Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(6):1096-1105. doi:10.7150/jca.23909 This issue Cite
Research Paper
Integrated Oncogenomic Profiling of Copy Numbers and Gene Expression in Lung Adenocarcinomas without EGFR Mutations or ALK Fusion
Yanzhuo Luo1,2, Bingjin Li3, Guangxin Zhang4, Yuxiao He5, Jeeyoo Hope Bae6, Fengping Hu6, Ranji Cui3, Runhua Liu6,7, Zhou Wang1 ![]() , Lizhong Wang6,7
, Lizhong Wang6,7 ![]()
1. Department of Thoracic Surgery, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, Shandong, 250021, China;
2. Department of Cardiac Surgery, The First Hospital Affiliated to Jiamusi University, Jiamusi, Heilongjiang, 154002, China;
3. Provincial Key Laboratory on Molecular and Chemical Genetic, Second Hospital of Jilin University, Changchun, 130041, Jilin, China;
4. Department of Thoracic Surgery, Second Hospital of Jilin University, Changchun, 130041, Jilin, China;
5. Department of Thoracic Surgery, Shengjing Hospital of China Medical University, Shenyang, Liaoning, 110003, China.
6. Department of Genetics, University of Alabama at Birmingham, Birmingham, Alabama, 35294, USA;
7. Comprehensive Cancer Center, University of Alabama at Birmingham, Birmingham, Alabama, 35294, USA.
Received 2017-11-17; Accepted 2018-1-29; Published 2018-2-28
Citation:
Luo Y, Li B, Zhang G, He Y, Bae JH, Hu F, Cui R, Liu R, Wang Z, Wang L. Integrated Oncogenomic Profiling of Copy Numbers and Gene Expression in Lung Adenocarcinomas without EGFR Mutations or ALK Fusion. J Cancer 2018; 9(6):1096-1105. doi:10.7150/jca.23909. https://www.jcancer.org/v09p1096.htm
Other stylesAbstract
Targeted therapies based on EGFR mutations or on the ALK fusion oncogene have become the standard treatment for certain patients with lung adenocarcinoma (LUAD). However, most LUAD patients have no EGFR mutation or ALK fusion, and their oncogenetic alterations remain to be characterized. Here we conducted an integrated analysis of public datasets to assess the genomic alterations of 23 highly lung cancer-associated genes. The copy numbers of these genes were measured in ten micro-dissected, paired tumors and normal lung tissues of LUAD patients without EGFR mutations or ALK fusion. The copy numbers of PTEN, RB1, HMGA2, and PTPRD were lower in tumors compared with those for normal tissues. Although there were reduced mRNA levels of PTEN and RB1 in tumors, there was a correlation between copy number and expression only for PTEN. In addition, analysis of the copy number alterations of these 23 genes revealed correlations between EMSY/CCND1, EMSY/PIK3CA, CCND1/CDKN2A, and CCND1/PIK3CA. Our exploration of integrated copy number and gene expression analysis gives priority to the PTEN-PIK3CA and RB1-CCND1 pathways in developing therapeutic strategies for LUAD patients without EGFR mutations or ALK fusion.
Keywords: Lung adenocarcinoma, Oncogenomic profiling, Genetic alteration, PTEN, RB1
Introduction
With the highest mortality rate among all cancers in China and many other countries, lung cancer annually causes approximately 1.38 million deaths worldwide [1, 2]. Lung cancer is histologically sub-classified into four categories: lung adenocarcinoma (LUAD), squamous cell carcinoma (LUSC), large cell carcinoma, and small cell carcinoma of the lung. LUAD, an epithelial cancer of glandular origin, is the most common pathological subtype of non-small cell lung cancer (NSCLC), even for never-smokers [3, 4]. Since LUAD is a heterogeneous tumor with diverse molecular, clinical, and pathological characteristics, the identification of oncogenic drivers has increased understanding of LUAD biology [5, 6].
Recognition of molecular alterations in LUAD has facilitated tailored therapy targeting these alterations and has ushered in the era of “personalized” oncologic medicine. For example, LUAD patients with activating mutations of the epidermal growth factor receptor gene (EGFR) have a better response to EGFR tyrosine kinase inhibitors (TKIs) than those without EGFR mutations [7], and rearrangement of the anaplastic lymphoma kinase gene (ALK) is the best predictor of LUAD response to the ALK TKI crizotinib [8-10]. These facts indicate that therapeutic effectiveness is linked to the presence of specific oncogenomic alterations. However, patients with EGFR mutations or ALK fusion account for only one-third of patients diagnosed with LUAD [11], which indicates the necessity of understanding of the genetic basis for LUAD without EGFR mutations or ALK fusion.
The molecular mechanisms underlying LUAD development are unclear, and the heterogeneous nature of lung cancer makes it difficult to achieve an understanding. It is not known if LUAD patients with EGFR/ALT alterations harbor distinct genetic characteristics compared to those without such alterations. Moreover, little is understood about the correlation of copy numbers and gene expression of top-ranked genes in lung cancer, especially for LUAD without EGFR mutations or ALK fusion. In addition, since the development and progression of LUAD are consequences of gene-gene interactions and regulatory coordination [12-14], the identification of therapeutic targets in regulatory pathways can provide insight into the etiology and pathogenesis of LUAD. Whether and how these lung cancer-associated genes are coordinated to affect cellular functions remain largely unexplored.
In the present study, we retrospectively analyzed genomic sequencing data for LUAD and LUSC patients. Oncogenomic alterations of 23 top candidate genes were assessed to explore the similarities and differences between these two lung cancer types. The copy numbers of these candidate genes were further evaluated in ten micro-dissected LUADs without EGFR mutations or ALK fusion. For a few genes, further exploration of the associations between copy number and gene expression was conducted. This study revealed a concordant change of PTEN at both genomic DNA and mRNA levels in LUAD without EGFR mutation or ALK fusion. The results provide an approach to validating molecules involved in lung carcinogenesis and a basis for identifying pathways as targets for the treatment of LUAD.
Materials and Methods
Human tissue specimens
Specimens, including tumor tissue and tumor-distant normal lung tissue from the same patient, were collected from ten patients with LUAD who underwent primary surgery between January 2009 and June 2012 at the First Hospital of Jiamusi University. This study involving human lung tumor tissues has been approved by the Institutional Review Board of the First Hospital of Jiamusi University. Informed consent was obtained from all human subjects in accordance with the requirements of the Institutional Review Board. At the time of LUAD diagnosis, all patients were naïve for chemo-, radio-, and targeted therapy. They did not receive any other treatment that could induce mutations. Patients were excluded if (1) they previously received radio-, chemo-, or targeted therapy; (2) their histological samples were insufficient for genetic testing; or (3) they were diagnosed with metastatic LUAD. Clinicopathological characteristics of the group are presented in Table 1. Two clinical pathologists conducted the pathological evaluations independently. All patients had histologically confirmed LUAD. The classification of LUAD subtypes was conducted following the 2011 International Association for the Study of Lung Cancer (IASLC)/the American Thoracic Society (ATS)/the European Respiratory Society (ERS) guidelines [15]. The pathological staging was reassessed with the new international tumor-node-metastasis (TNM) staging system for lung cancer approved by the American Joint Committee on Cancer (AJCC, 7th edition)[16]. Primary LUADs and tumor-distant normal lung tissue specimens were obtained from surgically resected lung tissues, which were preserved as formalin-fixed, paraffin-embedded sections, for biomarker and pathologic analyses.
Lung cancer specimens obtained from a clinical molecular diagnostic laboratory were tested for EGFR mutations and ALK fusion. Genomic DNA and total RNA were extracted from formalin-fixed and paraffin-embedded tissue sections. Mutations in EGFR were assessed on genomic DNA, whereas ALK fusions were determined with total RNA. The EGFR mutations were analyzed by fluorescent real-time PCR using Human EGFR Mutation Detection Kits (AmoyDx, Xiamen, China). ALK fusion variants were detected by multiplex One-step RT-PCR using Human ALK Gene Fusions Detection Kits (AmoyDx). RT-PCR was performed on a 7500 Real Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA), and the absence of EGFR mutations and ALK fusion was verified by direct sequencing of PCR and RT-PCR products as previously reported [17].
Microdissection of lung tissues
Sections (8-μm) of human lung specimens were cut and transferred to glass slides not coated with polylysine. The slides were stained with Harris hematoxylin for 50 seconds and eosin for 30 seconds and then dried in a laminar flow hood for 5 to 10 min prior to microdissection. For analysis of gene copy number and gene expression, cells (5 × 103) were laser-capture micro-dissected from target tissue sections using the Arcturus PixCell II system (Thermo Fisher Scientific) with an Olympus IX-50 microscope as described previously [18]. The time for procurement of micro-dissected tissue for RNA was less than 15 minutes. RNA was extracted with PicoPure RNA extraction kits (Thermo Fisher Scientific) and amplified by RT-PCR. Genomic DNA was extracted from micro-dissected tissue using PicoPure DNA Isolation kits (Thermo Fisher Scientific). The concentrations were measured with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific).
Quantification of gene copy numbers and mRNA levels
Quantitative analysis of copy numbers was conducted by QIAGEN qBiomarker Copy Number PCR assays based on a 7500 Real Time PCR System (Thermo Fisher Scientific) as previously described [19]. All qBiomarker Copy Number PCR Assays were designed for unique regions of the genome. A qBiomarker Multicopy Reference Copy Number PCR Assay (MRef) was included on each assay. The reference assay recognizes a stable sequence that appears in the human genome more than 40 times and whose copy number is not affected or minimally affected by local genomic changes. Relative gene copy numbers for each specimen were calculated as 2 × Tcopy number (tumor copy number/MRef copy number)/Ncopy number (tumor-distant normal tissue copy number/MRef copy number) from the same patient.
Quantitative RT-PCR (RT-qPCR) of gene expression was performed with a 7500 Real Time PCR System using Power SYBR Green Master Mixture (Thermo Fisher Scientific). Reverse transcription was accomplished with random hemaxmer primers and SuperScript II Reverse Transcriptase kits (Thermo Fisher Scientific). Fold changes were calculated according to the ΔΔCT method [20]. For relative gene expression assays, the endogenous control gene was GAPDH. The primers used for RT-qPCR are listed in S1 Table.
Retrieval of public genomic datasets
All datasets used were from publically available sources, including Broad [21] (Broad Institute of MIT and Harvard) and MSKCC [22] (Memorial Sloan Kettering Cancer Center) or from various projects, including TCGA [23, 24] (the Cancer Genome Atlas - Cancer Genome) and TSP [6] (the Tumor Sequencing Project). The whole-genome/exome or targeted sequencing data for tumors from LUAD and LUSC patients and the clinical and demographic information were extracted from these previous studies (all the sequencing data have been deposited online). For example, DNA sequencing data for the tumors and normal controls and the corresponding clinical information from LUAD patients with spirometry data available in the TCGA cohort were downloaded from gdac.broadinstitute.org.
Analysis of oncogenomic alterations
A cross-cancer alteration summary for 23 highly lung cancer-associated genes (selected by QIAGEN within Human Lung Cancer Copy Number PCR Array) was accomplished using c-Bioportal. Data mining was accomplished using cBioPortal [25, 26] for Cancer Genomics (cBioPortal for Cancer Genomics), available at http://www.cbioportal.org, to measure the incidence of conditions that are associated with alterations in these genes. The database query was based on deregulation (amplification, deletion, and mutants) of these genes. Tumor datasets were chosen in accordance with the publication guidelines (last updated on January 17, 2014) of TCGA (tcga@mail.nih.gov). Generally, sequence variations were mapped to the corresponding genomic coordinates and inspected using the genome browsers of Ensembl (www.ensembl.org) and NCBI (www.ncbi.nlm.nih.gov). Mutational signature analyses were performed as described previously [27]. In brief, mutational catalogues from the genomic sequencing data of primary tumors derived from cohorts of various origins were used to decipher the mutational signatures. The affected mutational process was determined by comparing the extracted signatures with signature sets identified previously (http://cancer.sanger.ac.uk/cosmic/signatures) [27, 28]. Boundaries for deletion, amplification, and complex rearrangements were annotated as previously described [6].
Statistical methods
Differences of gene copy numbers between paired tumor tissue and tumor-distant normal tissue were tested using the nonparametric Mann-Whitney U test. Differences of gene expression between paired tumor tissue and tumor-distant normal tissue were tested by use of the paired two-sample t test. The associations between DNA copy numbers and gene expression levels were evaluated by Pearson's correlation test. Standard false discovery rate (FDR) and Bonferroni corrections were applied for the analysis of oncogenomic alterations. A P value < 0.05 was considered as statistically significant, and all statistical tests were two-sided. The analyses were performed using SPSS 24, R package, and GraphPad Prism 6.0.
Figure 1
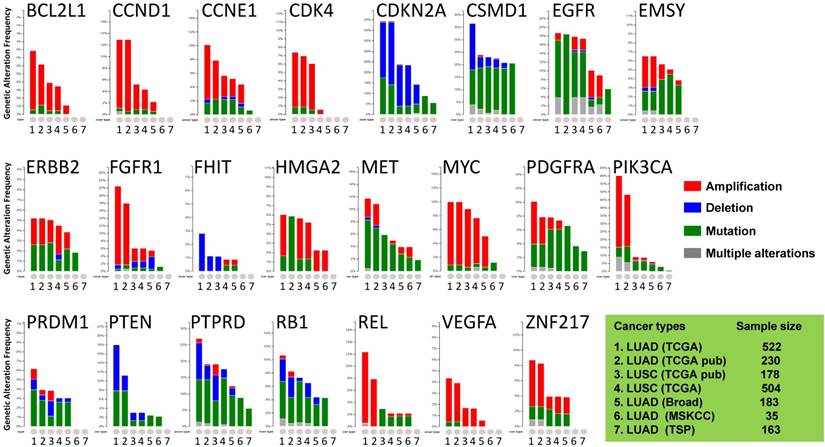
Public dataset-based analysis of somatic alterations for 23 lung cancer-associate genes associated with lung cancers. A cross-cancer alteration summary for 23 lung cancer-associated genes was prepared using c-Bioportal. Data mining was accomplished with cBioPortal [25, 26] for Cancer Genomics, a data portal (cBioPortal for Cancer Genomics) available at http://www.cbioportal.org, to measure the incidence of conditions that are associated with the alterations in these genes. The database query was based on deregulation (amplification, deletion, and mutation) of these genes. All datasets used were from publically available sources, including Broad [21] (Broad Institute of MIT and Harvard, 183 LUAD samples), MSKCC [22] (Memorial Sloan Kettering Cancer Center, 35 LUAD samples) or various projects, including TCGA [23, 24] (the Cancer Genome Atlas - Cancer Genome, 230 LUAD samples and 178 LUSC samples), TSP [6] (the Tumor Sequencing Project, 163 LUAD samples). "TCGA" means provisional TCGA datasets; "TCGA pub" means TCGA datasets used in the corresponding publications. False discovery rate (FDR) and Bonferroni corrections were applied.
Results
Analysis of somatic alterations for cancer-associated genes in lung cancers
Most of the cancer-associated genes affected by copy number alterations (CNAs) are genes in cancer-signaling pathways involved in carcinogenesis and tumor progression [29, 30]. Thus, we investigated the somatic genomic alterations of 23 lung-cancer related genes in publically available datasets derived from large cohorts of patients with LUAD or LUSC (Fig 1). Data mining was accomplished with cBioPortal for Cancer Genomics (available at http://www.cbioportal.org) to measure the incidence of conditions that are associated with alterations in these genes. Since the portal reduces molecular profiling data from cancer tissues and cell lines into readily understandable genetic, epigenetic, gene expression events [25], a graph representing a cross-cancer alteration (amplification, deletion, mutation and multiple alterations) for each gene was generated (Fig 1).
The focus was on genes whose alterations predispose to susceptibility for lung cancers. The results demonstrated that the oncogenetic profile of lung cancer varies among the histological subtypes, with differences between LUAD and LUSC. Almost half of the checked genes, including CCND1, CSMD1, EGFR, EMSY, MYC, PDGFRA, PIK3CA, PTPRD, RB1, REL, and ZNF217, displayed multiple alterations (amplification, deletion, and mutation occurring simultaneously). Although there were multiple alterations in the MYC gene only in LUSC, various alterations for genes CCND1, EMSY, PIK3CA, and ZNF217 were present in LUAD. LUAD harbored higher frequencies of amplification of CCND1, PIK3CA, FGFR1, REL, and ZNF217 than LUSC, whereas LUSC harbored higher frequencies of mutations of EMSY, PDGFRA, and REL. In LUAD, the PTEN gene, which has rare gene amplification as a tumor suppressor, exhibited higher incidences of mutation and deletion [5, 6, 23, 29]. The VEGFA gene, whose up-regulation is associated with tumor progression and angiogenesis [31], had mutations only in LUSC [24]. The genomic alteration patterns varied among the genes. BCL2L1, CCND1, CCNE1, CDK4, ERBB2, FGFR1, MYC, PIK3CA, REL, VEGFA, and ZNF217 predominated in gene amplifications; CSMD1, EGFR, MET, PDGFRA, PRDM1, PTPRD, and RB1 largely exhibited gene mutations; other genes (CDKN2A, FHIT, and PTEN) had more deletions. These results suggested that LUAD patients harbor distinct somatic alterations in a few genes, which can be exploited for personalized medical care of those patients.
Clinicopathological characteristics of the patients with LUAD without EGFR mutations or ALK fusion
Samples from ten Chinese LUAD patients were used assess the CNAs of the 23 genes. Their clinicopathological characteristics are listed in Table 1. These patients included five men and five women who had a median age of 55 years (range: 46-72 years). Six patients were never smokers and four were smokers. All tumors from these patients were negative for EGFR mutations and ALK fusion. The pathological stages were "Tumor Stage II" (n=7) and "Tumor Stage III" (n=3), with “Moderate" tumor grade predominating (n=5). Six patients had tumor invasion of local lymph nodes (Table 1).
Comparative quantification of gene copy numbers in LUAD tumors and corresponding normal lung tissues
To understand the association between the gene copy numbers and lung cancer risk, their association with gene expression levels in normal lung cells were investigated. For LUAD tumor tissues and paired tumor-distant normal lung epithelial tissues from ten LUAD patients, the copy numbers of these genes were quantified by real-time PCR (Table 2 and Fig 2). Most of the 23 genes did not show significant changes in CNAs, compared with those for normal lung tissues. VEGFA, which is up-regulated in many tumors and correlates with tumor stage and progression [31], showed higher copy numbers in LUAD, although without statistical significance (T/N=2.65±0.92, P=0.0844). CDKN2A, a stabilizer of the tumor suppressor protein p53 [32], had a lower abundance in LUAD (T/N=1.66±0.43, P=0.0906). In LUAD, the proto-oncogene MET, whose amplification and over-expression are associated with various human cancers, including NSCLC [33], had lower copy numbers (T/N=1.83±0.26, P=0.0603). Three tumor suppressor genes (PTEN, RB1, and PTPRD) and one oncogene (HMGA2) had lower copy numbers (all P<0.05) in LUAD tissues (Fig 2).
Figure 2
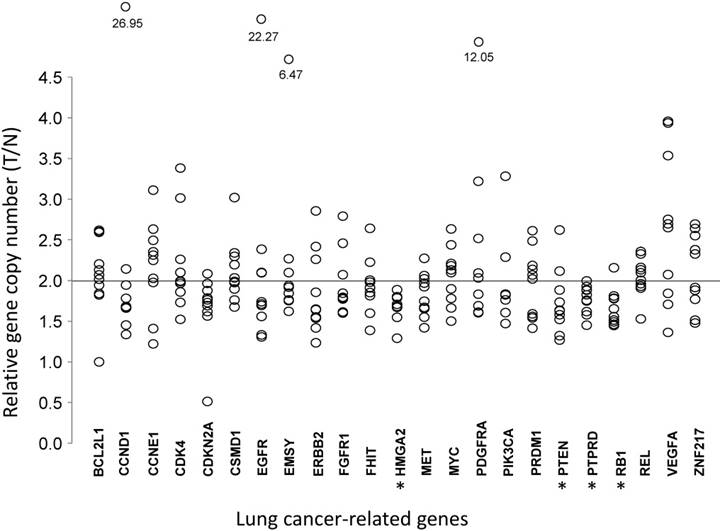
Comparative analysis of gene copy numbers in LUADs without EGFR mutations or ALK fusion. The copy numbers of 23 lung cancer-associate candidate genes were quantified in micro-dissected tumor cells and paired tumor-distant normal lung epithelial cells from ten LUAD patients without EGFR mutation or ALK fusion. The y-axis indicates the copy number index calculated by 2 × Tcopy number / Ncopy number, where T represents tumor cells and N is the tumor-distant normal lung epithelial cells. Each circle denotes one copy number index of the specified gene from one patient. * P < 0.05, tumor cells vs. paired tumor-distant normal epithelial cells, tested with the Mann-Whitney U test.
Table 1
The clinicopathological characteristics of the LUAD patients.
| Categories | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | Case 8 | Case 9 | Case 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Age at the time of diagnosis | 50 | 55 | 62 | 46 | 59 | 61 | 72 | 57 | 55 | 52 |
| Gender | Female | Male | Male | Female | Male | female | Male | Female | Male | Male |
| Race | Chinese | Chinese | Chinese | Chinese | Chinese | Chinese | Chinese | Chinese | Chinese | Chinese |
| Year of diagnosis | 2009 | 2010 | 2010 | 2010 | 2010 | 2011 | 2011 | 2011 | 2012 | 2012 |
| Pathologic type | AD | AD | AD | AD | AD | AD | AD | AD | AD | AD |
| EGFR mutation | No | No | No | No | No | No | No | No | No | No |
| ALK rearrangement | No | No | No | No | No | No | No | No | No | No |
| Tumor grade | Moderate | Well | Moderate | Poor | Moderate | Well | Poor | Poor | Moderate | Moderate |
| Tumor stage (TNM) | T2N2M0 | T2N0M0 | T2N0M0 | T3N3M0 | T2N1M0 | T3N1M0 | T3N0M0 | T2N2M0 | T2N0M0 | T2N1M0 |
Table 2
Relative gene copy numbers in LUAD cells compared with normal lung epithelial cells from the same patient.
| Gene | T1/N1 | T2/N2 | T3/N3 | T4/N4 | T5/N5 | T6/N6 | T7/N7 | T8/N8 | T9/N9 | T10/N10 | Mean | SD | p-Value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BCL2L1 | 2.60 | 2.59 | 1.84 | 2.07 | 2.13 | 2.02 | 2.62 | 1.94 | 1.82 | 2.20 | 2.18 | 0.31 | 0.1096 |
| CCND1 | 1.95 | 2.14 | 1.34 | 1.45 | 1.68 | 1.78 | 26.95 | 1.66 | 1.66 | 1.67 | 4.23 | 7.99 | 0.7085 |
| CCNE1 | 2.35 | 2.31 | 2.63 | 2.25 | 1.41 | 2.03 | 3.11 | 1.98 | 1.22 | 2.49 | 2.18 | 0.56 | 0.5746 |
| CDK4 | 3.38 | 2.26 | 1.99 | 1.99 | 1.97 | 2.10 | 3.01 | 1.86 | 1.52 | 1.73 | 2.18 | 0.58 | 0.4604 |
| CDKN2A | 2.08 | 1.88 | 1.73 | 1.62 | 1.76 | 1.69 | 0.51 | 1.79 | 1.56 | 1.96 | 1.66 | 0.43 | 0.0906 |
| CSMD1 | 2.03 | 2.30 | 2.19 | 3.02 | 1.67 | 1.76 | 2.34 | 1.90 | 1.97 | 1.99 | 2.12 | 0.38 | 0.4288 |
| EGFR | 1.33 | 1.71 | 2.10 | 2.38 | 1.31 | 2.09 | 22.27 | 1.69 | 1.56 | 1.74 | 3.82 | 6.49 | 0.6744 |
| EMSY | 2.27 | 2.10 | 1.62 | 1.76 | 1.84 | 1.75 | 6.47 | 1.94 | 1.75 | 1.92 | 2.34 | 1.46 | 0.6535 |
| ERBB2 | 1.54 | 2.26 | 1.24 | 1.64 | 1.42 | 2.42 | 2.85 | 1.55 | 1.65 | 1.86 | 1.84 | 0.51 | 0.2004 |
| FGFR1 | 1.79 | 2.07 | 1.61 | 1.79 | 1.77 | 1.60 | 2.79 | 1.85 | 1.79 | 2.46 | 1.95 | 0.38 | 0.5061 |
| FHIT | 2.01 | 1.91 | 1.98 | 2.64 | 1.85 | 1.60 | 1.39 | 1.92 | 2.23 | 1.81 | 1.93 | 0.34 | 0.4057 |
| HMGA2 | 1.67 | 1.80 | 1.71 | 1.79 | 1.29 | 1.67 | 1.89 | 1.67 | 1.55 | 1.68 | 1.67 | 0.16 | 0.0004 |
| MET | 2.02 | 2.06 | 1.67 | 1.75 | 1.42 | 1.66 | 2.27 | 1.92 | 1.97 | 1.55 | 1.83 | 0.26 | 0.0603 |
| MYC | 2.44 | 2.18 | 1.78 | 1.66 | 1.50 | 2.13 | 2.63 | 1.90 | 2.21 | 2.10 | 2.05 | 0.35 | 0.8151 |
| PDGFRA | 1.61 | 3.22 | 1.83 | 2.03 | 1.69 | 1.60 | 2.52 | 2.09 | 1.69 | 12.05 | 3.03 | 3.21 | 0.4031 |
| PIK3CA | 1.83 | 1.82 | 1.82 | 1.83 | 1.61 | 1.76 | 3.28 | 2.29 | 1.47 | 1.83 | 1.95 | 0.51 | 0.5144 |
| PRDM1 | 2.19 | 1.54 | 2.07 | 2.48 | 1.41 | 1.56 | 2.14 | 2.02 | 1.59 | 2.61 | 1.96 | 0.42 | 0.5693 |
| PTEN | 2.62 | 1.73 | 1.88 | 1.58 | 1.63 | 2.12 | 1.52 | 1.63 | 1.32 | 1.27 | 1.73 | 0.40 | 0.0371 |
| PTPRD | 1.88 | 1.93 | 1.89 | 1.76 | 1.58 | 1.83 | 1.45 | 1.75 | 1.62 | 2.00 | 1.77 | 0.17 | 0.0030 |
| RB1 | 2.16 | 1.56 | 1.65 | 1.81 | 1.78 | 1.49 | 1.45 | 1.78 | 1.46 | 1.51 | 1.67 | 0.22 | 0.0010 |
| REL | 2.33 | 2.00 | 2.13 | 2.10 | 1.96 | 1.91 | 2.36 | 2.16 | 1.53 | 1.93 | 2.04 | 0.24 | 0.7388 |
| VEGFA | 1.84 | 3.96 | 3.53 | 3.94 | 1.36 | 1.71 | 2.75 | 2.07 | 2.65 | 2.69 | 2.65 | 0.92 | 0.0844 |
| ZNF217 | 2.64 | 2.55 | 1.88 | 1.77 | 1.48 | 2.38 | 2.33 | 1.91 | 1.51 | 2.69 | 2.11 | 0.46 | 0.6502 |
Table 3
Correlation analysis of seven candidate genes based on their copy numbers in tumor tissues of LUAD patients without EGFR mutation or ALK fusion.
| Interacted Genes | Correlation | P-Value | 95% Lower | 95% Upper |
|---|---|---|---|---|
| EMSY, CCND1 | 0.994 | <0.0001 | 0.974 | 0.999 |
| EMSY, CCNE1 | 0.603 | 0.0647 | -0.043 | 0.894 |
| EMSY, CDKN2A | -0.887 | 0.0002 | -0.973 | -0.582 |
| EMSY, FGFR1 | 0.789 | 0.0047 | 0.318 | 0.948 |
| EMSY, HMGA2 | 0.482 | 0.1648 | -0.213 | 0.853 |
| EMSY, PIK3CA | 0.919 | <0.0001 | 0.688 | 0.981 |
| CCND1, CCNE1 | 0.587 | 0.0750 | -0.068 | 0.888 |
| CCND1, CDKN2A | -0.927 | <0.0001 | -0.983 | -0.715 |
| CCND1, FGFR1 | 0.769 | 0.0071 | 0.270 | 0.942 |
| CCND1, HMGA2 | 0.471 | 0.1762 | -0.226 | 0.849 |
| CCND1, PIK3CA | 0.913 | <0.0001 | 0.665 | 0.979 |
| CCNE1, CDKN2A | -0.397 | 0.2668 | -0.821 | 0.311 |
| CCNE1, FGFR1 | 0.584 | 0.0770 | -0.027 | 0.887 |
| CCNE1, HMGA2 | 0.805 | 0.0033 | 0.355 | 0.952 |
| CCNE1, PIK3CA | 0.695 | 0.0231 | 0.117 | 0.922 |
| CDKN2A, FGFR1 | -0.598 | 0.0680 | -0.892 | 0.051 |
| CDKN2A, HMGA2 | -0.392 | 0.2726 | -0.820 | 0.315 |
| CDKN2A, PIK3CA | -0.804 | 0.0033 | -0.952 | -0.353 |
| CDKN2A, PIK3CA | 0.459 | 0.1891 | -0.240 | 0.845 |
| FGFR1, PIK3CA | 0.731 | 0.0139 | 0.187 | 0.932 |
| HMGA2, PIK3CA | 0.589 | 0.0738 | -0.065 | 0.889 |
Correlations of copy numbers and mRNA levels for genes with lower copy numbers in LUAD tumors
Next, we focused on the lung cancer-associated genes with lower copy numbers in LUAD tissues and measured their expression in LUAD tumor tissues and in paired normal lung epithelial tissues from ten patients with LUAD. Consistent with previous studies [6], expressions of the tumor suppressor genes PTEN and RB1 were significantly lower (both P < 0.05) based on comparisons of mRNA levels in tumor and normal tissues (Figs 3A and 3B). However, for the tumor suppressor gene PTPRD and the oncogene HMGA2 (Figs 3C and 3D), there were no significant changes in gene expression in LUAD tumors relative to normal tissues (n=6, P=0.506 for PTPRD and n=8, P=0.462 for HMGA2). We determined if, in LUAD tumors, the expression of these four genes correlated with their copy numbers. For PTEN, there was a significant correlation between these two parameters (R2=0.827, P=0.003) (Fig 4A). The relative expression of RB1 showed only a marginal association (R2=0.2798, P=0.1159) with its relative copy numbers (Fig 4B), and the copy numbers of HMGA2 and PTPRD showed poor correlations with their expressions (R2=0.0514, P=0.5286 for HMGA2; R2=0.0713, P=0.4557 for PTPRD) (Figs 4C and 4D).
Interactions among lung cancer-associated genes based on copy numbers in LUAD without EGFR mutations or ALK fusion
Although various genes are associated with LUAD progression, little is known about the underlying regulatory mechanisms [12, 14]. Thus, Pearson correlation tests were performed to determine the relationship of the 23 lung cancer-associated genes based on their copy numbers in ten LUAD tumors without EGFR mutations or ALK fusion. The correlation matrix data, presented in S2 Table, contain the results of pairwise comparisons for all 23 genes. Table 3 displays correlations among seven genes that are most likely to interact with each other since they are implicated in tumorigenesis by their effects on cell cycle control and regulation of cellular growth. Most of the pairwise comparisons displayed significant correlations (P<0.05). The correlations of EMSY/CCND1, EMSY/PIK3CA, CCND1/ CDKN2A, and CCND1/PIK3CA were strong (all P< 0.001). The correlations of EMSY/CCND1, EMSY/ PIK3CA, and CCND1/PIK3CA were all positive (all matrix values >0.9), as these genes alter cell cycle progression, promote cellular growth, and contribute to tumorigenesis [34-38]. CDKN2A, which functions as a tumor suppressor gene, correlated negatively with CCND1 (matrix value= -0.927, P<0.001).
Figure 3
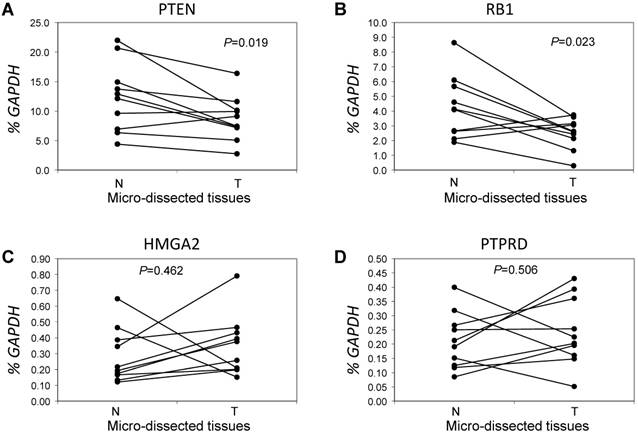
Comparative analysis of expression for genes with lower copy numbers in LUAD. Gene expressions were measured in tumor cells and paired tumor-distant normal lung epithelial cells from ten LUAD patients without EGFR mutation or ALK fusion. A paired t test was used to evaluate the correlations of genes expression for PTEN (A), RB1 (B), HMGA2 (C) and PTPRD (D) between tumor tissues (T) and normal lung tissues (N). Each line with paired dot ends indicates the mRNA level change of one patient for the specified gene. P values, tumor cells vs. paired tumor-distant normal epithelial cells, were tested using the paired two-sample t test.
Figure 4
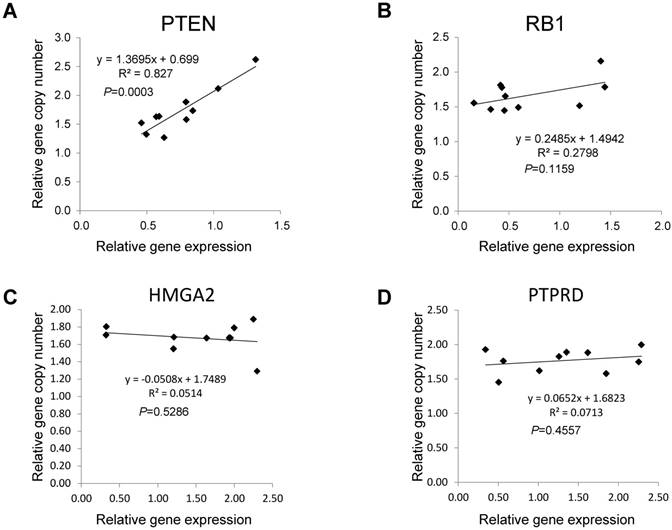
Associations between copy numbers and mRNA levels for PTEN, RB1, HMGA2, and PTPRD genes in LUAD without EGFR mutation or ALK fusion. The Pearson correlation test was employed to evaluate the association between copy number and gene expression for PTEN (A), RB1 (B), HMGA2(C) and PTPRD (D) in tumor tissues from ten LUAD patients without EGFR mutations or ALK fusion. Each dot represents one patient. P values for the association between DNA copy numbers and the gene expression levels were evaluated using the Pearson's correlation test.
Discussion
The discovery of driver oncogenes (e.g., EGFR, KRAS, and ALK) has changed the understanding of and the approach to treating lung cancer and has highlighted the importance of the genotype in therapy of lung cancer [7, 9, 10]. However, for LUAD patients without known genomic alterations, it is necessary to identify new therapeutic targets and provide more selective drugs for personal and precise medical care. In this context, the present study presented feasible approaches and introduced a reasonable framework for searching candidate genes and for validating them in a cohort of LUAD patients.
To gain insight into the genomic alterations of the lung cancer-associated genes in normal and malignant tissues without EGFR mutation or ALK fusion, we evaluated the copy numbers of 23 lung cancer-associated genes in micro-dissected LUAD tumor cells and in tumor-distant normal lung epithelial cells. There were lower copy numbers for four genes: PTEN, RB1, PTPRD, and HMGA2. In LUAD tumors, the lower copy numbers of tumor suppressor genes PTEN, RB1, and PTPRD (Fig 2 and Table 2) are consistent with their oncogenomic profiling results summarized for large cohorts of patients (Fig 1). Gene deletions and mutations (indicated by blue bars and green bars, respectively, in Fig 1,) but not gene amplification (indicated by red bars) predominated in the patterns of alterations for all three genes. Furthermore, the analysis suggested correlations between copy number and expression levels of PTEN and RB1 (Figs 4A and 4B), similar to results of previous studies that investigated LUAD tumors regardless of EGFR/ALK alteration [6]. This could be partially or fully attributed to no (for PTEN) or rare (for RB1) gene amplification found in the genomics of LUAD tumors (Fig 1). Our results strengthen the rationale for and importance of developing approaches targeting PTEN and RB1 for treating LUAD without genomic alterations such as EGFR mutations and ALK fusion. In addition, we also analyzed the copy number of PTEN, RB1, HMGA2, and PTPRD using c-Bioportal. Data mining in other categories of lung cancer. Of note, the copy numbers of these genes were also lower in LUSC (Fig. 1) but not in large cell carcinoma and small cell carcinoma of the lung.
In view of the lower PTPRD expression in LUAD tumors of a few patients (30%, Fig 3D) and no correlation between the copy numbers and gene expression levels of PTPRD (Fig 4D), gene deletion of PTPRD (Figs 1 and 2) is unlikely to contribute to its expression. Likewise, HMGA2 displayed reduced copy numbers in LUAD tumors (Table 2 and Fig 2), whereas HMGA2 had a high frequency of gene amplification in lung cancers from large cohorts of patients (Fig 1). For a variety of human cancers, HMGA2, a transcriptional factor, is positively associated with tumor progression [39-42]. Most LUAD tumors (80%) had higher levels of HMGA2 mRNA than normal lung tissues (Fig 3C), which is consistent with previous reports regarding the role of high HMGA2 expression in tumorigenicity and onco-transformation [43, 44]. It raises the question of whether HMGA2 is a contributor to LUAD tumors without EGFR/ALK alterations.
LUAD is a complex and heterogeneous disease involving various signaling pathways. Our correlation tests identified a relationship among four genes (EMSY, CCND1, PIK3CA, and CDKN2A) based on their copy numbers in ten LUAD tumors without EGFR mutations or ALK fusion (Table 3). EMSY, a BRCA2-interacting transcriptional repressor, is associated with tumor growth and metastasis [34, 35]. CCND1 interacts with its regulator RB1 to alter cell cycle progression and contributes to tumorigenesis in a variety of tumors [36, 37]. CDKN2A is a tumor suppressor gene involved in regulation of the G1/S phase transition of the cell cycle in human LUAD [23, 45]. In human cancers, PIK3CA and PTEN, signaling components of the PI3-kinase pathway, are frequently mutated [38]. The present studies confirmed previous findings on the roles of these genes in lung carcinogenesis, which is irrelevant to EGFR mutations and ALK rearrangements. Since the four strongest correlations (EMSY/CCND1, EMSY/PIK3CA, CCND1/CDKN2A, and CCND1/PIK3CA) cover either PIK3CA or CCND1, the results prioritize pathways related to PTEN-PIK3CA and RB1-CCND1 in developing therapeutic strategies for LUAD patients without EGFR mutation or ALK fusion.
The current investigation has strengths and limitations. Strengths are that it is the first to report on the oncogenomic aberrations in LUAD patients without EGFR mutations or ALK fusion and that it provides a framework for searching for and validating lung cancer-associated genes to facilitate implementation of personalized therapy. A limitation is its small sample size of 10 patients. Also, the patients were drawn from a single institution and thus were subject to referral bias. Furthermore, the methodology for quantitation of DNA copy numbers and mRNA levels cannot distinguish gene mutations, since the mutants, rather than deletions of some genes, such as RB1 (Fig 1), act in a dominant-negative manner and are involved in carcinogenesis. Given the inherent bias of intratumor heterogeneity and the presence of variables that we cannot account for, the smaller sample size may lead to missed associations among genomic features of the 23 genes. Nevertheless, although there was a lack of clinical information such as tumor size and tumor location, the current work is the first to quantify genetic aberrations of LUAD tumors that are negative for both EGFR mutation and ALK fusion. In addition, cigarette smoke and gender are risk factors for lung cancer and cigarette-smoking increases copy number alterations in NSCLC [46, 47], indicating that these factors may also affect copy numbers of these selected genes.
In conclusions, we conducted an analysis of public datasets to assess the genomic alterations of 23 lung cancer-associated genes and found clues to the etiology and pathogenesis of LUAD and LUSC. We also assessed the oncogenomic profiles of surgically resected tumors without EGFR mutations or ALK fusion and tumor-distant normal lung tissues from LUAD patients and evaluated possible associations between these candidate genes. Our findings have implications for the molecular stratification and therapeutic targeting of LUAD without EGFR mutations or ALK fusion. This information also advances understanding of lung carcinogenesis as it relates to oncogenomic CNAs and gene expression and facilitates the identification of personal therapeutic strategies for patients with LUAD.
Supplementary Material
Supplementary tables.
Acknowledgements
We thank Dr. Donald L. Hill for editorial assistance in preparing this manuscript. This work was supported by the USA National Institutes of Health/National Cancer Institute (CA179282 and CA118948), the USA Department of Defense (BC160808, PC130594, and PC140308), the Natural Science Foundation of China (No.31571342, 81772757, and 81773092).
Data Availability Statement
All relevant data are available from the paper, supporting information files, and public dataset (cBioPortal for Cancer Genomics at http://www.cbioportal.org).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893-917
2. Chen WQ, Zeng HM, Zheng RS, Zhang SW, He J. Cancer incidence and mortality in china, 2007. Chin J Cancer Res. 2012;24:1-8
3. Travis WD, Travis LB, Devesa SS. Lung cancer. Cancer. 1995;75:191-202
4. Subramanian J, Govindan R. Lung cancer in never smokers: a review. J Clin Oncol. 2007;25:561-70
5. Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R. et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893-8
6. Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K. et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069-75
7. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N. et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947-57
8. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG. et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693-703
9. Shaw AT, Yeap BY, Solomon BJ, Riely GJ, Gainor J, Engelman JA. et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011;12:1004-12
10. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S. et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561-6
11. Sholl LM. Biomarkers in lung adenocarcinoma: a decade of progress. Arch Pathol Lab Med. 2015;139:469-80
12. Hsu MK, Pan CL, Chen FC. Functional divergence and convergence between the transcript network and gene network in lung adenocarcinoma. Onco Targets Ther. 2016;9:335-47
13. Meng X, Lu P, Bai H, Xiao P, Fan Q. Transcriptional regulatory networks in human lung adenocarcinoma. Mol Med Rep. 2012;6:961-6
14. Lu J, Wang W, Xu M, Li Y, Chen C, Wang X. A global view of regulatory networks in lung cancer: An approach to understand homogeneity and heterogeneity. Semin Cancer Biol. 2017;42:31-8
15. Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, Yatabe Y. et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol. 2011;6:244-85
16. Goldstraw P, Crowley J, Chansky K, Giroux DJ, Groome PA, Rami-Porta R. et al. The IASLC Lung Cancer Staging Project: proposals for the revision of the TNM stage groupings in the forthcoming (seventh) edition of the TNM Classification of malignant tumours. J Thorac Oncol. 2007;2:706-14
17. Dong YU, Ren W, Qi J, Jin BO, Li Y, Tao H. et al. EGFR, ALK, RET, KRAS and BRAF alterations in never-smokers with non-small cell lung cancer. Oncol Lett. 2016;11:2371-8
18. Liu R, Yi B, Wei S, Yang WH, Hart KM, Chauhan P. et al. FOXP3-miR-146-NF-kappaB Axis and Therapy for Precancerous Lesions in Prostate. Cancer Res. 2015;75:1714-24
19. Ma L, Chung WK. Quantitative analysis of copy number variants based on real-time LightCycler PCR. Curr Protoc Hum Genet. 2014;80:Unit 7 21
20. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402-8
21. Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E. et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107-20
22. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ. et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124-8
23. Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543-50
24. Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519-25
25. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
26. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-4
27. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV. et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415-21
28. Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013;3:246-59
29. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J. et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899-905
30. Shlien A, Malkin D. Copy number variations and cancer. Genome Med. 2009;1:62
31. Goel HL, Mercurio AM. VEGF targets the tumour cell. Nat Rev Cancer. 2013;13:871-82
32. Roussel MF. The INK4 family of cell cycle inhibitors in cancer. Oncogene. 1999;18:5311-7
33. Song Z, Wang X, Zheng Y, Su H, Zhang Y. MET Gene Amplification and Overexpression in Chinese Non-Small-Cell Lung Cancer Patients Without EGFR Mutations. Clin Lung Cancer. 2017;18:213-9 e2
34. Zhao X, Zhou Y, Nie M, Xian S, Chen H, Wen Y. et al. EMSY promoted the growth and migration of ovarian cancer cells. Tumour Biol. 2015;36:3085-92
35. Madjd Z, Akbari ME, Zarnani AH, Khayamzadeh M, Kalantari E, Mojtabavi N. Expression of EMSY, a novel BRCA2-link protein, is associated with lymph node metastasis and increased tumor size in breast carcinomas. Asian Pac J Cancer Prev. 2014;15:1783-9
36. Lou X, Zhang J, Liu S, Xu N, Liao DJ. The other side of the coin: the tumor-suppressive aspect of oncogenes and the oncogenic aspect of tumor-suppressive genes, such as those along the CCND-CDK4/6-RB axis. Cell Cycle. 2014;13:1677-93
37. Mizuarai S, Machida T, Kobayashi T, Komatani H, Itadani H, Kotani H. Expression ratio of CCND1 to CDKN2A mRNA predicts RB1 status of cultured cancer cell lines and clinical tumor samples. Mol Cancer. 2011;10:31
38. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497-510
39. Tan L, Wei X, Zheng L, Zeng J, Liu H, Yang S. et al. Amplified HMGA2 promotes cell growth by regulating Akt pathway in AML. J Cancer Res Clin Oncol. 2016;142:389-99
40. Li T, Yang XD, Ye CX, Shen ZL, Yang Y, Wang B. et al. Long noncoding RNA HIT000218960 promotes papillary thyroid cancer oncogenesis and tumor progression by upregulating the expression of high mobility group AT-hook 2 (HMGA2) gene. Cell Cycle. 2017;16:224-31
41. Kaur H, Ali SZ, Huey L, Hutt-Cabezas M, Taylor I, Mao XG. et al. The transcriptional modulator HMGA2 promotes stemness and tumorigenicity in glioblastoma. Cancer Lett. 2016;377:55-64
42. Wu J, Zhang S, Shan J, Hu Z, Liu X, Chen L. et al. Elevated HMGA2 expression is associated with cancer aggressiveness and predicts poor outcome in breast cancer. Cancer Lett. 2016;376:284-92
43. Di Cello F, Hillion J, Hristov A, Wood LJ, Mukherjee M, Schuldenfrei A. et al. HMGA2 participates in transformation in human lung cancer. Mol Cancer Res. 2008;6:743-50
44. Meyer B, Loeschke S, Schultze A, Weigel T, Sandkamp M, Goldmann T. et al. HMGA2 overexpression in non-small cell lung cancer. Mol Carcinog. 2007;46:503-11
45. Romagosa C, Simonetti S, Lopez-Vicente L, Mazo A, Lleonart ME, Castellvi J. et al. p16(Ink4a) overexpression in cancer: a tumor suppressor gene associated with senescence and high-grade tumors. Oncogene. 2011;30:2087-97
46. Gasperino J. Gender is a risk factor for lung cancer. Med Hypotheses. 2011;76:328-31
47. Huang YT, Lin X, Liu Y, Chirieac LR, McGovern R, Wain J. et al. Cigarette smoking increases copy number alterations in nonsmall-cell lung cancer. Proc Natl Acad Sci U S A. 2011;108:16345-50
Author contact
![]() Corresponding authors: Department of Genetics, University of Alabama at Birmingham, Birmingham, Alabama, 35294, USA. Tel: +1-205-934-5948; E-mails: lwang12edu or Department of Thoracic Surgery, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, Shandong, 250021, China. Tel: +86-136-65312567; E-mails: wz620226com
Corresponding authors: Department of Genetics, University of Alabama at Birmingham, Birmingham, Alabama, 35294, USA. Tel: +1-205-934-5948; E-mails: lwang12edu or Department of Thoracic Surgery, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, Shandong, 250021, China. Tel: +86-136-65312567; E-mails: wz620226com
Citation styles
APA
Luo, Y., Li, B., Zhang, G., He, Y., Bae, J.H., Hu, F., Cui, R., Liu, R., Wang, Z., Wang, L. (2018). Integrated Oncogenomic Profiling of Copy Numbers and Gene Expression in Lung Adenocarcinomas without EGFR Mutations or ALK Fusion. Journal of Cancer, 9(6), 1096-1105. https://doi.org/10.7150/jca.23909.
ACS
Luo, Y.; Li, B.; Zhang, G.; He, Y.; Bae, J.H.; Hu, F.; Cui, R.; Liu, R.; Wang, Z.; Wang, L. Integrated Oncogenomic Profiling of Copy Numbers and Gene Expression in Lung Adenocarcinomas without EGFR Mutations or ALK Fusion. J. Cancer 2018, 9 (6), 1096-1105. DOI: 10.7150/jca.23909.
NLM
Luo Y, Li B, Zhang G, He Y, Bae JH, Hu F, Cui R, Liu R, Wang Z, Wang L. Integrated Oncogenomic Profiling of Copy Numbers and Gene Expression in Lung Adenocarcinomas without EGFR Mutations or ALK Fusion. J Cancer 2018; 9(6):1096-1105. doi:10.7150/jca.23909. https://www.jcancer.org/v09p1096.htm
CSE
Luo Y, Li B, Zhang G, He Y, Bae JH, Hu F, Cui R, Liu R, Wang Z, Wang L. 2018. Integrated Oncogenomic Profiling of Copy Numbers and Gene Expression in Lung Adenocarcinomas without EGFR Mutations or ALK Fusion. J Cancer. 9(6):1096-1105.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.