Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(7):1182-1187. doi:10.7150/jca.23649 This issue Cite
Research Paper
Hepatitis B Virus Infection Dampens CtIP Expression in Hepatoma Cell
Dongxin Zhang1,2,*, ![]() , Haojing Liu3,4,*, Jusheng Lin4, Duyun Ye2,
, Haojing Liu3,4,*, Jusheng Lin4, Duyun Ye2, ![]()
1. Department of Clinical Laboratory, Puai Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430033, People's Republic of China
2. Department of Pathophysiology, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, People's Republic of China
3. Department of Internal Medicine, Puai Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430033, People's Republic of China
4. Institute of Liver Diseases, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, People's Republic of China
*Dongxin Zhang and Haojing Liu contributed equally to this work.
Received 2017-11-3; Accepted 2018-1-28; Published 2018-3-8
Citation:
Zhang D, Liu H, Lin J, Ye D. Hepatitis B Virus Infection Dampens CtIP Expression in Hepatoma Cell. J Cancer 2018; 9(7):1182-1187. doi:10.7150/jca.23649. https://www.jcancer.org/v09p1182.htm
Other stylesAbstract
Hepatitis B virus (HBV) infection is a leading cause for hepatocellular carcinoma (HCC). Dysregulation of DNA double-strand break (DSB) repair may explain the pathogenesis of HBV-related HCC. Tumor suppressor CtIP plays a critical role in DSB repair. The purpose of present study was to clarify whether HBV affects CtIP expression in DSB repair of hepatoma cell. HepG2.2.15 was selected as the HBV positive hepatoma cell line, while HepG2 as the HBV negative hepatoma cell line. The two cell lines were treated with bleomycin to induce DSB. Bleomycin treatment could result in DSB by γ-H2AX detection. CtIP gene expression was significantly upregulated after DSB in both HepG2 and HepG2.2.15, while CtIP expression of HepG2.2.15 was higher than that observed in HepG2 before and after DSB. CtIP protein expression was the same pattern as its gene expression. Phosphorylated CtIP (p-CtIP, serine site) was even lower than detectable limit in both HepG2 and HepG2.2.15 before DSB. However, p-CtIP of HepG2.2.15 was significantly lower than that of HepG2 after DSB. These results suggest that HBV could interfere CtIP via enhancing its expression while dampening its phosphorylation, which may disrupt DSB repair pathways and implicate CtIP dysfunction in HBV-related HCC.
Keywords: CtIP, double-strand break, hepatitis B virus, hepatocellular carcinoma, phosphorylation.
Introduction
Primary liver cancer is the fifth most common cancer worldwide and the third most common cause of cancer mortality, and hepatocellular carcinoma (HCC) accounts for between 85% and 90% of primary liver cancers[1]. Most cases of HCC are associated with chronic hepatitis B virus (HBV) infection[2]. HBV could indirectly alter host gene expression and cellular phenotypes that are recognized as hallmarks of cancer, and further overcome senescence by inhibiting the expression and function of tumour suppressors[3]. Maintenance of genome stability depends on efficient, accurate repair of DNA damage, and DNA double-strand break (DSB) are among the most lethal types of DNA damage, with the potential to cause mutation, chromosomal rearrangement, and genomic instability that could contribute to cancer[4]. Increasing evidence demonstrates that HBV infection induces hepatic oxidative stress and increases oxidative DNA damage[5]. Recognition of damaged DNA and timely and precise repair of the lesions play a crucial role in the maintenance of chromosomal stability[5]. Inactivation of DSB repair is related to uncontrolled cell growth and increased cancer risk, and there has been compelling evidence that defective DSB repair accelerates liver carcinogenesis[5, 6]. It has been demonstrated that HBV could inhibit the repair of damaged hepatocyte DNA[7]. Mechanically, HBV upregulates DNA methyltransferases (DNMTs)[8], which decreases the expression of genes involved in DNA repair[9]. Additionally, HBV could alter cellular signalling pathways of hepatocyte to modulate biologically activity of transcriptional factors and target proteins involved in hepatocarcinogenesis[7].
DSB repair is tightly regulated during the cell cycle. DSB damage can be repaired by two common pathways nonhomologous end-joining (NHEJ) and homologous recombination (HR), while the former is error-prone and the latter error-free[10]. As the primary factor responsible for cell-cycle regulation of resection, CtIP is required for DSB repair[11]. The tumor suppressor protein CtIP controls the HR repair pathway of DSB damage via regulating the initiation of DSB end resection after integrating signals from the DNA damage checkpoint response and cell cycle cues[4]. CtIP is required not only for repair of DSB by HR in S/G2 phase but also for NHEJ in G1, whereas function of CtIP in HR, but not NHEJ, is dependent on the phosphorylation of serine residue and recruitment of BRCA1[10]. CtIP undergoes ATR-dependent hyperphosphorylation in response to DSB, which is required for activation of DSB processing[12]. Nonphosphorylatable CtIP does not bind to chromatin or initiate resection, and chromatin binding by modified CtIP precedes extensive resection and full checkpoint activation[12].
Hepatocellular DSB could lead to HCC development, while evidences have shown that HBV induces DSB while hinders DSB repair in host hepatocytes. CtIP plays a critical role in DSB repair. Whether HBV could affect CtIP expression of hepatoma cell remains to be unclear. The purpose of the present study is to confirm the detailed effects of HBV on CtIP of hepatoma cell.
Materials and Methods
Reagents and antibodies
Bleomycin was from Nippon Kayaku Co., Ltd. (Chiyoda-ku, Tokyo, Japan). G418 was from Roche. Anti γ-H2AX antibody was from Cell Signaling Technology (Danvers, MA, USA). Anti Lamin B (sc-374015) and β-actin (sc-1616) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti CtIP and phosphoserine antibodies were from Abcam (Cambridge, UK). Protein A Agarose was from Millipore (USA). RIPA Lysis Buffer and Nuclear and Cytoplasmic Protein Extraction Kit were from Beyotime Institute of Biotechnology (Shanghai, China). BCA protein assay kit was from Pierce (Rockford, IL, USA). Trizol was purchased from Life Technologies (Grand Island, NY, USA).
Cell culture and treatment
HepG2 and HepG2.2.15 cell lines were routinely cultured in our laboratory. The two cells were cultured at 37°C in a humidified atmosphere with 5% CO2 in DMEM high glucose medium supplemented with 10% heat-inactivated fetal bovine serum, 25 mM HEPES, 100 U mL-1 penicillin G and 100 U mL-1 streptomycin. Meanwhile, 0.5% G418 was added into HepG2.2.15 cultures to ensure stable transfection of HBV in this cell line. Cells were treated with indicated concentrations of bleomycin at different time for different assays.
Determination of HBsAg in HepG2 and HepG2.2.15 cultures
75 μl of negative control, positive control, blank control, HepG2 culture supernatant and HepG2.2.15 culture supernatant were added into the reaction hole of HBsAg ELISA plate respectively. After ELISA plate was sealed and incubated for 60 min at 37°C, 50 μl of enzyme conjugates were added into each reaction hole. After 10 s of oscillation, ELISA plate was incubated for 30 min at 37°C. Then, all reaction holes of the ELISA plate were washed five times with washing liquid, and immediately added with 50 μl of chromogenic agent A and 50 μl of chromogenic agent B. After oscillation, the ELISA plate was placed at 37°C for 30 min, and all reaction holes were added with 50 μl stop solution. Values of optical density (OD) were measured with a micro ELISA reader (Bio-rad, USA) at a wavelength of 450 nm.
DSB construction by bleomycin treatment
MTT assay was firstly performed to screen proper bleomycin concentration for DSB inducement with minimum effect on cell proliferation. 3 μg ml-1 was selected as proper concentration of bleomycin for DSB inducement finally. Furthermore, cells were treated with 3 μg ml-1 of bleomycin at different times (0, 12, 24, 48 and 72 hours) and, meanwhile, γ-H2AX was determined to observe the best time of bleomycin inducement for DSB model. Then, cells were treated with 3 μg ml-1 of bleomycin for different experiments according to the protocol.
RNA isolation and real-time PCR
Total RNA was isolated from cells using Trizol reagent according to manufacturer's protocol. The concentration and purity of RNA was determined by the UV spectrophotometer (Eppendorf, Hamburg, Germany). cDNA synthesis was performed using ReverTra Ace cDNA Synthesis kit (FSQ-101, TOYOBO, Osaka, Japan) according to manufacturer's instruction. Quantitative PCR analysis was carried out on a Bio-Rad CFX Connect Real-Time PCR Detection System using iTaq Universal SYBR Green Supermix (Bio-Rad Laboratories, Richmond, CA, USA). Each sample was quantified in triplicate (n=3). PCR conditions were 30 s at 95°C, followed by 40 cycles of 10 s at 95°C and 20 s at 60°C. Data were analyzed using the Bio-Rad CFX Manager 2.1 software, and normalized to β-actin. The sequences of primers were as follows: β-actin, forward 5'-GTTGCGTTACACCCTTTCTTG-3' and reverse 5'-GACTGCTGTCACCTTCACCGT-3'; CtIP, forward 5'-CAGGAACGAATCTTAGATGCACA-3' and reverse 5'-GCCTGCTCTTAACCGATCTTC T-3'.
Western blot
Nuclear and total proteins were extracted using Nuclear and Cytoplasmic Protein Extraction Kit and RIPA Lysis Buffer respectively. Protein concentrations were determined using a BCA protein assay kit. Then, regular western blotting assay was performed. Briefly, equal amounts of protein (40 μg) were subjected to 12% SDS-PAGE electrophoresis and transferred to PVDF membranes (Millipore, USA). The membranes were blocked with 5% (w/v) non-fat dried milk or BSA and incubated with primary antibodies overnight at 4°C, followed by incubation with a secondary HRP-conjugated antibody. The bound antibody was detected by an enhanced chemiluminescence kit (Millipore, USA) on X-ray film. Lamin B and β-actin served as internal controls for nuclear and total protein respectively.
Immunoprecipitation and western blot analysis
Cells were lysed in RIPA Lysis Buffer with protease inhibitors. Immunoprecipitation from the cell lysates was performed with anti-CtIP and Protein A-coupled Agarose beads and then washed in PBST buffer. Immunoprecipitates were analyzed by western blot with ECL reagents to detect p-CtIP. After detecting p-CtIP expression, the PVDF membrane was further washed with stripping buffer to detect total CtIP content.
Statistical analysis
All statistical analyses were done using the SPSS 19.0 software. The results were expressed as means ± S.E.M of multiple independent experiments. The means of the different groups were compared by employing either Student's t-test or one-way ANOVA followed by S-N-K post-hoc test. A value of p < 0.05 was considered significant.
Results
Determination of HBsAg in hepatoma cells
In the study, HepG2.2.15 was selected as the HBV positive hepatoma cell line, while HepG2 as the HBV negative hepatoma cell line. Thus, ELISA was conducted to confirm whether HBV infected the two cell lines. According to ELISA results, OD of negative control ≤ 0.1, OD of positive control ≥ 1.0, OD of HepG2/(OD of negative control+0.1) ≤ 1.0, OD of HepG2.2.15/(OD of negative control+0.1) ≥ 1.0, indicating positive expression of HBsAg in HepG2.2.15 and negative expression in HepG2.
DSB construction of hepatoma cells
To observe the effect of HBV on DNA repair protein CtIP expression, hepatoma cells were treated with low concentration of bleomycin to construct DSB model. After HepG2 and HepG2.2.15 were administered 3 μg/ml of bleomycin at 0, 12, 24, 48 and 72 hours respectively, nuclear protein was extracted and western blot was employed to detect expressions of DSB standard γ-H2AX. As shown in Fig. 1, compared to control group (0 h), γ-H2AX of HepG2 was significantly up-regulated at 24 h, while γ-H2AX of HepG2.2.15 was significantly enhanced at 12 h. However, γ-H2AX expression of both HepG2 and HepG2.2.15 peaked at 24 h. These results suggest that DSB model was successfully constructed, and the peak time occurred at 24 h.
Figure 1
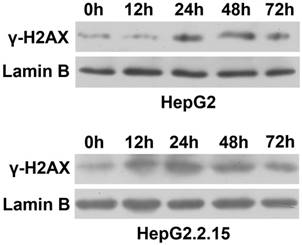
DSB construction of hepatoma cells by bleomycin. HepG2 and HepG2.2.15 were treated with 3 μg/ml bleomycin at 0, 12, 24, 48 and 72 hours. γ-H2AX expression was detected by western blot. Lamin B served as internal control for nuclear protein.
The effect of HBV on CtIP gene expression of hepatoma cells in DSB
RNA was extracted and realtime PCR was performed to detect CtIP gene expression of HepG2 and HepG2.2.15 before and after DSB. As shown in Fig. 2, CtIP gene expression of both HepG2 and HepG2.2.15 was significantly up-regulated after DSB, while CtIP gene expression of HepG2.2.15 was higher than that of HepG2 before and after DSB. These results suggest that DSB stimulation induces CtIP gene expression of hepatoma cells, and HBV infection further enhances its gene expression.
The effect of HBV on CtIP protein expression of hepatoma cells in DSB
Total protein was extracted and western blot was performed to detect CtIP protein expression of HepG2 and HepG2.2.15 before and after DSB. As shown in Fig. 3, CtIP protein expression of both HepG2 and HepG2.2.15 was significantly up-regulated after DSB, while CtIP protein expression of HepG2.2.15 was higher than that of HepG2 before and after DSB. These results suggest that DSB stimulation induces CtIP protein expression of hepatoma cells, and HBV infection further enhances its protein expression.
Figure 2
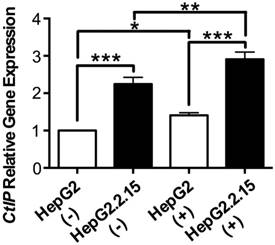
The effect of HBV on CtIP gene expression of hepatoma cells in DSB. HepG2 and HepG2.2.15 were treated with 3 μg/ml bleomycin at 24 hours. (+) represents bleomycin treatment, whereas (-) represents no bleomycin treatment. CtIP gene expression was examined by real-time PCR. Results are expressed as means ± SEM from six independent experiments. *P<0.05, **P<0.01 and ***P<0.001, one-way ANOVA with S-N-K posttest.
The effect of HBV on CtIP phosphorylation of hepatoma cells in DSB
Before and after DSB, total protein was extracted and IP was performed to acquire CtIP protein of HepG2 and HepG2.2.15. Then, western blot was performed to detect p-CtIP (Ser site) expression of HepG2 and HepG2.2.15 by anti-phosphoserine antibody. As shown in Fig. 4, p-CtIP expression of both HepG2 and HepG2.2.15 was below detection limit before DSB, while CtIP protein expression of HepG2.2.15 was higher than that of HepG2. There was no obvious difference in p-CtIP expression between HepG2 and HepG2.2.15 after DSB, while CtIP protein expression of HepG2.2.15 was higher than that of HepG2. As a result, p-CtIP/CtIP of HepG2.2.15 was significantly lower than that of HepG2 after DSB. These results suggest that HBV infection interveness CtIP phosphorylation upon DSB, thus dampening its biological function in DNA repair.
Discussion
Accumulating evidence suggest that HBV is involved in hepatocarcinogenesis via inducing DNA damage and gene mutations and disturbing the repair of hepatocyte DNA damage[3, 13, 14]. The tumor suppressor protein CtIP facilitates the transition from DSB sensing to DNA processing[15], and mediates cell cycle control of DNA end resection and DSB repair[16], which is required for efficient BRCA1 and MRN-mediated DSB repair through the complex formation[17]. CtIP emerges as a multivalent adaptor that connects cell cycle checkpoint control, transcriptional regulation and tumor suppression[18]. We demonstrate for the first time that HBV could lead to impaired CtIP phosphorylation of hepatoma cells in DSB, supporting that CtIP may represent an emerging key target for HBV-mediated HCC. Here, HBV-infected hepatoma cell line HepG2.2.15 was characterized by enhanced CtIP expression but inhibited CtIP phosphorylation when compared to HBV-negative hepatoma cell line HepG2, suggesting HCC may have occurred, at least in part, in response to impaired CtIP phosphorylation caused by HBV since CtIP is the key DNA repair protein.
Figure 3
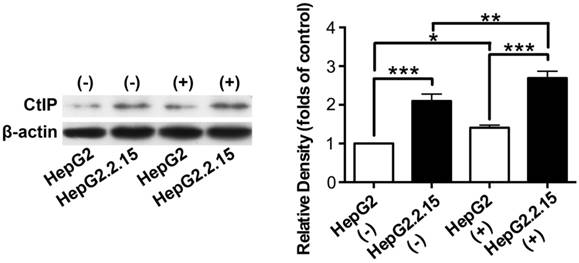
The effect of HBV on CtIP protein expression of hepatoma cells in DSB. HepG2 and HepG2.2.15 were treated with 3 μg/ml bleomycin at 24 hours. (+) represents bleomycin treatment, whereas (-) represents no bleomycin treatment. CtIP protein expression was examined by western blot. The histogram represents means ± SEM of the densitometric scans for protein bands from six independent experiments, normalized by comparison with β-actin and expressed as a percentage of HepG2 (-). *P<0.05, **P<0.01 and ***P<0.001, one-way ANOVA with S-N-K posttest.
Figure 4
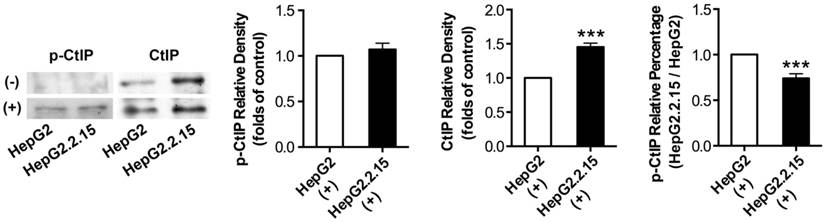
The effect of HBV on CtIP phosphorylation of hepatoma cells in DSB. HepG2 and HepG2.2.15 were treated with 3 μg/ml bleomycin at 24 hours. (+) represents bleomycin treatment, whereas (-) represents no bleomycin treatment. CtIP phosphorylation and stripped CtIP expression were examined by IP and western blot, and relative densitometric quantification for protein bands is also shown. p-CtIP relative percentage of HepG2.2.15/HepG2 is shown in last histogram of the Figure. ***P<0.001 versus control group, two-tailed Student's t-test.
The failure of hepatocyte DNA repair is the fundamental abnormality leading to HCC induced by HBV[7]. HR-mediated DSB repair is maximized by cyclin-dependent kinase (CDK) phosphorylation of CtIP[11], and its dysregulation is implicated in the pathogenesis of tumorigenesis[19, 20]. Whether HBV affects CtIP expression of hepatoma cells remains unclear. We demonstrated here increased CtIP expression in DSB model that may response to bleomycin-mediated DNA damage. Meanwhile, CtIP expression of HepG2.2.15 was higher than that of HepG2 regardless of DSB treatment. It is possible that secondary to bleomycin or HBV-mediated DNA damage, CtIP upregulation observed here may occur as a protective response against primary genome variation of liver cell. However, we reported CtIP phosphorylation inhibition in HBV-positive HepG2.2.15 when compared to HBV-negatvie HepG2, suggesting dysregulation of CtIP function by HBV. Thus, CtIP dysregulation by HBV provides an important mechanistic explanation for HBV-mediated DSB repair failure and further HCC. Further studies are planned to investigate the detailed reasons.
HBV-induced HCC is characterized by abnormal repair of DNA damage, resulting in hepatocyte injury, variation and further hepatocarcinogenesis. In attempting to interpret the pathologic feature found in HCC patients that were exposed to HBV, we suggest that HCC-related pathologic manifestations may be favored by HBV-mediated disturbance of DNA repair proteins that leads to hepatocyte genome variation. Indeed, we demonstrated that HBV enhanced the expression of CtIP but inhibited its phosphorylation in hepatoma cell. As the primary factor responsible for cell-cycle regulation of DNA resection, CtIP is crucial for DSB repair, and its function is dependent on the serine phosphorylation and recruitment of BRCA1[10]. CtIP undergoes ATR-dependent hyperphosphorylation in response to DSB, which is required for activation of DSB processing, while nonphosphorylatable CtIP does not bind to chromatin or initiate resection[12]. Thus, HBV-mediated inhibition of CtIP phosphorylation may be responsible for defective DSB repair, while HBV-mediated dysregulation of CtIP may represent a priming factor leading to HCC. Taken together, HBV dampened DSB repair via disturbing hepatocyte CtIP function, ultimately contributing to HCC.
In the study, HBV-transfective HepG2.2.15 was selected as the HBV positive hepatoma cell line, while HepG2 as the HBV negative hepatoma cell line. This is a well-designed in vitro model for studying the modulation of HBV on DSB repair of hepatoma cell. By using the model, we successfully found the molecular mechanism responsible for regulation of HBV on CtIP in DSB, providing an important clue for better understanding of the pathogenesis of HBV-induced HCC. The model was used in the study to simulate endogenous HBV infection of hepatocytes in patients. To a certain extent, the results from the study using HBV-transfective hepatoma cell line support that human hepatocyte CtIP is dysregulated by HBV in DSB, which represents a potential stimulus for hepatocyte variation and HCC development. However, this model may be different from the in vivo case of HBV patients, since there may exists complicated interaction mechanisms between HBV, CtIP and DSB in human liver. Thus, this study may be more relevant to how HBV regulate CtIP expression and contribute to abnormal CtIP phosphorylation in DSB of hepatoma cell line. To observe the effects of HBV on CtIP expression and phosphorylation, hepatoma cells were treated with low concentration of bleomycin to construct DSB model. γ-H2AX expression was examined as a marker of DSB. DSB standard γ-H2AX was detected to confirm the successful construction of DSB. Then, above model was performed based on the DSB condition.
It is noteworthy that there may be other causative factors in addition to dysregulated CtIP that are responsible for HBV-induced DSB repair failure and hepatocarcinogenesis, and may act in concert to stimulate DNA repair failure, hepatocyte variation and HCC development. HBV may intervene hepatocyte DSB repair through affecting other DNA repair-related proteins, including Mre11/Rad50/NBS1 (MRN) complex, ATM, ATR and BRCA1. Further studies are needed to study these potential molecular pathways and their interactions.
In conclusion, the change of CtIP expression and phosphorylation in DSB of hepatoma cells upon HBV infection provides strong experimental support for our hypothesis that CtIP is dysregulated by HBV in hepatocyte DSB. Relying on the results gathered by using bleomycin-induced DSB model and HBV-infective hepatoma cell model, the present study provides further evidence that HBV-mediated abnormality in CtIP function may be a potential causal factor for the pathogenesis of HCC. This study proposes a molecular pathway underlying the occurrence of HCC, providing new insights into the pathophysiology of HCC. Furthermore, our data describe a key target responsible for HBV-induced HCC, which may provide a clue for better understanding of the pathogenesis of HCC and contribute to develop potential therapeutic strategies in preventing HCC.
Acknowledgements
The authors acknowledge Dr. XingXing He for his technique assistance in IP experiment and for his expert assistance in manuscript preparation. This work was supported by the National Natural Science Foundation of China (grant numbers 81701469, 81671480); the Young and Mid-Aged Key Medical Personnel Training Project of Wuhan City (grant number 2016-59); and the Natural Science Foundation of Hubei Province (grant number 2015CFB179).
Competing Interests
The authors have declared that no competing interest exists.
References
1. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557-76
2. El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142:1264-73 e1
3. Arzumanyan A, Reis HM, Feitelson MA. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat Rev Cancer. 2013;13:123-35
4. You Z, Bailis JM. DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol. 2010;20:402-9
5. Na TY, Ka NL, Rhee H. et al. Interaction of hepatitis B virus X protein with PARP1 results in inhibition of DNA repair in hepatocellular carcinoma. Oncogene. 2016;35:5435-45
6. Li R, Yang Y, An Y. et al. Genetic polymorphisms in DNA double-strand break repair genes XRCC5, XRCC6 and susceptibility to hepatocellular carcinoma. Carcinogenesis. 2011;32:530-6
7. Arbuthnot P, Capovilla A, Kew M. Putative role of hepatitis B virus X protein in hepatocarcinogenesis: effects on apoptosis, DNA repair, mitogen-activated protein kinase and JAK/STAT pathways. J Gastroenterol Hepatol. 2000;15:357-68
8. Huang J, Wang Y, Guo Y. et al. Down-regulated microRNA-152 induces aberrant DNA methylation in hepatitis B virus-related hepatocellular carcinoma by targeting DNA methyltransferase 1. Hepatology. 2010;52:60-70
9. Toyota M, Suzuki H. Epigenetic drivers of genetic alterations. Adv Genet. 2010;70:309-23
10. Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009;459:460-3
11. Buis J, Stoneham T, Spehalski E. et al. Mre11 regulates CtIP-dependent double-strand break repair by interaction with CDK2. Nat Struct Mol Biol. 2012;19:246-52
12. Peterson SE, Li Y, Wu-Baer F. et al. Activation of DSB processing requires phosphorylation of CtIP by ATR. Mol Cell. 2013;49:657-67
13. Hsieh YH, Chang YY, Su IJ. et al. Hepatitis B virus pre-S2 mutant large surface protein inhibits DNA double-strand break repair and leads to genome instability in hepatocarcinogenesis. J Pathol. 2015;236:337-47
14. Zheng Y, Chen WL, Louie SG. et al. Hepatitis B virus promotes hepatocarcinogenesis in transgenic mice. Hepatology. 2007;45:16-21
15. You Z, Shi LZ, Zhu Q. et al. CtIP links DNA double-strand break sensing to resection. Mol Cell. 2009;36:954-69
16. Huertas P, Jackson SP. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem. 2009;284:9558-65
17. Chen L, Nievera CJ, Lee AY. et al. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J Biol Chem. 2008;283:7713-20
18. Wu G, Lee WH. CtIP, a multivalent adaptor connecting transcriptional regulation, checkpoint control and tumor suppression. Cell Cycle. 2006;5:1592-6
19. Chen PL, Liu F, Cai S. et al. Inactivation of CtIP leads to early embryonic lethality mediated by G1 restraint and to tumorigenesis by haploid insufficiency. Mol Cell Biol. 2005;25:3535-42
20. Chinnadurai G. CtIP, a candidate tumor susceptibility gene is a team player with luminaries. Biochim Biophys Acta. 2006;1765:67-73
Author contact
![]() Corresponding authors: Duyun Ye, Department of Pathophysiology, Tongji Medical College, Huazhong University of Science and Technology, No. 13, Hangkong Road, Wuhan 430030, People's Republic of China. E-mail: yedytjmu.edu.cn. Phone: +86-15607124302. Fax: +86-027-83692816 and Dongxin Zhang, Department of Clinical Laboratory, Puai Hospital, Tongji Medical College, Huazhong University of Science and Technology, No. 473, HanZheng Street, Wuhan 430033, People's Republic of China. E-mail: biozdxcom. Phone: +86-15927115788. Fax: +86-027-68834866.
Corresponding authors: Duyun Ye, Department of Pathophysiology, Tongji Medical College, Huazhong University of Science and Technology, No. 13, Hangkong Road, Wuhan 430030, People's Republic of China. E-mail: yedytjmu.edu.cn. Phone: +86-15607124302. Fax: +86-027-83692816 and Dongxin Zhang, Department of Clinical Laboratory, Puai Hospital, Tongji Medical College, Huazhong University of Science and Technology, No. 473, HanZheng Street, Wuhan 430033, People's Republic of China. E-mail: biozdxcom. Phone: +86-15927115788. Fax: +86-027-68834866.
Citation styles
APA
Zhang, D., Liu, H., Lin, J., Ye, D. (2018). Hepatitis B Virus Infection Dampens CtIP Expression in Hepatoma Cell. Journal of Cancer, 9(7), 1182-1187. https://doi.org/10.7150/jca.23649.
ACS
Zhang, D.; Liu, H.; Lin, J.; Ye, D. Hepatitis B Virus Infection Dampens CtIP Expression in Hepatoma Cell. J. Cancer 2018, 9 (7), 1182-1187. DOI: 10.7150/jca.23649.
NLM
Zhang D, Liu H, Lin J, Ye D. Hepatitis B Virus Infection Dampens CtIP Expression in Hepatoma Cell. J Cancer 2018; 9(7):1182-1187. doi:10.7150/jca.23649. https://www.jcancer.org/v09p1182.htm
CSE
Zhang D, Liu H, Lin J, Ye D. 2018. Hepatitis B Virus Infection Dampens CtIP Expression in Hepatoma Cell. J Cancer. 9(7):1182-1187.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.