Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(5):890-900. doi:10.7150/jca.21306 This issue Cite
Research Paper
Inhibition of PI3K/AKT Signaling Pathway Radiosensitizes Pancreatic Cancer Cells with ARID1A Deficiency in Vitro
Lin Yang1, Guanghai Yang2, Yingjun Ding3, Yuhong Dai1, Sanpeng Xu4, Qiuyun Guo1, Aini Xie5, Guangyuan Hu1 ![]()
1. Department of Oncology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei 430030, P.R. China.
2. Department of Thoracic Surgery, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei 430030, P.R. China.
3. Department of Physiology, University of Oklahoma Health Sciences Center, 975 N 10 th St, Oklahoma City, OK, US, 73104
4. Department of Pathology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei 430030, P.R. China.
5. Department of Cardiovascular Surgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei 430030, P.R. China.
Received 2017-6-3; Accepted 2018-1-29; Published 2018-2-25
Citation:
Yang L, Yang G, Ding Y, Dai Y, Xu S, Guo Q, Xie A, Hu G. Inhibition of PI3K/AKT Signaling Pathway Radiosensitizes Pancreatic Cancer Cells with ARID1A Deficiency in Vitro. J Cancer 2018; 9(5):890-900. doi:10.7150/jca.21306. https://www.jcancer.org/v09p0890.htm
Other stylesAbstract
Pancreatic cancer is among the most aggressive human cancers, and is resistant to regular chemotherapy and radiotherapy. The AT-rich interactive domain containing protein 1A (ARID1A) gene, a crucial chromatin remodeling gene, mutates frequently in a broad spectrum of cancers, including pancreatic cancer. Recent evidence suggests that ARID1A acts as tumor suppressor and plays an important role in DNA damage repair (DDR). However, the effect of ARID1A on the radiosensitivity of pancreatic cancer remains unclear. Herein, we investigated the involvement of ARID1A depletion in the radioresistance of pancreatic cancer cells, and explored the underlying mechanisms. The results reveal that knockdown of ARID1A enhances the radioresistance of pancreatic cancer cells through suppressing apoptosis, impairing G2-M checkpoint arrest, strengthening DDR, and accompanying activation of PI3K/AKT signaling pathway. Moreover, upon inhibition of PI3K/AKT pathway by PI3K-inhibitor LY294002 or AKT-inhibitor mk2206, the radiosensitivity of ARID1A-deficient pancreatic cancer cells is improved in vitro via increased apoptosis and weakened DDR. Taken together, these data suggest that loss of ARID1A expression enhances radioresistance of pancreatic cancer through activation of PI3K/AKT pathway, which maybe a promising target for radiosensitization of ARID1A-deficient pancreatic cancer.
Keywords: ARID1A, pancreatic cancer, PI3K/AKT pathway, radioresistance
Introduction
Pancreatic cancer, one of the most hyper-invasive human cancers, is the fourth leading cause of cancer-related deaths in the United States [1]. Over 250,000 people develop pancreatic cancer every year worldwide [2]. Despite 50 years of research and therapeutic development, the 5-year relative survival is only 8% [1].
More than 80% of pancreatic cancers patients are inoperable when diagnosed [3]. For such patients, chemotherapy only affords a median survival of less than one year [4, 5], even in combination with erlotinib [6]. Recent progress in radiotherapy has raised extensive concerns, including the application of new radiation technologies [7], and new sensitizing drugs [8, 9], especially for targeted agents [10, 11]. Pancreatic cancer displays a variety of gene mutations, which may suggest new therapeutic targets.
AT-rich interactive domain containing protein 1A (ARID1A), a chromatin remodeling gene, has been recently found to be mutated in various cancers, including ovarian clear cell carcinoma [12], endometrioid carcinoma [13], gastric cancer [14], breast cancer [15], liver cancer [16], and pancreatic cancer [17, 18]. ARID1A encodes the protein BRG1-associated factor 250a (BAF250a), a crucial noncatalytic subunit of the human switch/sucrose non-fermentable (SWI/SNF) complex [19], which participates in several important cell functions, including transcription, DNA replication, and DNA repair [20-22]. SWI/SNF was reported as a central tumor suppressive complex in pancreatic cancer, and it affected at least one-third of all pancreatic cancer cases [17].
Mutations of ARID1A usually cause down-regulated protein expression [23]. Moreover, functional data suggest that ARID1A is a bona fide tumor suppressor [24]. In pancreatic cancer, the incidence of ARID1A mutations varies from 6% to 9% [25, 26]. A review of the COSMIC database reveals that ARID1A is among the five most frequent somatic mutations in pancreatic cancer [25]. Furthermore, Witkiewicz AK et al. found that ARID1A protein deficiency was significantly associated with poor outcome in pancreatic cancer [18], although controversy over this finding still exists [27, 28]. These conclusions all suggest the potential roles of ARID1A in pancreatic cancer, of which currently little is known.
As ARID1A has been reported to take essential part in DNA damage repair (DDR) [21], and DNA damage is known to be the prime cause of radiation-induced cell death, we hypothesize that ARID1A may play a key role in radioresistance of pancreatic cancer. We conducted the present study to investigate the effect of ARID1A on radiosensitivity of pancreatic cancer. Likewise, the possible underlying mechanisms were explored, which may help identify new targets for overcoming the radioresistance.
Materials and Methods
Cell culture and reagents
Human pancreatic cancer cell lines PANC-1 and SW1990 were purchased from the American Type Culture Collection (ATCC, US). Cells were cultured in RPM1640 medium (Hyclone Laboratories, US) supplemented with 10% fetal bovine serum (Gibco Waltham, US) and incubated in a humidified incubator with 5% carbon dioxide at 37 °C. LY294002 and mk2206 were purchased from Selleckchem (TX, US) and dissolved in DMSO.
Short hairpin RNAs (shRNAs)-mediated ARID1A knockdown
Lentivirus-ARID1A-RNAi vector and the corresponding empty vector were obtained from Shanghai GeneChem, Co., Ltd. (China). Two independent shRNA constructs (sequence: shARID1A#1, 5'-GCCTGATCTATCTGGTTCAAT-3'; shARID1A#2, 5'-CCTCTCTTATACACAGCAGAT-3') were designed and subcloned into the lentivirus vector. After confirmation of the constructed plasmid by DNA sequencing, lentiviral vector and packaging mix with lipofectamin2000 (Invitrogen, CA, US) were transfected into HEK293FT cells. 48 hours later, the supernatant containing lentivirus was collected, purified and supplemented with 8μg/ml polybrene (Santa Cruz biotech, CA, US). The target cells were infected using the virus solution. After culturing for another 72 h, the stably transfected cells were selected on 2 μg/ml puromycin (Sangon biotech, Shanghai, China) for 1 week. The protein expression level was examined by western blot.
Western blot
Cells were collected and lysed in urine buffer supplemented with 1% protease and 1% phosphorylation inhibitors (Roche Diagnostics, US). Equal amount of total protein (30μg) for each sample was loaded to gradient SDS-PAGE gel. After electrophoresis, protein was transferred onto polyvinylidene fluoride membranes (Millipore, US). The membranes were blocked in TBS with 0.1% Tween-20 and 5% non-fat dried milk for 1 hour at room temperature. After washed for three times, the membranes were probed with primary antibodies overnight at 4°C. The following antibodies were used in this study: ARID1A (1:500, Bethyl Laboratory); ATM, p-ATM, CHK1, p-CHK1 (Ser345) (1:500, Cell Signaling); PTEN, PI3K, AKT, p-AKT (Ser473) (1:500, Abcam, MA); H2AX, γH2AX antibodies (1:1000, Millipore) and β-actin antibody (1:2000, Sigma, US). On the next day, membranes were washed for three times, and incubated with HRP conjugated anti-mouse or anti-rabbit secondary antibodies (1:5000, Invitrogen, US) for 1 hour. Target proteins were visualized using an ECL detection kit (Thermo Fisher Scientific, US).
Clonogenic assay
Briefly, cells were seeded into 6-well culture plates. The next day, cells were treated with indicated conditions and cultured for 2 weeks. Clones were fixed with 4% polymerised formaldehyde, stained with 0.01% crystal violet (Sangon biotech, Shanghai, China) and counted.
Apoptosis Assay
Cells were planted in 6-well culture plates with about 40% fusion. 24 hours later, the cells were exposed to ionizing radiation. 72 hours later, cells were harvested and resuspended in binding buffer containing Annexin V-PE, then propidium iodide (PI) was added, using Annexin V/PI apoptosis kit (Invitrogen, US). Flow cytometry analysis (FACScan, Beckman Coulter, US) was conducted to detect the apoptosis. In each result figure (Fig. 2A and 5A), the percentage of cells in the upper right (including necrotic or late apoptotic cells) and lower right corners (including early apoptotic cells) was added as the apoptotic percentage.
Small interfering RNA (siRNAs)-mediated ARID1A knockdown
Cells were seeded to 6cm cell culture dish with 30% density and cultured overnight, then transfected with ARID1A or non-target siRNA (GeneChem, Shanghai, China) using oligofectamine transfection reagent (Invitrogen, CA, US) according to the protocol provided by the manufacture. The following siRNA sequences were used [29]:
ARID1A:
sense: 5'-GCCCUAACAUGGCCAAUAUTT-3'.
antisense: 5'-AUAUUGGCCAUGUUAGGGCTT-3'.
non-target control:
sense: 5'-UUCUCCGAACGUGUCACGUTT-3'.
antisense: 5'- ACGUGACACGUUCGGAGAATT-3'.
Cell cycle analysis
After indicated treatment, cells were collected and fixed with 70% ethanol at 4°C overnight. Then cells were washed, suspended and stained with propidium iodide (PI) staining solution (50μg/ml PI and 1mg/ml RNase in PBS). And cell cycle analysis was performed using Flow cytometer FACScan (Beckman Coulter, US).
Immunofluorescent staining
Cells grown on coverslips were fixed with 3% paraformaldehyde at room temperature for 20 min and then permeabilized with PBS containing 0.5% Triton X-100 for 5 min. The coverslips were blocked with PBS containing 5% goat serum for 30 min, and immunostained with anti-γH2AX (Millipore) antibody at 4°C overnight, then washed for at least three times and incubated with secondary antibody, Alexa Fluor 488 conjugated goat anti-rabbit IgG (1:50, Cell Signaling, US), for 1 hour at room temperature. DAPI (4, 6-diamidino-2-phenylindole, Sangon biotech, Shanghai, China) was used to counterstain the nuclei. Photographs were captured by Olympus Laser scanning confocal microscopy (Olympus optical Co., Tokyo, Honshu, Japan). At least 50 cells were scored for each treatment group, and the number of γH2AX foci was counted.
Immunohistochemistry (IHC)
Twenty pancreatic cancer primary tissue samples were collected between January 2016 and June 2017 with informed consent from the patients under institutional review board-approved protocols. This study was approved by the institutional research ethics committee of our university. Formalin-fixed and paraffin-embedded tissue sections (5μm thick) were deparaffinized and rehydrated. Subsequently, the sections were blocked of endogenous peroxidase and subjected to antigen retrieval. Then, 5% bovine serum albumin (BSA) was used to incubate the sections for 30 min at room temperature, followed by the primary antibody dilution (ARID1A, 1:2000; PI3K, 1:200; p-Akt, 1:100) overnight at 4ºC. The slides were washed with Tris-buffered saline (TBS) and incubated with the secondary antibody dilution (anti-rabbit IgG and anti-mouse IgG, 1:200, Proteintech, Wuhan, China) for 2 hours at room temperature. 3,3'-diaminobenzidine (DAB) was used to develop visualization signal. Finally, the sections were counterstained with hematoxylin. The slides were viewed by two senior pathologists blinded to the clinical information. The staining scores were calculated based on the percentage of positive cells (0, <10% positive cells; 1, 10-25% positive cells; 2, 26-50% positive cells; and 3, >50% positive cells), and staining intensity (0, no staining; 1, weak staining; 2, moderate staining and 3 for strong staining). The total IHC score was calculated with the value of percent positive score×staining intensity score and ranged from 0 to 9. The cut-off was defined as a score < 4 which represents low expression and a score ≥4 indicates high expression.
Statistical analysis
Each experiment was repeated at least three times, and the data were presented as mean±standard deviation (SD) and were analyzed by SPSS 17.0 software. The student's t-test or ANOVA was used with a probability level of 0.05 indicating significant difference.
Results
Depletion of ARID1A enhances the radioresistance of pancreatic cancer cells
To explore whether ARID1A affects the radiosensitivity of pancreatic cancer cells, human pancreatic cancer cell lines PANC-1 and SW1990 were transfected with ARID1A shRNA. The efficacy of ARID1A knockdown was evaluated by western blot analysis (Fig. 1A). The results indicated that both of the two ARID1A shRNA induced significant down-regulation of ARID1A protein in PANC-1 and SW1990 cells, while control shRNA (luciferase) did not show any effect on protein expression.
Subsequently, the above cells were exposed to the indicated doses of ionizing radiation (IR). A clonogenic assay was used to identify the radiosensitivity. The results (Fig. 1B) and statistical data (Fig. 1C) revealed that radiation induced significant less reduction of clone counts in ARID1A-depleted PANC-1 and SW1990 cells compared to that of the control, suggesting that ARID1A-deficient pancreatic cancer cells were more radioresistant.
Silencing ARID1A suppresses apoptosis and impairs G2-M checkpoint arrest after IR
To identify the possible underlying mechanism of radioresistance in pancreatic cancer cells with depletion of ARID1A, the apoptosis assay was conducted. The results revealed that the control cells presented a significant increase of apoptosis after IR (6Gy). The apoptosis of PANC-1 and SW1990 cells with ARID1A depletion (shARID1A) also increased after IR, but the magnitude of increase was significantly lower than that of the control (Fig. 2A and 2B).
Subsequently, we examined the distribution of cell cycle. First, the expression of ARID1A was knocked down in PANC-1 cells by siRNA, and the efficacy of silencing was evaluated with western blot (Fig. 2C). The cells were then exposed to IR of 6Gy, and the cell cycle distribution was assessed at the indicated time points. Results showed that ARID1A-depleted PANC-1 cells accumulated less significantly in the G2-M phase compared to that of control at both 4 and 8 hours after IR (Fig. 2D), which was verified by statistical analysis (Fig. 2E). These findings indicate that pancreatic cancer cells with ARID1A deficiency have impaired G2-M checkpoint arrest after IR-induced DNA damage, which leads to a quicker entrance into the mitosis phase and contributes to the radioresistance.
Figure 1
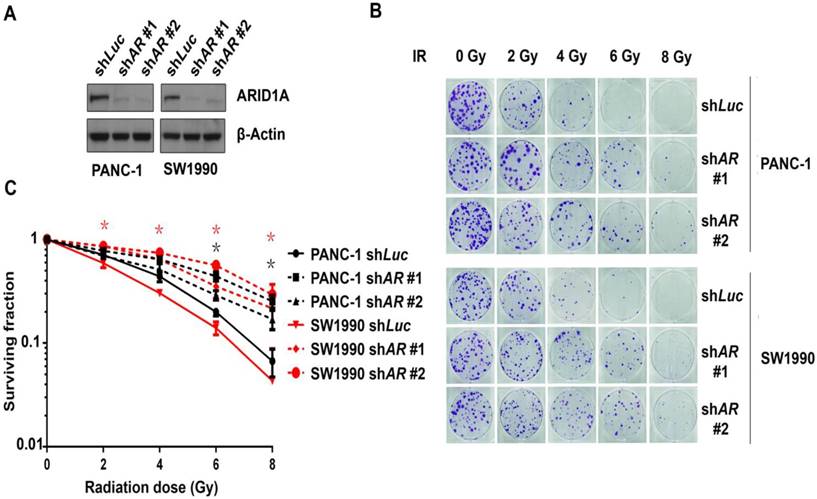
Depletion of ARID1A enhanced the radioresistance of pancreatic cancer cells. (A) Verification of knockdown efficacy. ARID1A was stably knocked down in PANC-1 and SW1990 cell lines by shARID1A #1 and shARID1A #2. Control cells were treated with shLuc. (B) Clonogenic assays were conducted in ARID1A knocked down and control PANC-1 and SW1990 cells with IR of indicated dose. (C) Survival curves represented the radiosensitivity of control (shLuc) and ARID1A depleted (shARID1A #1, #2) PANC-1 and SW1990 cells. Three independent experiments were performed with quantitative results representing the mean ± SD. The asterisk * represented p < 0.05. (AR, ARID1A)
Figure 2
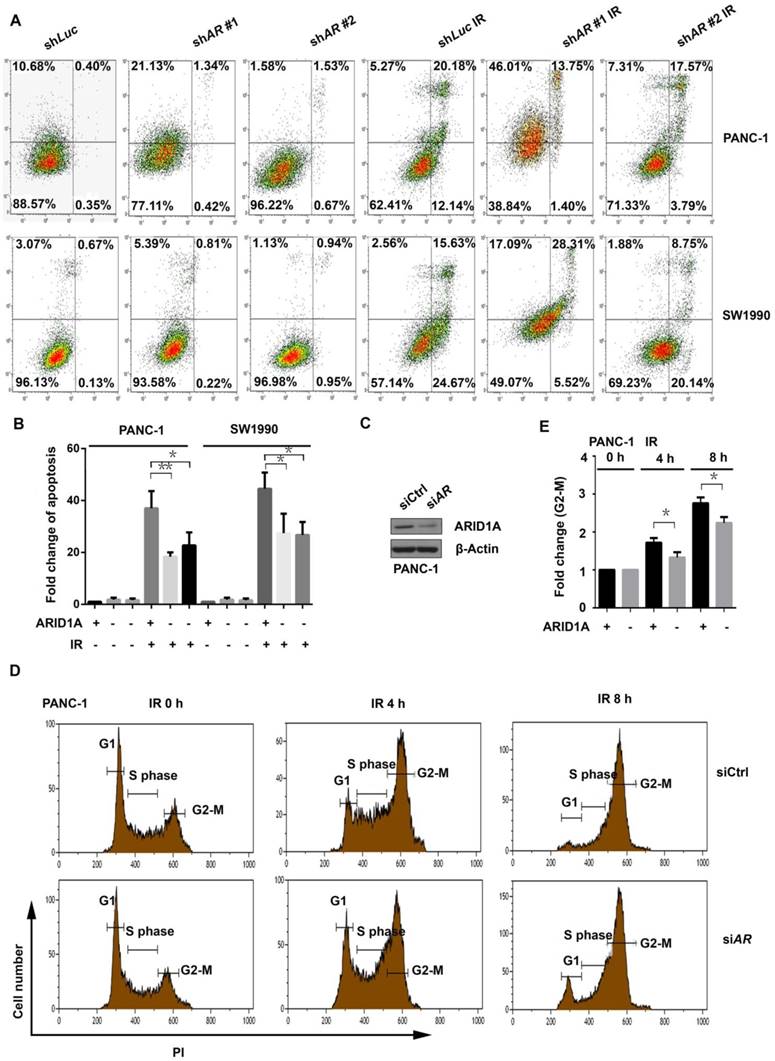
Silencing ARID1A suppressed IR-induced apoptosis and impaired G2-M checkpoint arrest after IR. (A) Apoptosis was detected with or without IR (6Gy) by Flow cytometry in ARID1A stably knockdown and control PANC-1 and SW1990 cells. (C) Western blot was used to detect ARID1A expression in transiently transfected PANC-1 cells with siCtrl or siARID1A. (D) The cell cycle analysis was conducted in control (siCtrl) and ARID1A silencing (siARID1A) PANC-1 cells after IR (6Gy) at indicated time points. (B) and (E) Quantitative results representing the mean ± SD of three independent experiments. (The asterisk * represented p < 0.05, ** represented p < 0.01; PI, propidium iodide; AR, ARID1A)
Figure 3
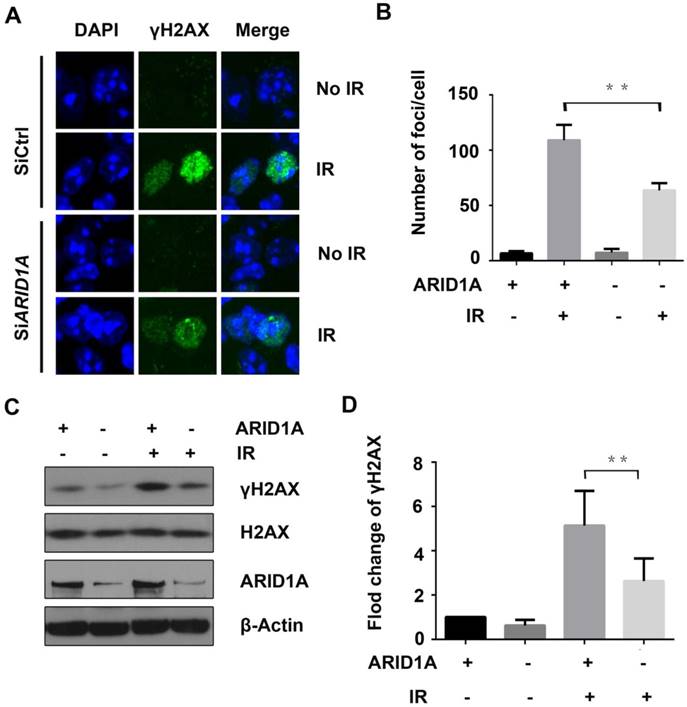
ARID1A knockdown strengthens DDR after IR. Control and AIRD1A depleted PANC-1 cells were exposed to IR (6Gy), and then (A) immunofluorescent staining for γH2AX foci and (C) western blot for γH2AX protein expression were conducted. Number of γH2AX foci and expression of γH2AX protein increased less significantly with ARID1A depletion compared to that of control after IR. (B) and (D) Quantitative results representing mean ± SD of three independent experiments. (The asterisk * represented p < 0.05, ** p < 0.01)
ARID1A knockdown strengthens DDR after IR
As ARID1A has been reported to play an essential role in DDR, which is important for radioresistance, we next evaluated the DNA damage marker, γH2AX, using immunofluorescence and western blot assays. PANC-1 cells transiently transfected with siARID1A or siCtrl were exposed to IR of 6Gy. Two hours later, γH2AX was assessed. The results revealed that IR significantly increased the γH2AX foci (Fig. 3A) and the protein expression of γH2AX (Fig. 3C) in control cells. However, the foci and protein expression of γH2AX were significantly lower in ARID1A-silenced PANC-1 cells compared to that of the control (Fig. 3B and 3D), inferring that the DDR after IR was enhanced with ARID1A deficiency.
ARID1A depletion activates PI3K/AKT pathway, which participates in the radioresistance
DDR-related proteins were then evaluated by western blot assay, including ATM, p-ATM, CHK1, p-CHK1, PTEN, PI3K, AKT, and p-AKT (Ser473), to identify the underlying target signaling proteins. The results showed that the expression of PI3K and p-AKT proteins significantly increased after IR in ARID1A-depleted PANC-1 cells compare to that of the control (Fig. 4A and 4B), whereas the expression level of other DDR-related proteins did not change notably (Fig. 4A).
Subsequently, the relation between the expression of ARID1A and PI3K or p-AKT in pancreatic cancer patients were evaluated using IHC. Twenty sets of human pancreatic cancer tissue samples were collected. As shown in Fig. 4C, the expression of ARID1A is significantly negatively correlated with the expression of PI3K (R = -0.535, p < 0.05) or p-AKT (R = -0.462, p < 0.05). There were 75% (3/4) of the tumors with low expression of ARID1A showed high expression of PI3K or p-AKT, and 56.3% (9/16) of the tumors with high expression of ARID1A exhibited high expression of PI3K, or p-AKT (43.8%, 7/16).
To explore whether the activated PI3K/AKT signaling pathway was involved in the radioresistance, a clonogenic assay was addressed after IR of 6Gy with PI3K-inhibitor LY294002 or AKT-inhibitor mk2206. As demonstrated in Fig. 4D, in ARID1A-knocked down PANC-1 and SW1990 cells (shARID1A), PI3K-inhibitor LY294002 or AKT-inhibitor mk2206 could rescue the radiosensitivity, which was proved by significantly decreased clone counts after IR. However, in control cells (shLuc), the above inhibitors did not change clone counts significantly (Fig. 4E). Such results indicate that the activated PI3K/AKT signaling pathway participates in the radioresistance induced by ARID1A depletion, and inhibition of PI3K/AKT signaling pathway sensitizes radiotherapy.
Figure 4
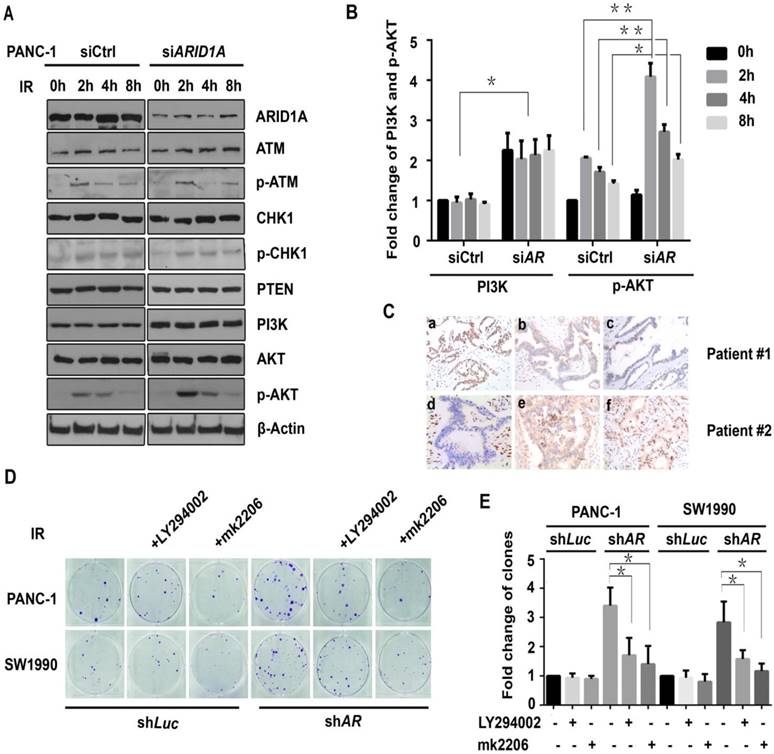
ARID1A depletion activates PI3K/AKT pathway, which participates in the radioresistance. (A) Western blot analysis for DDR-related proteins was performed in control (siCtrl) and ARID1A silencing (siARID1A) PANC-1 cells after IR (6Gy) at indicated time points. (C) Immunohistochemical staining of ARID1A (a, d), PI3K (b, e) and p-AKT (c, f) in representative pancreatic cancer specimens (magnification, ×200). (D) Clonogenic assay was used in ARID1A depleted PANC-1 and SW1990 cells with or without inhibitors (LY294002 or mk2206) after IR. (B) and (E) Qantitative results representing the mean ± SD of three independent experiments. (The asterisk * represented p < 0.05, ** represented p < 0.01; AR, ARID1A).
Figure 5
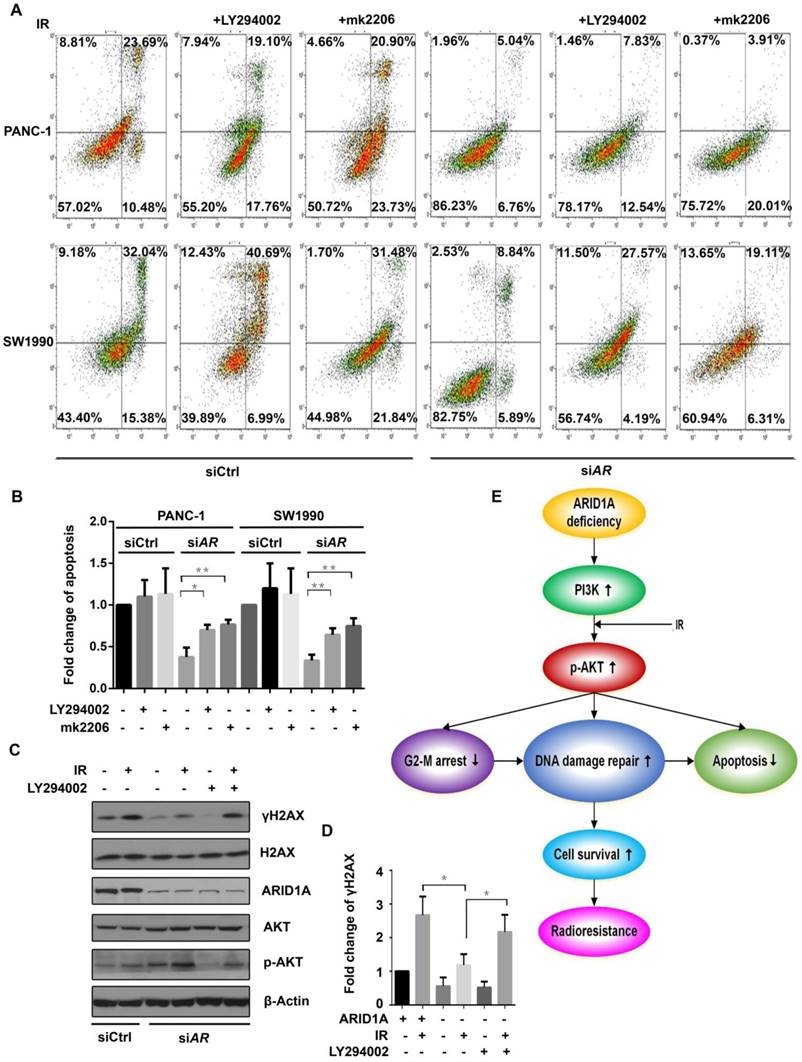
Inhibitors of PI3K/AKT pathway radiosensitize ARID1A deficient pancreatic cancer cells by increasing apoptosis and weakening DDR. (A) The apoptosis was checked after IR in ARID1A depleted PANC-1 and SW1990 cells (siARID1A) and control, with or without inhibitors (LY294002 or mk2206). (C) Western blot was performed to detect the expression of γH2AX in PANC-1 cells with ARID1A deficiency and control at 2 hours after IR, with or without PI3K-inhibitor LY294002. (B) and (D) Quantitative results representing the mean ± SD of three independent experiments. (E) The mechanistic model of ARID1A regulation of the PI3K/AKT signaling pathway that involved in radioresistance of pancreatic cancer. (The asterisk * represented p < 0.05, ** represented p < 0.01; AR, ARID1A).
Inhibitors of PI3K/AKT pathway radiosensitize ARID1A-deficient pancreatic cancer cells by increasing apoptosis and weakening DDR
Apoptosis was evaluated in ARID1A-knocked down PANC-1 and SW1990 cells after IR of 6Gy. The results showed that LY294002 or mk2006 significantly increased the IR induced apoptosis in ARID1A depleted cells. However, in control cells, PI3K/AKT inhibitors did not increase the apoptosis significantly (Fig. 5A and 5B).
Consequently, γH2AX was assessed by western blot analysis. ARID1A knockdown significantly attenuated IR induced elevation of γH2AX expression in PANC-1 cells, which was abrogated upon treatment with LY294002 (Fig. 5C and 5D), suggesting the enhanced DDR by ARID1A deficiency could be reversed by inhibition of the PI3K/AKT pathway.
Discussion
ARID1A, a bona fide tumor suppressor, has been observed to regulate cell proliferation and migration in gynecologic [24] and gastric [29, 30] cancers. However, knockdown of ARID1A did not affect cell growth in pancreatic cancer [17]. Controversy still exists surrounding the prognostic role of ARID1A deficiency in pancreatic cancer [18, 27, 28]. Despite these findings, to the best of our knowledge, no further reports regarding the role of ARID1A on radioresistance of pancreatic cancer have been published.
Here, in the present study, our results reveal that knockdown of ARID1A enhances the radioresistance of pancreatic cancer cells by suppressing apoptosis, impairing G2-M checkpoint arrest, strengthening DDR and accompanying activation of PI3K/AKT signaling pathway. PI3K-inhibitor LY294002 or AKT-inhibitor MK-2206 could alleviate the radioresistance through increasing apoptosis and weakening DDR. Taken together, these data suggest that depletion of ARID1A enhances the radioresistance of pancreatic cancer cells by activation of the PI3K/AKT pathway, which affords a promising target to sensitize radiotherapy in pancreatic cancer.
It has been reported that ARID1A regulates the cell cycle by modulating related genes, such as transcription factor E2F1 [31], CCNE1 [32], and c-MYC [33]. Shen et al found that ARID1A deficiency led to impaired G2-M checkpoint initiation and maintenance [21]. These were in accordance with our findings. Pancreatic cancer cells with deficient ARID1A proceed faster from G2-M checkpoint arrest into the mitosis phase after IR, which contributes to the radioresistance.
ARID1A has been reported to participate in DDR in several studies [21, 34]. Watanabe et al. found that cancer cells lacking in the expression of certain SWI/SNF factors, including ARID1A, were deficient in DDR. They considered that such cancer cells were potentially vulnerable to DNA damage [34]. However, in several other studies, ARID1A-mutated ovarian clear cell carcinoma has been reported to be resistant to conventional platinum-based chemotherapy regimens [35-37]. Our findings also demonstrate that pancreatic cancer cells with depleted ARID1A are more radioresistant. These findings suggest that down-stream signaling pathways regulated by ARID1A deficiency may compensate for the impaired DDR and lead to resistance to chemotherapy and radiotherapy.
Interestingly, recent studies reveal interdependency between ARID1A mutations and PI3K/AKT pathway activation, which is one of the major survival pathways in cancer cells [38]. In several cancers, including endometrial cancer [39], ovarian clear cell carcinoma [40], colon cancer [41], and gastric cancer [30], it has been shown that loss of ARID1A expression up-regulates the phosphorylation of AKT. Zhang et al reported that ARID1A-involved SWI/SNF complex inhibited PIK3CA transcription by direct binding to its promoter [30]. Furthermore, ARID1A mutations were discovered to occur frequently in a synergistic fashion with mutations in PIK3CA [32, 39, 40, 42], which lead to an activation of the PI3K/AKT pathway. These findings indicate that cancer cells with ARID1A deficiency may depend more on the activation of PI3K/AKT pathway.
Thus, cancers with loss of ARID1A expression should be more vulnerable to PI3K/AKT pathway inhibition [43]. Currently, two studies have verified this viewpoint. First, Samartzis et al reported a synthetic lethal interaction between loss of ARID1A expression and inhibition of the PI3K/AKT pathway in breast cancer cells and ovarian clear cell carcinoma cell lines [44]. Subsequently, Zhang et al revealed that ARID1A depletion activated PI3K/AKT signaling pathway in gastric cancer, and inhibitors targeting AKT and PI3K could inhibit cellular growth in vitro and xenograft tumor growth in vivo efficiently [30]. However, in our pilot experiment, inhibition of PI3K/AKT pathway does not significantly affect the cell growth in pancreatic cancer with ARID1A depletion (Fig. S1). As AKT is directly involved in the control of DDR and radioresistance [45], we explored AKT-targeting inhibitors as effective strategies to overcome PI3K/AKT-dependent radioresistance. Our findings indicate that the radioresistance of ARID1A-deficient pancreatic cancer cells is PI3K/AKT dependent, and down-regulated phosphorylation of AKT by LY294002 or mk2206 could sensitize them to radiotherapy.
Our current experiments reveal the role of PI3K/AKT signaling pathway on the radioresistance of pancreatic cancer cells induced by ARID1A deficiency in vitro. However, the results of in vitro studies have limit to extend to clinical significance, because microenvironment is important to the radiosensitivity. Although our IHC staining on patients' pancreatic cancer tissue showed the correlation between the expression of ARID1A and PI3K or p-AKT in vivo, more in vivo experiments are warranted in the future.
Collectively, our results demonstrate that depletion of ARID1A significantly enhances the radioresistance of pancreatic cancer cells and activates the PI3K/AKT signaling pathway. Moreover, PI3K- or AKT-inhibitors could radiosensitize pancreatic cancer cells with ARID1A deficiency via increasing apoptosis and weakening DDR. Our findings suggest that the PI3K/AKT-pathway is crucial for overcoming the radioresistance of ARID1A-deficient cancers, which is deserved to be exploited therapeutically in the future.
Abbreviations
ARID1A: AT-rich interactive domain containing protein 1A; DDR: DNA damage repair; IR: ionizing radiation; Luc: Luciferase.
Supplementary Material
Supplementary figure S1.
Acknowledgements
This study was supported by the Natural Science Foundation of Hubei Province (2015CFB541) and the Research Project of Hubei provincial health and Family Planning Commission (WJ2017M114).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7-30
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5-29
3. Grover S, Syngal S. Hereditary pancreatic cancer. Gastroenterology. 2010;139:1076-80
4. Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR. et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403-13
5. Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y. et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817-25
6. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S. et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960-6
7. Petrelli F, Comito T, Ghidini A, Torri V, Scorsetti M, Barni S. Stereotactic Body Radiation Therapy for Locally Advanced Pancreatic Cancer: A Systematic Review and Pooled Analysis of 19 Trials. Int J Radiat Oncol Biol Phys. 2017;97:313-22
8. Loehrer PJ Sr, Feng Y, Cardenes H, Wagner L, Brell JM, Cella D. et al. Gemcitabine alone versus gemcitabine plus radiotherapy in patients with locally advanced pancreatic cancer: an Eastern Cooperative Oncology Group trial. J Clin Oncol. 2011;29:4105-12
9. Herman JM, Wild AT, Wang H, Tran PT, Chang KJ, Taylor GE. et al. Randomized phase III multi-institutional study of TNFerade biologic with fluorouracil and radiotherapy for locally advanced pancreatic cancer: final results. J Clin Oncol. 2013;31:886-94
10. Yang SH, Kuo TC, Wu H, Guo JC, Hsu C, Hsu CH. et al. Perspectives on the combination of radiotherapy and targeted therapy with DNA repair inhibitors in the treatment of pancreatic cancer. World J Gastroenterol. 2016;22:7275-88
11. Azad A, Yin Lim S, D'Costa Z, Jones K, Diana A, Sansom OJ. PD-L1 blockade enhances response of pancreatic ductal adenocarcinoma to radiotherapy. EMBO Mol Med. 2017;9:167-80
12. Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R. et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228-31
13. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H. et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67-73
14. Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S. et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43:1219-23
15. Mamo A, Cavallone L, Tuzmen S, Chabot C, Ferrario C, Hassan S. et al. An integrated genomic approach identifies ARID1A as a candidate tumor-suppressor gene in breast cancer. Oncogene. 2012;31:2090-100
16. Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH. et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760-4
17. Shain AH, Giacomini CP, Matsukuma K, Karikari CA, Bashyam MD, Hidalgo M. et al. Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc Natl Acad Sci U S A. 2012;109:E252-9
18. Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J. et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744
19. Roberts CW, Orkin SH. The SWI/SNF complex-chromatin and cancer. Nat Rev Cancer. 2004;4:133-42
20. Mao TL, Shih Ie M. The roles of ARID1A in gynecologic cancer. J Gynecol Oncol. 2013;24:376-81
21. Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L. et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015;5:752-67
22. Nagl NG Jr, Patsialou A, Haines DS, Dallas PB, Beck GR Jr, Moran E. The p270 (ARID1A/SMARCF1) subunit of mammalian SWI/SNF-related complexes is essential for normal cell cycle arrest. Cancer Res. 2005;65:9236-44
23. Wu JN, Roberts CW. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov. 2013;3:35-43
24. Guan B, Wang TL, Shih Ie M. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011;71:6718-27
25. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D. et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945-50
26. Jiao Y, Yonescu R, Offerhaus GJ, Klimstra DS, Maitra A, Eshleman JR. et al. Whole-exome sequencing of pancreatic neoplasms with acinar differentiation. J Pathol. 2014;232:428-35
27. Sausen M, Phallen J, Adleff V, Jones S, Leary RJ, Barrett MT. et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun. 2015;6:7686
28. Numata M, Morinaga S, Watanabe T, Tamagawa H, Yamamoto N, Shiozawa M. et al. The clinical significance of SWI/SNF complex in pancreatic cancer. Int J Oncol. 2013;42:403-10
29. Yan HB, Wang XF, Zhang Q, Tang ZQ, Jiang YH, Fan HZ. et al. Reduced expression of the chromatin remodeling gene ARID1A enhances gastric cancer cell migration and invasion via downregulation of E-cadherin transcription. Carcinogenesis. 2014;35:867-76
30. Zhang Q, Yan HB, Wang J, Cui SJ, Wang XQ, Jiang YH. et al. Chromatin remodeling gene AT-rich interactive domain-containing protein 1A suppresses gastric cancer cell proliferation by targeting PIK3CA and PDK1. Oncotarget. 2016;7:46127-41
31. Van Rechem C, Boulay G, Leprince D. HIC1 interacts with a specific subunit of SWI/SNF complexes, ARID1A/BAF250A. Biochem Biophys Res Commun. 2009;385:586-90
32. Zang ZJ, Cutcutache I, Poon SL, Zhang SL, McPherson JR, Tao J. et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. 2012;44:570-4
33. Nagl NG Jr, Zweitzig DR, Thimmapaya B, Beck GR Jr, Moran E. The c-myc gene is a direct target of mammalian SWI/SNF-related complexes during differentiation-associated cell cycle arrest. Cancer Res. 2006;66:1289-93
34. Watanabe R, Ui A, Kanno S, Ogiwara H, Nagase T, Kohno T. et al. SWI/SNF factors required for cellular resistance to DNA damage include ARID1A and ARID1B and show interdependent protein stability. Cancer Res. 2014;74:2465-75
35. Yokoyama Y, Matsushita Y, Shigeto T, Futagami M, Mizunuma H. Decreased ARID1A expression is correlated with chemoresistance in epithelial ovarian cancer. J Gynecol Oncol. 2014;25:58-63
36. Lyu C, Zhang Y, Zhou X, Lang J. ARID1A gene silencing reduces the sensitivity of ovarian clear cell carcinoma to cisplatin. Exp Ther Med. 2016;12:4067-71
37. Katagiri A, Nakayama K, Rahman MT, Rahman M, Katagiri H, Nakayama N. et al. Loss of ARID1A expression is related to shorter progression-free survival and chemoresistance in ovarian clear cell carcinoma. Mod pathol. 2012;25:282-8
38. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627-44
39. Liang H, Cheung LW, Li J, Ju Z, Yu S, Stemke-Hale K. et al. Whole-exome sequencing combined with functional genomics reveals novel candidate driver cancer genes in endometrial cancer. Genome Res. 2012;22:2120-9
40. Chandler RL, Damrauer JS, Raab JR, Schisler JC, Wilkerson MD, Didion JP. et al. Coexistent ARID1A-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nat Commun. 2015;6:6118
41. Xie C, Fu L, Han Y, Li Q, Wang E. Decreased ARID1A expression facilitates cell proliferation and inhibits 5-fluorouracil-induced apoptosis in colorectal carcinoma. Tumour Biol. 2014;35:7921-7
42. Huang HN, Lin MC, Huang WC, Chiang YC, Kuo KT. Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations and ZNF217 amplification in ovarian clear cell carcinoma. Mod Pathol. 2014;27:983-90
43. Samartzis EP, Noske A, Dedes KJ, Fink D, Imesch P. ARID1A mutations and PI3K/AKT pathway alterations in endometriosis and endometriosis-associated ovarian carcinomas. Int J Mol Sci. 2013;14:18824-49
44. Samartzis EP, Gutsche K, Dedes KJ, Fink D, Stucki M, Imesch P. Loss of ARID1A expression sensitizes cancer cells to PI3K- and AKT-inhibition. Oncotarget. 2014;5:5295-303
45. Toulany M, Rodemann HP. Phosphatidylinositol 3-kinase/Akt signaling as a key mediator of tumor cell responsiveness to radiation. Semin Cancer Biol. 2015;35:180-90
Author contact
![]() Corresponding author: Guangyuan Hu, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, Hubei province, China. Fax: +86 027 83662834; Tel: +86 027 83663408; Email: h.g.y.121com
Corresponding author: Guangyuan Hu, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, Hubei province, China. Fax: +86 027 83662834; Tel: +86 027 83663408; Email: h.g.y.121com
Citation styles
APA
Yang, L., Yang, G., Ding, Y., Dai, Y., Xu, S., Guo, Q., Xie, A., Hu, G. (2018). Inhibition of PI3K/AKT Signaling Pathway Radiosensitizes Pancreatic Cancer Cells with ARID1A Deficiency in Vitro. Journal of Cancer, 9(5), 890-900. https://doi.org/10.7150/jca.21306.
ACS
Yang, L.; Yang, G.; Ding, Y.; Dai, Y.; Xu, S.; Guo, Q.; Xie, A.; Hu, G. Inhibition of PI3K/AKT Signaling Pathway Radiosensitizes Pancreatic Cancer Cells with ARID1A Deficiency in Vitro. J. Cancer 2018, 9 (5), 890-900. DOI: 10.7150/jca.21306.
NLM
Yang L, Yang G, Ding Y, Dai Y, Xu S, Guo Q, Xie A, Hu G. Inhibition of PI3K/AKT Signaling Pathway Radiosensitizes Pancreatic Cancer Cells with ARID1A Deficiency in Vitro. J Cancer 2018; 9(5):890-900. doi:10.7150/jca.21306. https://www.jcancer.org/v09p0890.htm
CSE
Yang L, Yang G, Ding Y, Dai Y, Xu S, Guo Q, Xie A, Hu G. 2018. Inhibition of PI3K/AKT Signaling Pathway Radiosensitizes Pancreatic Cancer Cells with ARID1A Deficiency in Vitro. J Cancer. 9(5):890-900.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.