Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(7):1259-1266. doi:10.7150/jca.23992 This issue Cite
Review
Reassessing the Potential of Myb-targeted Anti-cancer Therapy
Xiaofeng Liu1, Yunxiao Xu1, Liping Han2, Yan Yi1 ![]()
1. Department of Hematology, the Second Xiangya Hospital, Central South University, Changsha, Hunan Province, P.R. China
2. School of Life Science, Changchun Normal University, Changchun, Jilin Province, P.R. China
Received 2017-11-23; Accepted 2018-2-28; Published 2018-3-15
Citation:
Liu X, Xu Y, Han L, Yi Y. Reassessing the Potential of Myb-targeted Anti-cancer Therapy. J Cancer 2018; 9(7):1259-1266. doi:10.7150/jca.23992. https://www.jcancer.org/v09p1259.htm
Other stylesAbstract
Transcription factor MYB is essential for the tumorigenesis of multiple cancers, especially leukemia, breast cancer, colon cancer, adenoid cystic carcinoma and brain cancer. Thus, MYB has been regarded as an attractive target for tumor therapy. However, pioneer studies of antisense oligodeoxynucleotides against MYB, which were launched three decades ago in leukemia therapy, were discontinued because of their unsatisfactory clinical outcomes. In recent years, the roles of MYB in tumor transformation have become increasingly clear. Moreover, the regulatory mechanisms of MYB, such as the vital effects of MYB co-regulators on MYB activity and of transcriptional elongation on MYB expression, have been unveiled. These observations have underpinned novel approaches in inhibiting MYB. This review discusses the structure, function and regulation of MYB, focusing on recent insights into MYB-associated oncogenesis and how MYB-targeted therapeutics can be explored. Additionally, the main MYB-targeted therapies, including novel genetic therapy, RNA interference, microRNAs and low-molecular-weight compounds, which are especially promising inhibitors that target MYB co-regulators and transcriptional elongation, are described, and their prospects are assessed.
Keywords: MYB, MYB inhibitor, therapeutic strategy, targeted therapy, anti-cancer therapy
Introduction
Cellular MYB (c-Myb) is the homologue of the viral MYB (v-Myb), which is expressed by the avian myeloblastosis virus (AMV) and avian leukemia virus E26, respectively. Aberrant expression of MYB was observed in myeloid and erythroid leukemias [1]. Subsequently, it was identified as a transcription factor that was critical for cell proliferation and differentiation [2]. MYB belongs to a family of transcription factors that comprises two highly homologous proteins, MYBL1/A-Myb and MYBL2/B-Myb that play critical roles in various mammalian tissues [3]. This review will focus on MYB/c-Myb.
Apart from hematopoietic malignancies, aberrant expression of MYB is also associated with many malignant solid tumors [4]. MYB is required at relatively high levels for tumor survival. Therefore, MYB seems to be a target for anti-cancer therapeutics with a strategy to differentiate the excessively proliferating tumor cells [5].
MYB-targeted therapeutics for the treatment of leukemia has been intensely investigated for three decades [6]. However, the use of MYB-targeted therapeutics has been hindered by problems associated with the delivery, efficacy and toxicity of RNA interference approach [7]. Moreover, transcription factors have been considered traditionally almost impossible for targeted therapy. Recent years, new emerging technological advances on targeting of transcription factors with small molecules and new insights regarding the MYB itself have led to exploring novel MYB-targeted therapeutics [7]. It is hopeful that MYB-targeted therapy shed new light on anti-cancer therapeutics.
Structure and Function of Myb
The MYB gene (Figure 1A) is located on chromosome 6q23.3 and encodes a 75-kDa transcription factor with an N-terminal DNA-binding domain (DBD), a central transactivation domain (TAD) and a C-terminal negative regulatory domain (NRD) [8] (Figure 1B). The TAD is required for the activation of its target genes [5, 7]. The v-Myb protein is truncated at both N- and C- termini. It induces leukemia by transforming immature hematopoietic cells [9]. Truncations at either N- or C-termini of the MYB gene are sufficient to induce transformation [10].
Highly conserved DBD includes three tandem repeats (R1, R2, and R3), the first of which is deleted in the AMV and E26 oncoproteins [4] (Figure 1B and Figure 1C). R1 stabilizes the MYB-DNA complex. R2 and R3 form the core DNA binding domain [11]. Apart from DNA binding, DBD is involved in protein-protein interactions and activation of the target genes, which was identified by studying the activation of the first known MYB-regulated gene, mim-1. Mim-1 was activated by MYB, AMV and E26 proteins by binding to its promoter in reporter gene assays. However, AMV failed to activate the endogenous mim-1 gene embedded in cellular chromatin; while MYB and E26 proteins activated the endogenous gene. The critical reason is that three amino acid changes only in DBD of AMV (Figure 1C) disrupt its interaction with C/EBPbeta protein [12].
The C-terminal NRD suppresses the transforming activity of MYB [13]. NRD contains EVES motif, which mediates inter- and intra-molecular regulation of MYB [14] (Figure 1B). The N- and C- termini of the MYB protein interact with each other through EVES motif, resulting in transcriptional inactivation. Interestingly, a competing EVES motif is present in p100, which is a ubiquitously expressed transcriptional co-activator in diverse species. MYB adopts an open configuration by interacting with p100, which enables its interaction with additional co-activators [6]. In case of v-Myb, the auto-regulation is absent because of the C-terminal truncation resulting in absence of the EVES motif (Figure 1C).
Interactions between Myb and its Co-regulators
As shown in Figure 1B, MYB transcription factor, interacts with various co-regulators including C/EBPbeta and p100 [7, 13]. Protein-protein interactions determine the specificity of MYB as well as its ability to induce proliferation or differentiation [9]. For example, as mentioned above, MYB and C/EBPbeta interaction is necessary to activate MYB target genes [12], and the interaction between MYB and p100 regulates MYB activity [6].
CREB-binding protein (CBP) and p300 are also co-activators of MYB, which bind to the MYB TAD and acetylate MYB (Figure 1B), subsequently altering the protein-protein interactions and specificity of MYB [13]. In mice harboring the MYB M303V mutation, the disruption of MYB and p300 interaction leads to increased number of hematopoietic stem cells and blocks the development of blood cells [15]. This suggests that MYB and p300 interaction is critical for the normal proliferation and differentiation of hematopoietic stem and progenitor cells.
C-Ski, N-CoR, and mSin3A are 3 co-repressors that bind to the MYB DBD and form a complex with TIF1beta (Figure 1B). TIF1beta binds to the C-terminus and recruits the histone deacetylase complex to MYB, thereby negatively regulating MYB activity. Mutations or deletions in the NRD or DBD decrease the interaction of MYB with its co-repressors and increase its oncogenic transactivation [2]. The c-Ski co-repressor protein competes with CBP for binding to MYB [9]. These studies indicate that interaction with co-activators or co-repressors is critical for MYB-dependent transcription.
Moreover, NRD contains numerous sites for post-translational modifications like acetylation, phosphorylation, sumoylation and ubiquitinylation [13] (Figure 1B). For example, it contains the binding site of Pin1 (Figure 1B), an isomerase that binds to phosphorylated Ser/Thr-Pro motif and upregulates MYB activity [16]. Wnt-1 also induces phosphorylation of MYB at multiple NRD sites, which is followed by ubiquitination and proteasome-dependent degradation of MYB [9]. Therefore, co-regulators and post-translational modifications regulate the activity or stability of MYB. The oncogenic v-Myb protein lacks phosphorylation and ubiquitinylation sites resulting in resistance to Wnt-1 induced degradation. This partially explains the transformational capacity of v-Myb.
A substantial number of MYB target genes are involved in diverse cellular functions like proliferation, differentiation, cell cycle, apoptosis, cell signaling, angiogenesis and cell adhesion [2]. These include cyclin genes (CCNA1, CCNE1, CCNB1), proto-oncogenes (MYC and KIT) and survival gene (BCL-2), which are also critical for tumorigenesis [9]. Other MYB target genes linked to tumorigenesis include COX-2, BCL-XL, MIM1, CD4, CCNB1, HSPA5, HSP70 and GATA3 [2].
Regulation of Myb Expression by MicroRNAs
The 3'untranslated region (3'UTR) of MYB contains binding sites for several miRNAs that regulate the turnover and translation of MYB mRNA [17]. For example, miR-150 binds to the MYB 3'-UTR and down-regulates MYB expression in a stage-specific manner in B-cell development as well as differentiation of other hematopoietic lineages [18]. Granulocytes from patients with primary myelofibrosis (PMF) demonstrate low miR-150 levels associated with MYB overexpression [19]. Other tumor suppressor miRNAs associated with MYB include miRNA-193b-3p and miRNA-103a [20, 21].
Role of Myb in Normal Cells
MYB expression is high in hematopoietic progenitor cells and decreases during differentiation [2]. Antisense knockdown of MYB expression in human bone marrow cells decreases the size of colonies and the number of the mononuclear cells [22]. Homozygous MYB mutant mice die by embryonic day 15 due to hematopoietic failure [23]. Studies with conditional MYB knockout mice show that precise expression of MYB is necessary for T- and B- cell development, myelopoiesis, erythropoiesis and HSC self-renewal [7]. Therefore, MYB is essential for maintaining the proliferative state of hematopoietic progenitor cells as well as distinct differentiation steps in most hematopoietic cell lineages.
MYB expression has also been identified to be critical for the normal differentiation and progenitor cell homeostasis of colonic crypts and neurogenic region of the adult brain [2, 24, 25]. Furthermore, recent conditional knockout studies showed evidence that MYB is globally expressed in many tissues and cells and is required for their normal development [4].
Role of Myb in Leukemias and Solid Tumors
Elevated MYB expression has been reported in many cases of leukemias, colon and breast cancers [7, 26-29]. Recurrent translocations, duplications and C-terminal deletions have also been reported in leukemia, colon cancer, breast cancer, adenoid cystic carcinoma and brain cancer [2, 9, 30-34] (Figure 1C and Table 1).
Moreover, high MYB expression has been proved to block differentiation and promote proliferation in most human leukemias and brain cancer [7, 35], and to be required for the maintenance of proliferation in primary human leukemia cells [36]. Furthermore, recently, Liu et al demonstrated the tumorigenic potential of MYB in zebrafish model [37]. In their study, transgenic zebrafish with hyperactive MYB (MYBhyper) exhibited abnormal granulocyte expansion resembling human myelodysplastic syndrome (MDS). And few MYBhyper adult fish developed acute myeloid or lymphoid- and leukemia-like disorders with age. Moreover, the MYB target drug, Flavopiridol, relieved MDS-like symptoms in both MYBhyper embryos and adult fish.
It has been suggested that tissues with essential roles for MYB in development and/or homeostasis like bone marrow, colon, brain and mammary gland are also susceptible to MYB-dependent oncogenesis [2]. Additionally, alternative splicing of human MYB transcripts produces multiple forms of the MYB protein that have the same DBD, but different C-termini (Figure 1C). This results in differential MYB activities that correlate with cancer prognosis [38]. For example, MYB9A expression correlates with poor survival for leukemia patients [35].
Table 1
Human malignancies associated with MYB
| Study | Tumor entity | Abnormality |
|---|---|---|
| Ferrari et al 1985 | AML and ALL | MYB overexpression |
| Barletta et al 1987 | Leukemia and lymphoma carrying 6q- deletions | MYB overexpression |
| Tomita et al 1998 | TK-6 cell line (CML in T cell blast crisis) | MYB truncation |
| Clappier et al 2007 | T-cell acute leukemia | Recurrent chromosomal translocation and genomic duplication of MYB locus |
| Murati et al 2009 | Acute myelomonocytic leukemia | Genomic gain of the MYB locus |
| Quelen et al 2011 | Acute basophilic leukemia | Recurrent translocation involving MYB |
| Nakano et al 2016 | Adult T-cell Leukemia | MYB (unbalanced MYB-9A) overexpression |
| Alitalo et al 1984 | Colon cancer | MYB overexpression; amplified MYB oncogene |
| Thompson et al 1997 | Colon cancer | MYB overexpression; microsatellite deletions in MYB transcriptional attenuator region |
| Hugo et al 2006 | Colon cancer | MYB overexpression; mutations in MYB intron 1 regulatory sequence |
| Guérin et al 1990 | Breast cancer | MYB expression associated with oestrogen-receptor expression |
| Kauraniemi et al 2000 | Hereditary breast cancer | MYB overexpression; amplified MYB oncogene |
| Persson et al 2009 | Adenoid cystic carcinoma | Recurrent translocation involving MYB |
| Drier et al 2016 | Adenoid cystic carcinoma | Recurrent translocation involving MYB |
| Zhang et al 2013 | Brain tumor | Recurrent translocation involving MYB |
| Ramkissoon et al 2017 | Brain tumor | Recurrent translocation involving MYB |
Abbreviations: AML, acute myeloid leukemia; ALL, acute lymphocytic leukemia; CML, chronic myelogenous leukemia
Figure 1
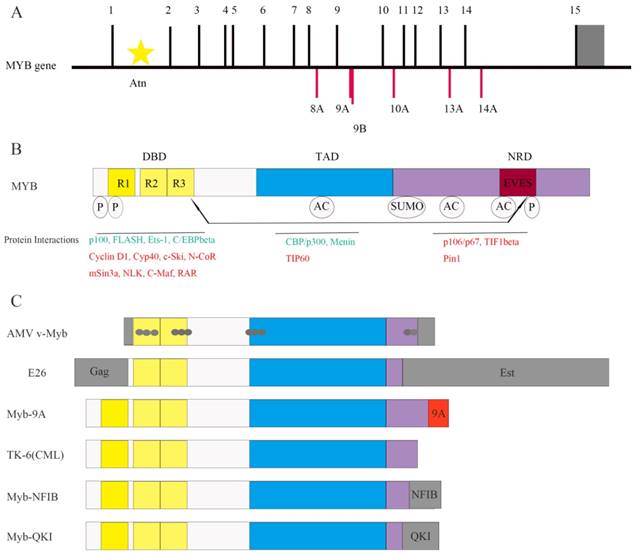
Structure of MYB and its co-regulators. (A) The MYB proto-oncogene consists of 15 normal exons and many alternative spliced exons such as 8A, 9A, 9B, 10A, 13A and 14A. The 3' untranslated region is shown in gray. Transcription is initiated from multiple start sites and is regulated by the attenuator sequence (Atn) within intron 1.(B) The normal MYB protein contains the DNA-binding domain (DBD), transactivation domain (TAD) and negative regulatory domain (NRD). The post-translational modifications such as phosphorylation (P), acetylation (AC) and sumoylation (SUMO) as well as the EVES peptide sequence that is involved in intra- and intermolecular protein-protein interactions are also shown. The MYB co-activators are listed in green and the co-repressors are listed in red. Arrows indicate potential intramolecular interactions. (C) Schematic representation of MYB variants expressed in leukemia and solid tumors. Both AMV v-Myb and E26 proteins are truncated at both the N- and C-termini of c-MYB. The AMV v-Myb protein contains 6 amino acids derived from the retroviral Gag protein fused to amino acids 72-442 of MYB followed by 13 novel amino acids at the C-terminus (shaded gray). It also has 11 point mutations that cause amino acid changes (gray dots). The E26 protein is a Gag-MYB-Ets fusion protein with 272 amino acids of the retroviral Gag protein fused to MYB, which in turn is fused to 491 amino acids from Ets-1transcription factor at the C-terminus. Aberrant expression of MYB-9A in adult T-cell leukemia correlates with poor outcomes. C-terminal truncated protein has also been identified inTK-6, a chronic myeloid leukemia (CML) cell line. The fusion of MYB and NFIB genes (translocation 6; 9) generates a MYB-NFIB fusion protein in adenoid cystic carcinoma (ACC) that resembles truncated MYB protein. In brain tumors, MYB-QKI fusion gene generates similar MYB fusion protein lacking C-terminal domains.
Co-operation between Myb and Other Oncogenes
MYB is an essential downstream effector for MLL-ENL and homeobox-mediated transformation in MLL-associated leukemia [39, 40]. In a mouse model of human MLL-ENL leukemia, short-term siRNA suppression of MYB effectively inhibited leukemiagenesis by MLL-ENL [39]. Moreover, in mouse model of MLL-AF9 related acute myeloid leukemia (AML), MYB knockdown decreased the number of leukemia stem cells (LSCs) [41].
MYB is also a pivotal regulator of oncogenic super-enhancers, which are large regulatory elements that drive the expression of critical oncogenes. In T-cell acute lymphoblastic leukemias, MYB recruits histone H3 lysine 27 (H3K27) acetylase-binding partners like CBP, RUNX1 and GATA-3, which act as a super-enhancer driving the expression of the TAL1 oncogene [42]. In MLL-associated leukemogenesis, MYB was also identified to bind upstream of MLL through menin and contribute to MLL-mediated methylation of histone H3 at lysine 4 (H3K4) [43].
MYB is also involved in leukemogenesis triggered by other oncogenes like E2A-HLF, BCR-ABL, AML1-ETO and Setbp1 missense mutants [7, 40]. These oncogenes aberrantly modulate the MYB-related transcriptional program resulting in leukemiogenesis. Therefore, MYB is indispensable for the initiation and/or maintenance of leukemia [7] and is an attractive target for leukemic therapy.
Figure 2
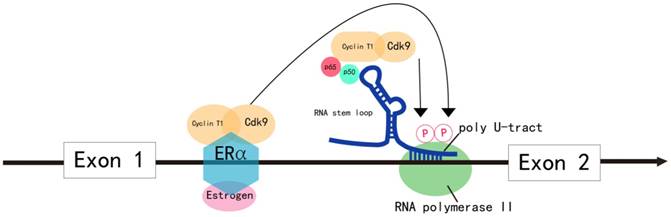
Model for the regulation of MYB transcriptional elongation. MYB transcriptional elongation is regulated by sequences within intron 1, which encode a RNA stem loop and a poly U tract of 19 uridines that stall RNA polymerase II (RNA Pol II). RNA Pol II overcomes transcription blockade through the attenuation region to express full-length MYB transcripts in normal cells. In colon cancer, the sequences encoding the stem loop or poly U tract are mutated, which disrupts transcriptional attenuation. In ER+ breast cancer, estrogen receptor α (ERα) and the PTEFB complex are recruited to the transcriptional attenuation region upon estrogen stimulation. Transcriptional block is relieved by the phosphorylation of the Ser-2 residue of RNA Pol II by the CDK9 subunit of PTEFB. In human leukemia cells, MYB transcriptional elongation is facilitated by the binding of the NFκB p50-p65 heterodimer to the stem loop, which mediates the recruitment of PTEFB and subsequent Ser-2 phosphorylation of RNA Pol II by CDK9.
Elongation Control Regulates Myb Transcription
In most malignancies, MYB overexpression is due to increased transcriptional rate. MYB transcription is predominantly regulated by elongation control sequences within intron 1 that generate a RNA stem loop and a poly U tract, which stalls RNA polymerase II (RNA Pol II). RNA Pol II overcomes this transcription blockade by recruiting positive elongation transcription factor B (PTEFB) to the transcriptional attenuation region or due to mutations in the elongation control sequences of intron 1 (Figure 2). This results in increased transcription of MYB in tumors [2].
In colon cancer, MYB expression is regulated by controlling transcriptional elongation by the attenuator sequences within intron 1, which are mutated in many instances [27] (Figure 2). The mutations facilitate transcriptional elongation and increase read-through, thereby increasing MYB expression in cancer cells [2].
In estrogen receptor positive (ER+) breast cancer, B lymphoma and erythroleukemia cells, the activity of PTEFB is essential within the transcription attenuation region of the MYB gene to resume transcriptional elongation [44-47]. In ER+ breast cancer cells, ER recruits PTEFB when bound to estrogen [44] (Figure 2). The ER/PTEFB complex interacts with the MYB gene, phosphorylates RNA Pol II and relieves the attenuation of elongation. In leukemia cells, PTEFB is recruited when the NFκB heterodimer binds to the stem loop resulting in subsequent phosphorylation of RNA Pol II, which facilitates MYB transcription [45] (Figure 2). Therefore, suppressing PTEFB activity is a potential strategy to inhibit MYB expression in malignancies [48].
Myb as a Target for Anti-cancer Therapy
Several approaches have been attempted to inhibit aberrant MYB expression in cancer cells. These strategies include disrupting co-regulator interactions, promoting MYB transcription attenuation, targeting MYB upstream or downstream effectors, cancer vaccines, antisense oligodeoxynucleotides (short-hairpin RNA), overexpressing miRNAs that target MYB directly and low-molecular-weight compounds.
Antisense ODNs and Short Hairpin RNAs
Three decades ago, Gewirtz et al used antisense ODNs or shRNAs against MYB to effectively suppress growth and proliferation of primary patient derived leukemia cell lines in vitro [6]. And leukemia cells were found to be more sensitive to MYB inhibition than normal hematopoietic cells [49]. The MYB-specific antisense ODNs were also identified to be effective in a K562 leukemia model in SCID mice [50]. However, MYB-specific antisense ODNs showed limited efficacy in the pilot study of chronic myelogenous leukemia patients [51]. On the other hand, the antisense RNA and MYB ODNs have been proved to have the capacity to strongly inhibit proliferation of human colorectal cancer cells and transformed neuroectodermal cell lines in vitro. Moreover, the antisense MYB ODNs was shown to potentiate the anti-proliferative effects of conventional chemotherapeutic drugs such as taxol, 5-fluorouracil, vinblastine and doxorubicin [52, 53].
Over the last decade, many studies have attempted RNA interference (RNAi) to suppress MYB. In a mouse model of MLL-AF9 leukemia, the MYB specific shRNAs silenced MYB effectively and achieved complete remission and eradication of the aggressive leukemia without preventing normal myelopoiesis [41]. Furthermore, in mouse model of pre-B-cell acute lymphoblastic leukemia, the MYB shRNA decreased proliferation of leukemia cells and increased their sensitivity to cytotoxic agents, thereby significantly delaying disease onset [54].
Many studies explored combining MYB antisense ODNs with other targeted therapies. For example, MYB antisense ODNs synergized with GD2-specific antibodies achieved long term survival in murine xenograft models of neuroblastoma [55]. The liposomes were coated with the anti-GD2 antibodies (targeted liposomes) and MYB antisense ODNs. The GD2-targeted liposomes successfully delivered the ODNs to the target cells and enhanced their toxicity without affecting the normal cells. Additionally, the CpG motifs stimulated the immune cells. Therefore, MYB RNAi can effectively inhibit the proliferation of leukemia and other tumor cells and cooperate with other cytotoxic agents or targeted therapies. But, MYB RNAi still requires improvements in effective delivery, stability and toxicity for implementation in clinical applications [7].
Myb DNA Vaccine
Overexpression of MYB occurs in 80% of colorectal cancer (CRC) and is associated with aggressive disease and poor prognosis [56]. Ramsay et al tested MYB DNA vaccine in a mouse model of colorectal cancer. The MYB DNA vaccine suppressed colorectal tumor growth by inducing the T cell-mediated immunity and increasing the infiltration of immune effector cells at the tumor site [56]. Moreover, in the CRC mice model, the MYB DNA vaccine in combination with anti-PD-1 antibody or low dose cyclophosphamide effectively killed tumor cells [57]. Pre-clinical and clinical studies are needed to explore the utility of MYB DNA vaccine in clinical applications.
Inhibition of Myb-coregulator Interactions
P300 has been investigated as a therapeutic target because it is an important co-activator of MYB [7]. In the study on Booreana mice carry a mutant allele of MYB, which disrupts its interaction with p300, irradiated recipient mice transplanted with Booreana hematopoietic cells transduced with AML1-ETO9a or MLL-AF9 retroviruses did not develop leukemia [58]. This indicates that disruption of the MYB and p300 interaction is a potential therapeutic strategy in malignancies relying on MYB [59].
The therapeutic efficacy of many small-molecule inhibitors of the MYB/p300 interaction have been investigated [60]. Among these studies, Celastrol, a potent low molecular weight inhibitor, was shown to suppress the proliferative potential of AML cells without affecting normal hematopoietic progenitor cells. Moreover, Celastrol enhanced survival of the mice model of HoxA9/Meis1-driven AML [61].
Also, MYB is recruited to the MLL histone methyl transferase complex through its interaction with menin, and contributes to MLL-associated leukemogenesis [43]. Therefore, the interaction of MYB with menin has been explored as an anti-cancer strategy. Grembecka et al demonstrated that several small-molecule inhibitors of the menin-MLL complex blocked transformation by MLL fusion proteins in bone marrow cells (BMC) and relieved the differentiation block in MLL-associated human leukemia cells [62].
Therapeutic Targeting of Myb Elongation
Since PTEFB activity correlates with elevated MYB expression, suppressing PTEFB activity is an attractive strategy to inhibit aberrant MYB expression [45, 48]. Flavopiridol is an inhibitor of the Cdk9 subunit of PTEFB, which suppresses the expression of MYB and its target genes like BCL-2 and CCNB1 in ER + breast cancer cells. Moreover, Flavopiridol is at least 10 times more effective in eliminating ER+ breast cancer cell lines than ER-MYB- cells. The sensitivity of ER+ breast cancer cells could be reversed by ectopic MYB expression. This demonstrated that Flavopiridol is a potential therapeutic agent for ER+ breast cancers and other MYB-dependent cancers [63]. In MYBhyper zebrafish studies, Flavopiridol relieved MDS-like symptoms in both MYBhyper embryos and adult fish [37]. However, clinical trials of Flavopiridol in solid tumors were discontinued because of adverse events and low efficacy [64]. Meanwhile, Flavopiridol shows clinical activity in several hematological malignancies such as chronic lymphocytic leukemia, but, the relevance between MYB suppression and clinical activity is ambiguous [65]. Therefore, new, specific and more effective inhibitors of MYB elongation need to be developed.
Myb Specific MicroRNAs
Several MYB-targeting miRNAs demonstrate potential as anti-cancer therapeutics. Restoring miR-150 expression in EBV-positive Burkitt lymphoma cell lines decreased MYB levels, which reduced proliferation and induced differentiation in the lymphoma cells. Moreover, MYB knockdown showed similar characteristics to overexpressing miR-150 in the lymphoma cell lines [66]. This showed therapeutic potential of miR-150 and other MYB targeting miRNAs in future anti-cancer therapy.
Other Approaches for Targeting Myb
Drugs with multiple targets are more effective than specific target drugs in the treatment of complex diseases such as cancer. The bromodomain and extra-terminal (BET) protein BRD4 has been identified to regulate the expression and activity of MYB in several modes. BRD4 regulates MYB expression through recruiting PTEFB, which promotes MYB elongation [67]. It also regulates downstream effectors of MYB such as MYC, and MYB co-regulators including p300/CBP [68, 69]. Hence, BRD4 inhibition is potentially an effective strategy for suppressing MYB. In fact, BRD4 inhibition is being tested as a therapy for acute leukemia and breast cancer with high-level MYB expression [70-72].
Additionally, since the positive feedback loop of MYB and Hoxa/Meis is critical for regulating leukemogenesis, Hox is a potential therapeutic target [43]. Also, regulators of MYB degradation like the Wnt-1 signaling pathway have shown therapeutic potential.
Klempnauer et al reported several sesquiterpene lactones that suppressed MYB-dependent gene expression specifically [73]. In their studies, mexicanin-I suppressed the proliferation of all MYB-expressing tumor cell lines and inhibited the colony growth of blasts from multiple AML patients; while tumor cell lines without MYB expression are less sensitive to mexicanin-I [74]. Further investigations are in progress to evaluate the clinical efficacy of these novel low-molecular-weight MYB inhibitors.
More recently, the widely used mebendazole was identified to induce c-MYB degradation via the proteasome by interfering with the heat shock protein 70 (HSP70) chaperone system, which is sufficient to inhibit colony formation by AML cells, but not normal cord blood-derived cells. Moreover, mebendazole is effective at impairing AML progression in mouse experiments [75]. It seems that mebendazole is also a safe and novel therapeutic approach for AML. Further research is ongoing.
Conclusions
The MYB transcription factor is a suitable target for tumor therapy because its aberrant expression is pivotal for the growth and progression of multiple solid tumors and leukemias. While MYB-targeted antisense ODNs have not been satisfactory in clinical studies, RNAi and miRNAs targeting MYB have shown great promise in preliminary studies. Moreover, better understanding of the regulation of the activity and expression of MYB and efficient inhibitor screens have opened up novel therapeutic avenues for low-molecular-weight inhibitors that target MYB co-regulators or its transcriptional elongation. Multiple inhibitors are effective in inhibiting tumors with elevated MYB expression in laboratory studies [59]. Further investigations are in progress to evaluate their efficacy in pre-clinical and clinical studies. The focus of future studies include studying the efficacy of these inhibitors, their toxicity in animal models or preclinical studies, and screening promising inhibitors individually or in combination with conventional chemotherapeutics or other targeted drugs in clinical trials. Since MYB plays a central role in tumorigenesis, new and effective MYB inhibitors will contribute to improving the survival of patients with MYB-related tumors.
Acknowledgements
This study was supported in part by the National Natural Science Foundation of China (grant no. 81100360).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Beug H, von Kirchbach A, Döderlein G. et al. Chicken hematopoietic cells transformed by seven strains of defective avian leukemia viruses display three distinct phenotypes of differentiation. Cell. 1979;18:375-390
2. Ramsay RG, Gonda TJ. MYB function in normal and cancer cells. Nat Rev Cancer. 2008;8:523-534
3. Oh IH, Reddy EP. The myb gene family in cell growth, differentiation and apoptosis. Oncogene. 1999;18:3017-3033
4. Zhou Y, Ness SA. Myb proteins: angels and demons in normal and transformed cells. Front Biosci (Landmark Ed). 2011;16:1109-1131
5. George OL, Ness SA. Situational awareness: regulation of the myb transcription factor in differentiation, the cell cycle and oncogenesis. Cancers (Basel). 2014;6:2049-2071
6. Gewirtz AM. Myb targeted therapeutics for the treatment of human malignancies. Oncogene. 1999;18:3056-3062
7. Pattabiraman DR, Gonda TJ. Role and potential for therapeutic targeting of MYB in leukemia. Leukemia. 2013;27:269-277
8. Biedenkapp H, Borgmeyer U, Sippel AE. et al. Viral myb oncogene encodes a sequence-specific DNA-binding activity. Nature. 1988;335:835-837
9. Zhou Y, Ness SA. Myb proteins: angels and demons in normal and transformed cells. Front Biosci (Landmark Ed). 2011;16:1109-1131
10. Grässer FA, Graf T, Lipsick JS. Protein truncation is required for the activation of the c-myb proto-oncogene. Mol Cell Biol. 1991;11:3987-3996
11. Tanikawa J, Yasukawa T, Enari M. et al. Recognition of specific DNA sequences by the c-myb protooncogene product: role of three repeat units in the DNA-binding domain. Proc Natl Acad Sci U S A. 1993;90:9320-9324
12. Introna M, Golay J, Frampton J. et al. Mutations in v-myb alter the differentiation of myelomonocytic cells transformed by the oncogene. Cell. 1990;63:1289-1297
13. George OL, Ness SA. Situational awareness: regulation of the myb transcription factor in differentiation, the cell cycle and oncogenesis. Cancers (Basel). 2014;6:2049-2071
14. Dash AB, Orrico FC, Ness SA. The EVES motif mediates both intermolecular and intramolecular regulation of c-Myb. Genes Dev. 1996;10:1858-1869
15. Sandberg ML, Sutton SE, Pletcher MT. et al. c-Myb and p300 regulate hematopoietic stem cell proliferation and differentiation. Dev Cell. 2005;8:153-166
16. Pani E, Menigatti M, Schubert S. et al. Pin1 interacts with c-Myb in a phosphorylation-dependent manner and regulates its transactivation activity. Biochim Biophys Acta. 2008;1783:1121-1128
17. Feng J, Yang Y, Zhang P. et al. miR-150 functions as a tumour suppressor in human colorectal cancer by targeting c-Myb. J Cell Mol Med. 2014;18:2125-2134
18. Xiao C, Calado DP, Galler G. et al. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell. 2007;131:146-159
19. Guglielmelli P, Tozzi L, Pancrazzi A. et al. MicroRNA expression profile in granulocytes from primary myelofibrosis patients. Exp Hematol. 2007;35:1708-1718
20. Mets E, Van der Meulen J, Van Peer G. et al. MicroRNA-193b-3p acts as a tumor suppressor by targeting the MYB oncogene in T-cell acute lymphoblastic leukemia. Leukemia. 2015;29:798-806
21. Liang J, Liu X, Xue H. et al. MicroRNA-103a inhibits gastric cancer cell proliferation, migration and invasion by targeting c-Myb. Cell Prolif. 2015;48:78-85
22. Gewirtz AM, Calabretta B. A c-myb antisense oligodeoxynucleotide inhibits normal human hematopoiesis in vitro. Science. 1988;242:1303-1306
23. Mucenski ML, McLain K, Kier AB. et al. A functional c-myb gene is required for normal murine fetal hepatic hematopoiesis. Cell. 1991;65:677-689
24. Malaterre J, Carpinelli M, Ernst M. et al. c-Myb is required for progenitor cell homeostasis in colonic crypts. Proc Natl Acad Sci U S A. 2007;104:3829-3834
25. Malaterre J, Mantamadiotis T, Dworkin S. et al. c-Myb is required for neural progenitor cell proliferation and maintenance of the neural stem cell niche in adult brain. Stem Cells. 2008;26:173-181
26. Barletta C, Pelicci PG, Kenyon LC. et al. Relationship between the c-myb locus and the 6q-chromosomal aberration in leukemias and lymphomas. Science. 1987;235:1064-1067
27. Thompson MA, Flegg R, Westin EH. et al. Microsatellite deletions in the c-myb transcriptional attenuator region associated with over-expression in colon tumour cell lines. Oncogene. 1997;14:1715-1723
28. Alitalo K, Winqvist R, Lin CC. et al. Aberrant expression of an amplified c-myb oncogene in two cell lines from a colon carcinoma. Proc Natl Acad Sci U S A. 1984;81:4534-4538
29. Guérin M, Sheng ZM, Andrieu N. et al. Strong association between c-myb and oestrogen-receptor expression in human breast cancer. Oncogene. 1990;5:131-135
30. Hugo H, Cures A, Suraweera N. et al. Mutations in the MYB intron I regulatory sequence increase transcription in colon cancers. Genes Chromosomes Cancer. 2006;45:1143-1154
31. Kauraniemi P, Hedenfalk I, Persson K. et al. MYB oncogene amplification in hereditary BRCA1 breast cancer. Cancer Res. 2000;60:5323-5328
32. Drier Y, Cotton MJ, Williamson KE. et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat Genet. 2016;48:265-272
33. Zhang J, Wu G, Miller CP. et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. 2013;45:602-612
34. Ramkissoon SH, Bandopadhayay P, Hwang J. et al. Clinical targeted exome-based sequencing in combination with genome-wide copy number profiling: precision medicine analysis of 203 pediatric brain tumors. Neuro Oncol. 2017;19:986-996
35. Nakano K, Uchimaru K, Utsunomiya A. et al. Dysregulation of c-Myb Pathway by Aberrant Expression of Proto-oncogene MYB Provides the Basis for Malignancy in Adult T-cell Leukemia/lymphoma Cells. Clin Cancer Res. 2016;22:5915-5928
36. Selvakumaran M, Liebermann DA, Hoffman-Liebermann B. Deregulated c-myb disrupts interleukin-6- or leukemia inhibitory factor-induced myeloid differentiation prior to c-myc: role in leukemogenesis. Mol Cell Biol. 1992;12:2493-2500
37. Liu W, Wu M, Huang Z. et al. c-myb hyperactivity leads to myeloid and lymphoid malignancies in zebrafish. Leukemia. 2017;31:222-233
38. O'Rourke JP, Ness SA. Alternative RNA splicing produces multiple forms of c-Myb with unique transcriptional activities. Mol Cell Biol. 2008;28:2091-2101
39. Somervaille TC, Matheny CJ, Spencer GJ. et al. Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell. 2009;4:129-140
40. Hess JL, Bittner CB, Zeisig DT. et al. c-Myb is an essential downstream target for homeobox-mediated transformation of hematopoietic cells. Blood. 2006;108:297-304
41. Zuber J, Rappaport AR, Luo W. et al. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev. 2011;25:1628-1640
42. Mansour MR, Abraham BJ, Anders L. et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346:1373-1377
43. Jin S, Zhao H, Yi Y. et al. c-Myb binds MLL through menin in human leukemia cells and is an important driver of MLL-associated leukemogenesis. J Clin Invest. 2010;120:593-606
44. Mitra P, Pereira LA, Drabsch Y. et al. Estrogen receptor-α recruits P-TEFb to overcome transcriptional pausing in intron 1 of the MYB gene. Nucleic Acids Res. 2012;40:5988-6000
45. Dooley S, Seib T, Welter C. et al. c-myb intron I protein binding and association with transcriptional activity in leukemic cells. Leuk Res. 1996;20:429-439
46. Bender TP, Thompson CB, Kuehl WM. Differential expression of c-myb mRNA in murine B lymphomas by a block to transcription elongation. Science. 1987;237:1473-1476
47. Suhasini M, Reddy CD, Reddy EP. et al. cAMP-induced NF-kappaB (p50/relB) binding to a c-myb intronic enhancer correlates with c-myb up-regulation and inhibition of erythroleukemia cell differentiation. Oncogene. 1997;15:1859-1870
48. Vantaggiato C, Tocchetti M, Cappelletti V. et al. Cell cycle dependent oscillatory expression of estrogen receptor-α links Pol II elongation to neoplastic transformation. Proc Natl Acad Sci U S A. 2014;111:9561-9566
49. Calabretta B, Sims RB, Valtieri M. et al. Normal and leukemic hematopoietic cells manifest differential sensitivity to inhibitory effects of c-myb antisense oligodeoxynucleotides: an in vitro study relevant to bone marrow purging. Proc Natl Acad Sci U S A. 1991;88:2351-2355
50. Ratajczak MZ, Kant JA, Luger SM. et al. In vivo treatment of human leukemia in a scid mouse model with c-myb antisense oligodeoxynucleotides. Proc Natl Acad Sci U S A. 1992;89:11823-11827
51. Luger SM, O'Brien SG, Ratajczak J. et al. Oligodeoxynucleotide-mediated inhibition of c-myb gene expression in autografted bone marrow: a pilot study. Blood. 2002;99:1150-1158
52. Abaza MS, Al-Attiyah RJ, Al-Saffar AM. et al. Antisense oligodeoxynucleotide directed against c-myb has anticancer activity and potentiates the antiproliferative effect of conventional anticancer drugs acting by different mechanisms in human colorectal cancer cells. Tumour Biol. 2003;24:241-257
53. Raschellà G, Negroni A, Skorski T. et al. Inhibition of proliferation by c-myb antisense RNA and oligodeoxynucleotides in transformed neuroectodermal cell lines. Cancer Res. 1992;52:4221-4226
54. Sarvaiya PJ, Schwartz JR, Hernandez CP. et al. Role of c-Myb in the survival of pre B-cell acute lymphoblastic leukemia and leukemogenesis. Am J Hematol. 2012;87:969-976
55. Brignole C, Pastorino F, Marimpietri D. et al. Immune cell-mediated antitumor activities of GD2-targeted liposomal c-myb antisense oligonucleotides containing CpG motifs. J Natl Cancer Inst. 2004;96:1171-1180
56. Williams BB, Wall M, Miao RY. et al. Induction of T cell-mediated immunity using a c-Myb DNA vaccine in a mouse model of colon cancer. Cancer Immunol Immunother. 2008;57:1635-1645
57. Cross RS, Malaterre J, Davenport AJ. et al. Therapeutic DNA vaccination against colorectal cancer by targeting the MYB oncoprotein. Clin Transl Immunology. 2015;4:e30
58. Pattabiraman DR, McGirr C, Shakhbazov K. et al. Interaction of c-Myb with p300 is required for the induction of acute myeloid leukemia (AML) by human AML oncogenes. Blood. 2014;123:2682-2690
59. Uttarkar S, Frampton J, Klempnauer KH. Targeting the transcription factor Myb by small-molecule inhibitors. Exp Hematol. 2017;47:31-35
60. Uttarkar S, Piontek T, Dukare S. et al. Small-Molecule Disruption of the Myb/p300 Cooperation Targets Acute Myeloid Leukemia Cells. Mol Cancer Ther. 2016;15:2905-2915
61. Uttarkar S, Dassé E, Coulibaly A. et al. Targeting acute myeloid leukemia with a small molecule inhibitor of the Myb/p300 interaction. Blood. 2016;127:1173-1182
62. Grembecka J, He S, Shi A. et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol. 2012;8:277-284
63. Mitra P, Yang RM, Sutton J. et al. CDK9 inhibitors selectively target estrogen receptor-positive breast cancer cells through combined inhibition of MYB and MCL-1 expression. Oncotarget. 2016;7:9069-9083
64. Asghar U, Witkiewicz AK, Turner NC. et al. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14:130-146
65. Phelps MA, Lin TS, Johnson AJ. et al. Clinical response and pharmacokinetics from a phase 1 study of an active dosing schedule of flavopiridol in relapsed chronic lymphocytic leukemia. Blood. 2009;113:2637-2645
66. Chen S, Wang Z, Dai X. et al. Re-expression of microRNA-150 induces EBV-positive Burkitt lymphoma differentiation by modulating c-Myb in vitro. Cancer Sci. 2013;104:826-834
67. Yang Z, Yik JH, Chen R. et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;19:535-545
68. Roe JS, Mercan F, Rivera K. et al. BET Bromodomain Inhibition Suppresses the Function of Hematopoietic Transcription Factors in Acute Myeloid Leukemia. Mol Cell. 2015;58:1028-1039
69. Delmore JE, Issa GC, Lemieux ME. et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904-917
70. Zuber J, Shi J, Wang E. et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524-528
71. Knoechel B, Roderick JE, Williamson KE. et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014;46:364-370
72. Feng Q, Zhang Z, Shea MJ. et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014;24:809-819
73. Bujnicki T, Wilczek C, Schomburg C. et al. Inhibition of Myb-dependent gene expression by the sesquiterpene lactone mexicanin-I. Leukemia. 2012;26:615-622
74. Schomburg C, Schuehly W, Da Costa FB. et al. Natural sesquiterpene lactones as inhibitors of Myb-dependent gene expression: structure-activity relationships. Eur J Med Chem. 2013;63:313-320
75. Walf-Vorderwülbecke V, Pearce K, Brooks T. et al. Targeting acute myeloid leukemia by drug-induced c-MYB degradation. Leukemia. 2017 [Epub ahead of print]
Author contact
![]() Corresponding author: Department of Hematology, the Second Xiangya Hospital, Central South University, 139 Middle Renmin Road, Changsha, Hunan Province, 410011, P.R. China. Fax: +86-731-85533525; E-mail: yiyancsuedu.cn
Corresponding author: Department of Hematology, the Second Xiangya Hospital, Central South University, 139 Middle Renmin Road, Changsha, Hunan Province, 410011, P.R. China. Fax: +86-731-85533525; E-mail: yiyancsuedu.cn
Citation styles
APA
Liu, X., Xu, Y., Han, L., Yi, Y. (2018). Reassessing the Potential of Myb-targeted Anti-cancer Therapy. Journal of Cancer, 9(7), 1259-1266. https://doi.org/10.7150/jca.23992.
ACS
Liu, X.; Xu, Y.; Han, L.; Yi, Y. Reassessing the Potential of Myb-targeted Anti-cancer Therapy. J. Cancer 2018, 9 (7), 1259-1266. DOI: 10.7150/jca.23992.
NLM
Liu X, Xu Y, Han L, Yi Y. Reassessing the Potential of Myb-targeted Anti-cancer Therapy. J Cancer 2018; 9(7):1259-1266. doi:10.7150/jca.23992. https://www.jcancer.org/v09p1259.htm
CSE
Liu X, Xu Y, Han L, Yi Y. 2018. Reassessing the Potential of Myb-targeted Anti-cancer Therapy. J Cancer. 9(7):1259-1266.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.