Patients with IBC typically present with a rapid-onset redness and thickening (edema/swelling), involving more than 1/3 of the breast in a short period. The diffuse redness in the breast skin is initially pink, quickly becomes bleeding-like purple and erysipelas-like changes as skin edema, thickening and hardening. The ipsilateral breast is enlarged, and shows an orange peel-like appearance with clear skin edges but unclear tumor boundary due to edema and swelling. There is no clear masse in about one-third of the cases with only inflammation-like changes. The temperature of ipsilateral skin is often higher than the opposite breast skin. The breast may also be tender and painful or itchy. More than half of the patients have enlarged palpable axillary or supraclavicular lymph nodes at the time of diagnosis, and some patients even have bone, lung, liver, brain and other distant metastases with relevant clinical manifestations [7]. It should be noted that the breast redness, tenderness, warmth and pain or itching are common symptoms of a breast infection or inflammation such as mastitis, but antibiotic treatment will not improve the disease. Once IBC is suspected based on clinical manifestations, imaging tests such as mammogram, breast ultrasound and MRI may be needed, but only a breast biopsy can make a definite diagnosis. IBC has no special histological changes and various pathological types of breast cancer have been observed. However, the intradermal lymphatic tumor thrombus is considered as a characteristic pathological finding [8]. Based on the biopsy, the cancer cells may be graded and further molecular pathological examination of cancer cells such as HER2 and estrogen receptor will aid in clinical management.
Epidemiology of IBC
Hance et al [9] have analyzed 180,224 cases with diagnosed breast cancer from 1988 to 2000 in the SEER database. A total of 3648 cases were classified as IBC, 3636 cases as locally advanced breast cancer (LABC), and 172,940 cases as non-T4 stage breast cancer. IBC was accounted for approximately 2% of all breast cancers. During the period of study, the incidence (per 100,000 woman/year) of IBC was significantly increased from 2.0 to 2.5 while the incidence of LABC was significantly decreased from 2.5 to 2.0 and the non-T4 breast cancer from 108 to 101. The incidence of IBC in African American women was significantly higher than in Caucasian women (3.1 vs 2.2/100,000 women/-year). The median survival times were 2.9 years for IBC, 6.4 years for LABC, and more than 10 years for non-T4 breast cancer, respectively. The median survival times of IBC and LABC were significantly shorter in black women than in white women. Compared to the 2% incidence, IBC accounted for 7% (1936/27747) of mortality for all breast cancer, so the mortality rate of IBC was significantly higher than other types of breast cancer [9]. The incidence of IBC has a large geographical difference with a 2% incidence in China [10], and a 6-10% incidence in the countries of North Africa, particularly in Morocco, Algeria, Tunisia, and Egypt [11]. Robertson et al. [12] reported that the five-year survival rate of the patients with IBC was less than 5% when treated with single surgical resection or chemotherapy. However, Baldini et al. [13] performed a prospective study in 68 cases of IBC patients treated with a combination of surgery, chemotherapy and radiotherapy, and found that the 5-year and 10-year disease free survival rates were 29% and 20%, respectively, with a median disease free survival time of 2.2 years; and the 5-year and 10-year overall survival rates were 44% and 32%, respectively, with a median overall survival time of 4 years [13] (Table 1). Taken together, these studies indicate that the effectiveness of a multimodal approach with a combination of surgery, chemotherapy and radiotherapy is significantly superior to a monotherapy. Even so, the therapeutic efficacy of IBC is still unsatisfactory and remains to be improved.
Table 1
The survival rate of breast cancer (13)
| Disease free survival rate | 5-year 29% |
| 10-year 20% | |
| Overall survival rate | 5-year 44% |
| 10-year 32% | |
| Median disease free survival time | 2.2 years |
| Median overall survival time | 4.0 years |
The etiology and pathogenesis of IBC
The etiology and pathogenesis of IBC are poorly understood. Studies over the years have suggested that P53, nuclear factor kappa B (NF-кB), TNF, IL-6, IFN and other substances that regulate the processes of inflammation, carcinogenesis, cancer development, and cell apoptosis may be involved in IBC development and progression [1]. NF-кB, a family of the transcription factor proteins, is involved in various immune regulation and inflammation through controlling the expression of many cytokines, chemokines, growth factors, and adhesion factors [14], plays a key role in cell proliferation, differentiation, apoptosis, invasion and metastasis [15]. NF-кB has been shown to be activated in a variety of tumors [16]. Mann et.al., [17] have demonstrated that the overexpression of transglutaminase (TG2) can activate NF-кB, while TG2 knockdown by small interfering RNAs (siRNAs) eliminates NF-кB activation in multiple cell lines and tumor tissues, suggesting that TG2 may be a useful molecular target for anticancer therapy. Moreover, Lerebours et al. [18] have studied the NF-кB related gene expression in tumor tissues from 35 IBC and 22 non-IBC patients and identified six genes including TNFAIP3/A20, SELE, COX2, CXCL12, CCND3 and IER3L as a molecular signature to distinguish IBC from non-IBC. In addition, it has reported that an activation of NF-кB in IBC is related to a decrease in estrogen receptor levels [19]. These data collectively suggest that NF-кB is key factor in IBC formation, development and progression.
P53, a tumor suppressor gene, can inhibit cell proliferation and promote cell apoptosis, which is completely opposite to the roles of NF-кB in the cells and NF-кB activation is contributed to cancer development and progression [16]. Sawaki et al have reported that IBC patients whose tumors were p53-positive had a better outcome compared to those with p53-negative tumors in a small single-center retrospective study. In contrast, Saydam et al [20] have shown that IBC patients whose tumors were p53-negative had similar survival outcome compared to p53-negative non-IBC, while patients with p53-positive IBC had worse survival outcome compared to p53-positive non-IBC. This finding indicates that p53 might be an indicator of poor prognosis in IBC. These apparent conflict reports suggest that the significance of p53 in IBC is not clear and remains to be elucidated. Further analysis of p53 gene mutation and functionality may shed some light for these controversial findings.
IL-6, a pro-inflammatory cytokine, is involved in cancer cells proliferation, survival and metastasis. Studies have demonstrated that serum IL-6 levels, which are regulated by protein kinase CK2, were significantly increased in patients with IBC compared to those with non-IBC [21]. Wolfe et al [22] have recently demonstrated that IBC cells co-cultured with bone marrow derived mesenchymal stem cells (MSCs) showed enhanced tumor formation and invasion, which was blocked by anti-IL-6 and statin treatment, indicating a significant role of IL-6 in tumor formation and metastasis.
A number of studies have indicated that inflammatory cells attracted to the sites of tumor lesions promote rather than inhibit tumor growth [23]. Further analyses have found that a variety of inflammatory cells such as macrophages, neutrophils, eosinophils, dendritic cells, and lymphocytes, etc., were involved in the control of tumor development and progression [24, 25]. Growing evidence indicates that tumor cells could regulate the infiltration and activation of the host cells to promote their own survival and progression, and the white blood cells presented in the tumor microenvironment could promote tumor growth, angiogenesis, and metastasis via producing a series of growth factors, proteases, and angiogenic mediators [24]. Mast cells are one of the important immune regulatory cells to induce colitis-related colorectal cancer (CRC). Since the inner mucosal mast cells (MMCs) and connective tissue mast cells (CTMCs) play different intestinal barrier functions in the case of stability of internal environment and inflammation, it is difficult to determine which type of mast cells plays the key role in the development of CRC from colitis to colon cancer. Xu et al., [25] found that both numbers and gene expression levels of MMCs were increased in the mouse model of experimental induced CRC. After MMC activation, the production of mast cell protease 1 not only caused the accumulation of CD11b+Gr1+ inflammatory cells in the colon tissues but also regulated the activity of CD11b+Gr1+ cells to support the growth of tumor cells and to inhibit the activation of T cells. Prevention of the activation of mice MMC (which can lead to the abnormality of colitis-associated epithelial structure), could reduce the infiltration of CD11b+Gr1+ cells and inhibit the development of CRC. The results from the study suggest that the activation of MMCs can increase and regulate CD11b+Gr1+ cells to promote the formation of CRC; therefore, MMCs can be the target cells in the prevention and treatment of CRC. Many cancers occur in the sites of body suffering from infection, chronic irritation and inflammation. It is relatively clear that the tumor microenvironment is the essential condition for the differentiation, survival and metastasis of tumors and inflammatory cells are heavily involved in the regulation of tumor microenvironment. On the other hand, tumor cells also can regulate some signaling molecules of the body's natural immune system, such as selecting proteins (selectins), chemokines and their receptors to provide services for the tumor cells transfer, migration and invasion. Right now, people have widely recognized that there is a causal relationship among inflammatory reaction, the body's natural immune system and cancer. The important condition for the inflammatory cells to develop cancer is the occurrence of continuous cell differentiation in the microenvironment of being rich in inflammatory cells, growth factors, activated matrixes, and DNA-damaged materials. This environment will certainly increase the risk of cancer [26]. The normal inflammatory response is self-limited because the anti-inflammatory cytokines will soon be produced after the emergence of pro-inflammatory cytokines. However, chronic inflammatory response may be out of control in the present of the above-mentioned harmful substances in the environment. In addition, the tumor cells themselves can produce a variety of cytokines and chemokines to attract the migration of leukocytes to the site of the tumor. The neoformation in the developmental stage contains a variety of white blood cells, such as neutrophils, eosinophils, dendritic cells, mast cells, macrophages and lymphocytes. All of these cells can produce a series of cytokines, cytotoxic mediators such as reactive oxygen species, cysteine and serine proteases, matrix metalloproteinases, membrane penetration agents, and soluble cell killing media, such as TNF-α, interleukin and interferon. These materials under unfavorable microenvironment (such as damaged DNA) or with the presence of the tumor cells, can promote tumor formation or maintain the growth and survival of the tumor. There are two kinds of pathways to connect inflammation and cancer, endogenous and exogenous. The exogenous pathway is caused by inflammation or infection and the endogenous pathway is caused by the activation of oncogenes, the two pathways promote the formation of tumor microenvironment. It is apparent that many cancers are originated in the sites of infection, chronic irritation and inflammation. It is now realized that inflammatory cells are heavily involved in tumor microenvironment and tumor microenvironment is essential for tumor initiation, survival, growth and metastasis. Furthermore, tumor cells are able to interact with immune system to facilitate tumor cell transfer, migration and invasion. It is now documented that there is a close association among inflammation, immune system and cancer. An important prerequisite for inflammatory cells to stimulate cancer development is the occurrence of continuous cell proliferation/differentiation in the microenvironment rich in inflammatory cells, growth factors, activated matrixes, and DNA-damaged materials [26]. It showed that only few of tumors were induced by germline mutations, while mostly (90%) were caused by somatic mutations and environmental factors [27]. In inflammatory microenvironment, there are three basic stages for tumor formation and development: initiation, promotion, and metastasis. It is involved with inflammatory cells, immune cells, and a variety of induced bio-active substances. Grivennikov et al. [27] have described the relationship between immune cells, inflammatory cells and cancer, and stated that the tumor microenvironment contains innate immune cells (including macrophages, neutrophils, mast cells, myeloid-derived suppressor cells, dendritic cells, and natural killer cells) and adaptive immune cells (T and B lymphocytes) in addition to the cancer cells and their surrounding stroma (including fibroblasts, endothelial cells, pericytes, and mesenchymal cells). These diverse cells communicate with each other through either direct cell contact or autocrine and paracrine actions of cell-derived cytokines and chemokines to control the direction of the microenvironment conditions in favor of tumor-promoting inflammation or antitumor immunity [27]. This complicated relationship among inflammatory cells, immune cells, cancer cells and extracellular matrix in the tumor microenvironment are schematically illustrated in Figure 1.
Figure 1
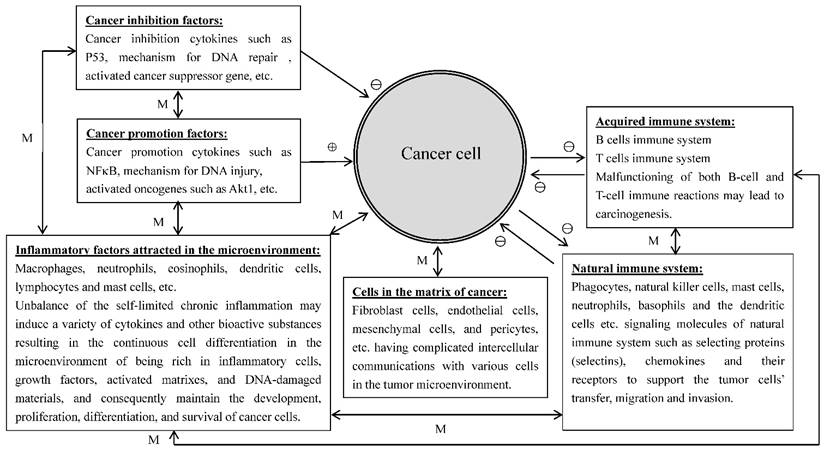
The relationships of the inflammatory cells, immune cells, cancer cells and extracellular matrix in the tumor microenvironment. ⊕: promotion; ⊖: Inhibition; ↔: mutual reaction; M: depends on the mutual reaction in specific context.
Molecular and cellular biological Mechanisms of IBC
Some inflammatory mediators secreted by the cells released to the plasma function in the distance through the blood circulation, and the others function at the local tissues or the cells themselves which produce inflammatory mediators through autocrine and paracrine of the cells. The common inflammatory mediators include vasoactive amines, arachidonic acid metabolites, cytokines and chemokines [28]. The expressions of chemokines CCL2 and CCL5, TNF-α, and IL-1β were significantly higher in the breast cancer than in the normal breast tissues [29]. NF-κB is a family of the transcription factor proteins mediating the development of inflammation, promoting of the evolution of chronic inflammation [14], and enhancing the invasion and metastasis of tumors[15]. It related to the decrease of estrogen receptor levels in IBC [19]. Although growing evidence indicates that various factors in the tumor microenvironment interact with IBC cells and promote the invasiveness and metastasis of IBC cells, the mechanism related to this change is unclear. Cohen et al. [30] hypothesized that the soluble cytokines secreted by the activated immune cells and inflammatory cells can cause epithelial to mesenchymal transition (EMT) of the IBC cells, resulting in a facilitation of cancer cell transfer, migration and invasion. EMT refers to a process by which epithelial cells lose the cell polarity and cell-cell adhesion, and obtain migratory and invasive properties to become mesenchymal stem cells, which is characterized by the absence of epithelial phenotype and exhibition of mesenchymal cell phenotype. They [30] have also observed a correlation between the circulating levels of TNF-α and EMT biomarkers in patients with breast cancer. Moreover, they have demonstrated that multiple cytokines originated from the activated T cells stimulated the expressions of such EMT-related genes as fibronectin (FN1), vimentin (VIM), tissue transglutaminase (TGM2) and zinc-finger E-box binding homeobox1 (ZEB1) in several IBC cell lines. This data suggests that EMT may play a significant role in cytokine promotion of IBC cell invasion and metastasis. It is also interesting to note that SUM149 IBC cells went through epithelial interstitial transformation with increased E-cadherin expression and enhanced cell invasion and migration under the stimulation of immune factors, whereas this phenomenon did not occur in non-IBC cells, and E-cadherin expression is generally reduced in other tumor cells by such immune factors [30]. When both cell migration and E-cadherin expression are simultaneously activated, IBC cells may form tight cell clusters known as "tumor emboli", leading to an occlusion of lymphatic vessels and resulting in the inflammation-like clinical manifestations observed in IBC [30]. Joglekar et al. [31] have investigated the effect of caveolin-1 on the invasion of IBC cells and demonstrated that the caveolin-1 induced invasion of IBC cells was mediated through activation of Akt1, which phosphorylated RhoC GTPase, a metastatic switch responsible for tumor phenotype. Yoshimura et al. [31] studied the expression of COX1 and COX2 in the breast cancer tissues and found that the expression of COX1 was relatively weak while the expression of COX2 was much higher in the breast cancer tissues compared to benign breast tumors such as fibroadenomas and breast cystic hyperplasia. COX2 overexpression has also been observed in the IBC tissues and IBC metastatic sites. Moreover, Robertson et al. [32] have demonstrated that the administration of GW627368X, a selective antagonist of prostaglandin E2 receptor 4 (EP4), and the knockdown of EP4 by shRNA significantly inhibited cell proliferation and invasion in SUM149 IBC cells. Taken together, these data suggest that COX2 is overexpressed in breast cancer tissues and COX2/prostanoid signal pathway plays a significant role in IBC growth and metastasis, presumably mediated through EP4[32-34]
The S100 proteins, originally isolated by Moore et al. in 1965, are a family of low-molecular-weight (9-14 kD) acidic proteins characterized by two calcium-binding sites that have helix-loop-helix conformation. There are currently at least 21 different S100 proteins encoded by distinctive genes. Studies over the years have shown that S100 proteins may possess significant functions in the pathological process of inflammations, tumorigenesis, tumor growth, tumor metastasis and angiogenesis [35, 36]. Hansen et al. [37] have shown in breast cancer models that the administration of S100A4 induced the expressions of acute-phase serum amyloid A (SAAs), protein molecules that play key roles in inflammation, and cytokines via Toll-like receptor 4 (TLR4)/nuclear factor-κB signaling, and the aberrant expressions of acute-phase SAA1 and SAA3 in the tumor cells strongly promoted widespread metastasis accompanied by a massive infiltration of immune cells, suggesting that SAAs are effectors of S100A4 for promoting tumor metastasis, and may serve as a link between inflammation and tumor progression.
The proteins S100A8 and S100A9, two members in S100 family that form a heterodimer called calprotectin, are pro-inflammatory response proteins present in the tumor microenvironment, also play a significant role in the cell growth, proliferation, differentiation, invasion, and metastasis in breast cancer [38]. S100A8 and S100A9 overexpression was observed in the patients with high pathological grade, estrogen receptor (ER) negative, or basal-like breast cancer, and strongly associated with a poor prognosis [39-41], but not in metastatic lymph nodes [42]. S100A8 and S100A9 proteins have been shown to promote tumor metastasis in the brain in a xenograft 4T1 animal model, presumably through attracting myeloid derived suppressor cells and metastatic cancer cells [43]. S100A8 and S100A9 have been reported to enhance the cell invasion and metastasis via binding to the receptor of advanced glycation endproducts (RAGE) in MCF-7 and MDA-MB-231 breast cancer cells [44]. Moreover, S100A8 and S100A9 are closely related to inflammatory cytokines as IL-1β and TNFα have been revealed to upregulate S100A8 and S100A9 expressions in SKBR3 and MCF7 breast cancer cells [45]. It has been shown that some of the major inflammatory signaling pathways such as NF-κB, COX-2 and JAK/STAT signaling systems may be in a constantly activated state, and play a major role in IBC initiation; some inflammatory molecules such as IL-6, TNF-α and γ-interferon (γ-IFN) may mainly function in the process of malignant transformation of IBC, while others such as transforming growth factor-β (TGF-β), IL-8, IL-1β and TNF-α are involved in cancer cell proliferation, survival, EMT, invasion, and metastasis [46]. In summary, the cumulative evidence up-to-date strongly indicates that the diverse cells, bioactive molecules and their related signal transduction pathways in the tumor microenvironment of IBC communicate and interact with each other, forming a complicated network, presumably mediated through various molecular and cellular biological mechanisms in the control of IBC formation, development and progression as illustrated in Figure 2.
Conclusion
IBC is a rare and very aggressive disease with a poor prognosis. Although IBC has been described over a centenary and the mechanisms of IBC pathogenesis and progression are far from clear, significant progress in understanding the disease such as the role of microenvironment, has only emerged in the last 2 decades. In the tumor microenvironment, a variety of cells such as inflammatory cells, immune cells, matrix cells and cancer cells and the bioactive molecules produced by these cells commute and interact with each other, forming a complex signal transduction network to regulate IBC formation, development and progression. The aberrant expressions of inflammatory factors and immune factors may promote carcinogenesis, tumor cell proliferation, migration, invasion and metastasis. Since IBC lacks specific histopathological features to non-IBCs, we hypothesize that IBC cells are similar to non-IBC cells except their capacity of migration and invasion are dramatically enhanced by the stimulation of inflammatory and immune factors in the tumor microenvironment. In addition, the ability of IBC cells to form cancer emboli in the lymphatic and/or blood vessels under the actions of inflammatory and immune factors leading to a specific clinical presentation in IBC patients. Although the use of multimodal therapy has improved the outcome of IBC patients, the therapeutic efficacy is far from satisfactory. With the growing understanding of the critical role of tumor microenvironment, particularly inflammatory and immune cytokines and hormones, in IBC development and progression, the effects of proinflammatory cytokine blockade and immunotherapy have not been explored in IBC therapy and warrant further investigation either as a monotherapy or as a combination therapy regimen.
Abbreviations
IBC, inflammatory breast cancer; LABC, locally advanced breast cancer; NF-кB, nuclear factor kappa B; TG2, tissue transglutaminase; CRC, colitis-related colon cancer; MMCs, mucosal mast cells; EMT, epithelial to mesenchymal transition; COX, cyclooxygenase; SAA, serum amyloid A; ER, estrogen receptor; IL, interleukin; TNF, tumor necrosis factor; IFN, interferon; TGF, transforming growth factor; CD, cluster of differentiation.
Figure 2
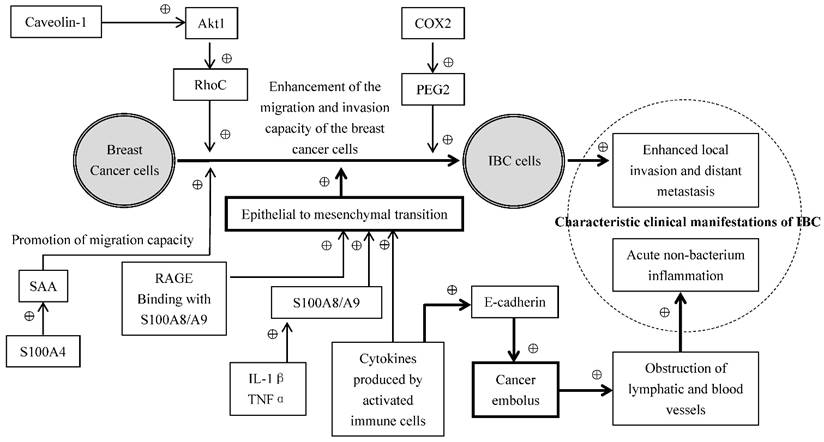
Proposed molecular and cellular biological mechanisms of IBC. ⊕: Promotion; Bold line and arrowheads: emphasized route or approach.
Acknowledgements
This work was supported by a grant from the Hunan Provincial Innovation Foundation for Postgraduate (CX2014B112). The authors wish to thank Dr. Yuanshan Zhu (Weill Cornell Medical College, Cornell University, New York, NY, USA) for critical reviewing, proof-reading and editing the manuscript.
Competing Interests
The authors have declared that no competing interests exist.
References
1. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539-45
2. Hussain SP, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer. 2007;121:2373-80
3. Ma Z, Li F, Zhang W. The diagnosis and prevention of inflammatory breast cancer. Journal of Nongken Medicine. 2012:44-8
4. Matro JM, Li T, Cristofanilli M, Hughes ME, Ottesen RA, Weeks JC. et al. Inflammatory breast cancer management in the national comprehensive cancer network: the disease, recurrence pattern, and outcome. Clin Breast Cancer. 2015;15:1-7
5. van Uden DJ, van Laarhoven HW, Westenberg AH, de Wilt JH, Blanken-Peeters CF. Inflammatory breast cancer: an overview. Crit Rev Oncol Hematol. 2015;93:116-26
6. Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17:1471-4
7. Zhou J, Tang L. Status and development of inflammatory breast cancer. Chinese Archives of General Surgery (Electronic Edition). 2013:307-10
8. Vermeulen PB, van Golen KL, Dirix LY. Angiogenesis, lymphangiogenesis, growth pattern, and tumor emboli in inflammatory breast cancer: a review of the current knowledge. Cancer. 2010;116:2748-54
9. Hance KW, Anderson WF, Devesa SS, Young HA, Levine PH. Trends in inflammatory breast carcinoma incidence and survival: the surveillance, epidemiology, and end results program at the National Cancer Institute. J Natl Cancer Inst. 2005;97:966-75
10. Shao Z, Liu G, Ding J. [Establishment and characterization of human inflammatory breast carcinoma neoplasm transplantation in nude mice]. Zhonghua Wai Ke Za Zhi. 2001;39:796-8
11. Soliman AS, Kleer CG, Mrad K, Karkouri M, Omar S, Khaled HM. et al. Inflammatory breast cancer in north Africa: comparison of clinical and molecular epidemiologic characteristics of patients from Egypt, Tunisia, and Morocco. Breast Dis. 2011;33:159-69
12. Robertson FM, Bondy M, Yang W, Yamauchi H, Wiggins S, Kamrudin S. et al. Inflammatory breast cancer: the disease, the biology, the treatment. CA Cancer J Clin. 2010;60:351-75
13. Baldini E, Gardin G, Evagelista G, Prochilo T, Collecchi P, Lionetto R. Long-term results of combined-modality therapy for inflammatory breast carcinoma. Clin Breast Cancer. 2004;5:358-63
14. Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7-11
15. Luo JL, Maeda S, Hsu LC, Yagita H, Karin M. Inhibition of NF-kappaB in cancer cells converts inflammation- induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell. 2004;6:297-305
16. Philip M, Rowley DA, Schreiber H. Inflammation as a tumor promoter in cancer induction. Semin Cancer Biol. 2004;14:433-9
17. Mann AP, Verma A, Sethi G, Manavathi B, Wang H, Fok JY. et al. Overexpression of tissue transglutaminase leads to constitutive activation of nuclear factor-kappaB in cancer cells: delineation of a novel pathway. Cancer Res. 2006;66:8788-95
18. Lerebours F, Vacher S, Andrieu C, Espie M, Marty M, Lidereau R. et al. NF-kappa B genes have a major role in inflammatory breast cancer. BMC Cancer. 2008;8:41
19. Van Laere SJ, Van der Auwera I, Van den Eynden GG, van Dam P, Van Marck EA, Vermeulen PB. et al. NF-kappaB activation in inflammatory breast cancer is associated with oestrogen receptor downregulation, secondary to EGFR and/or ErbB2 overexpression and MAPK hyperactivation. Br J Cancer. 2007;97:659-69
20. Saydam BK, Goksel G, Korkmaz E, Zekioglu O, Kapkac M, Sanli UA. et al. Comparison of inflammatory breast cancer and noninflammatory breast cancer in Western Turkey. Med Princ Pract. 2008;17:475-80
21. Drygin D, Ho CB, Omori M, Bliesath J, Proffitt C, Rice R. et al. Protein kinase CK2 modulates IL-6 expression in inflammatory breast cancer. Biochem Biophys Res Commun. 2011;415:163-7
22. Wolfe AR, Trenton NJ, Debeb BG, Larson R, Ruffell B, Chu K. et al. Mesenchymal stem cells and macrophages interact through IL-6 to promote inflammatory breast cancer in pre-clinical models. Oncotarget. 2016
23. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57-70
24. Lin EY, Pollard JW. Role of infiltrated leucocytes in tumour growth and spread. Br J Cancer. 2004;90:2053-8
25. Xu L, Yi HG, Wu Z, Han W, Chen K, Zang M. et al. Activation of mucosal mast cells promotes inflammation-related colon cancer development through recruiting and modulating inflammatory CD11b(+)Gr1(+) cells. Cancer Lett. 2015;364:173-80
26. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860-7
27. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883-99
28. Korniluk A, Koper O, Kemona H, Dymicka-Piekarska V. From inflammation to cancer. Ir J Med Sci. 2016
29. Soria G, Ofri-Shahak M, Haas I, Yaal-Hahoshen N, Leider-Trejo L, Leibovich-Rivkin T. et al. Inflammatory mediators in breast cancer: coordinated expression of TNFalpha & IL-1beta with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition. BMC Cancer. 2011;11:130
30. Cohen EN, Gao H, Anfossi S, Mego M, Reddy NG, Debeb B. et al. Inflammation Mediated Metastasis: Immune Induced Epithelial-To-Mesenchymal Transition in Inflammatory Breast Cancer Cells. PLoS One. 2015;10:e0132710
31. Joglekar M, Elbazanti WO, Weitzman MD, Lehman HL, van Golen KL. Caveolin-1 mediates inflammatory breast cancer cell invasion via the Akt1 pathway and RhoC GTPase. J Cell Biochem. 2015;116:923-33
32. Robertson FM, Simeone AM, Mazumdar A, Shah AH, McMurray JS, Ghosh S. et al. Molecular and pharmacological blockade of the EP4 receptor selectively inhibits both proliferation and invasion of human inflammatory breast cancer cells. J Exp Ther Oncol. 2008;7:299-312
33. Yoshimura N, Sano H, Okamoto M, Akioka K, Ushigome H, Kadotani Y. et al. Expression of cyclooxygenase-1 and -2 in human breast cancer. Surg Today. 2003;33:805-11
34. Zhu S, Yang J, Guan Y. The latest progress in E-prostanoid (EP) receptor subtypes EP2 and EP4. Clinical Medical Engineering. 2009:109-12
35. Bresnick AR, Weber DJ, Zimmer DB. S100 proteins in cancer. Nat Rev Cancer. 2015;15:96-109
36. Li C, Zhang F, Wang Y. S100A proteins in the pathogenesis of experimental corneal neovascularization. Mol Vis. 2010;16:2225-35
37. Hansen MT, Forst B, Cremers N, Quagliata L, Ambartsumian N, Grum-Schwensen B. et al. A link between inflammation and metastasis: serum amyloid A1 and A3 induce metastasis, and are targets of metastasis-inducing S100A4. Oncogene. 2015;34:424-35
38. Cormier K, Harquail J, Ouellette RJ, Tessier PA, Guerrette R, Robichaud GA. Intracellular expression of inflammatory proteins S100A8 and S100A9 leads to epithelial-mesenchymal transition and attenuated aggressivity of breast cancer cells. Anticancer Agents Med Chem. 2014;14:35-45
39. Arai K, Takano S, Teratani T, Ito Y, Yamada T, Nozawa R. S100A8 and S100A9 overexpression is associated with poor pathological parameters in invasive ductal carcinoma of the breast. Curr Cancer Drug Targets. 2008;8:243-52
40. Parris TZ, Kovacs A, Aziz L, Hajizadeh S, Nemes S, Semaan M. et al. Additive effect of the AZGP1, PIP, S100A8 and UBE2C molecular biomarkers improves outcome prediction in breast carcinoma. Int J Cancer. 2014;134:1617-29
41. McKiernan E, McDermott EW, Evoy D, Crown J, Duffy MJ. The role of S100 genes in breast cancer progression. Tumour Biol. 2011;32:441-50
42. Arai K, Teratani T, Kuruto-Niwa R, Yamada T, Nozawa R. S100A9 expression in invasive ductal carcinoma of the breast: S100A9 expression in adenocarcinoma is closely associated with poor tumour differentiation. Eur J Cancer. 2004;40:1179-87
43. Liu Y, Kosaka A, Ikeura M, Kohanbash G, Fellows-Mayle W, Snyder LA. et al. Premetastatic soil and prevention of breast cancer brain metastasis. Neuro Oncol. 2013;15:891-903
44. Yin C, Li H, Zhang B, Liu Y, Lu G, Lu S. et al. RAGE-binding S100A8/A9 promotes the migration and invasion of human breast cancer cells through actin polymerization and epithelial-mesenchymal transition. Breast Cancer Res Treat. 2013;142:297-309
45. Bode G, Luken A, Kerkhoff C, Roth J, Ludwig S, Nacken W. Interaction between S100A8/A9 and annexin A6 is involved in the calcium-induced cell surface exposition of S100A8/A9. J Biol Chem. 2008;283:31776-84
46. Fouad TM, Kogawa T, Reuben JM, Ueno NT. The role of inflammation in inflammatory breast cancer. Adv Exp Med Biol. 2014;816:53-73
Author contact
![]() Corresponding author: Dr. Lili Tang, Department of Breast Surgery, Xiangya Hospital, Central South University, 87 Xiangya Road, Changsha, Hunan 410008, China. Tel: +86 13574139088; E-mail: tlli77com.
Corresponding author: Dr. Lili Tang, Department of Breast Surgery, Xiangya Hospital, Central South University, 87 Xiangya Road, Changsha, Hunan 410008, China. Tel: +86 13574139088; E-mail: tlli77com.
Citation styles
APA
Huang, A., Cao, S., Tang, L. (2017). The tumor microenvironment and inflammatory breast cancer. Journal of Cancer, 8(10), 1884-1891. https://doi.org/10.7150/jca.17595.
ACS
Huang, A.; Cao, S.; Tang, L. The tumor microenvironment and inflammatory breast cancer. J. Cancer 2017, 8 (10), 1884-1891. DOI: 10.7150/jca.17595.
NLM
Huang A, Cao S, Tang L. The tumor microenvironment and inflammatory breast cancer. J Cancer 2017; 8(10):1884-1891. doi:10.7150/jca.17595. https://www.jcancer.org/v08p1884.htm
CSE
Huang A, Cao S, Tang L. 2017. The tumor microenvironment and inflammatory breast cancer. J Cancer. 8(10):1884-1891.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.