Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664
- Current Issue
- Volume 17; 2026
- Volume 16; 2025
- Volume 15; 2024
- Volume 14; 2023
- Volume 13; 2022
- Archive
- Editorial Board
- Cover Images
- Index & Coverage
- Special Issues
Top
Hypothesis
General Considerations
Horizontal vs. Vertical...
Familial Cancer and Directly...
An Alternative Viewpoint on...
Stem Cells, Clonal Dominance,...
Kin Selection, Soft vs. Hard...
Metastatic Colonization Genes...
Game Theory, Cancer...
Viral-Induced Tumorigenesis
Molecular and Cellular Profiles:...
Transmission Modes, Evolutionary...
Conclusion
Abbreviations
Acknowledgements
References
Hypothesis
General Considerations
Horizontal vs. Vertical...
Familial Cancer and Directly...
An Alternative Viewpoint on...
Stem Cells, Clonal Dominance,...
Kin Selection, Soft vs. Hard...
Metastatic Colonization Genes...
Game Theory, Cancer...
Viral-Induced Tumorigenesis
Molecular and Cellular Profiles:...
Transmission Modes, Evolutionary...
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(10):1707-1724. doi:10.7150/jca.24679 This issue Cite
Review
Hypothesis: Cancer Is a Disease of Evolved Trade-Offs Between Neoplastic Virulence and Transmission
Michael Bordonaro ![]()
Department of Basic Sciences, Geisinger Commonwealth School of Medicine, 525 Pine Street, Scranton, PA 18509, USA.
Received 2018-1-1; Accepted 2018-2-10; Published 2018-4-19
Citation:
Bordonaro M. Hypothesis: Cancer Is a Disease of Evolved Trade-Offs Between Neoplastic Virulence and Transmission. J Cancer 2018; 9(10):1707-1724. doi:10.7150/jca.24679. https://www.jcancer.org/v09p1707.htm
Other stylesAbstract
Virulence is defined as the ability of a pathogen to cause morbidity and/or mortality in infected hosts. The relationship between virulence and transmissibility is complex; natural selection may promote decreased virulence to enhance host mobility and increase the probability for transmission, or transmissibility may be enhanced by increased virulence, leading to higher pathogen load and, in some cases, superior evasion from host defenses. An evolutionary trade-off exists between the ability of pathogens to maintain opportunities for long-term transmission via suppressed virulence and increased short-term transmission via enhanced virulence. We propose an analogy between transmissibility and virulence in microbial pathogens and in cancer. Thus, in the latter case, the outcome of invasive growth and metastasis is analogous to transmissibility, and virulence is defined by high rates of proliferation, invasiveness and motility, potential for metastasis, and the extent to which the cancer contributes to patient morbidity and mortality. Horizontal and vertical transmission, associated with increased or decreased pathogen virulence respectively, can also be utilized to model the neoplastic process and factors that would increase or decrease tumor aggressiveness. Concepts of soft vs. hard selection and evolutionary game theory can optimize our understanding of carcinogenesis and therapeutic strategies. Therefore, the language of transmissibility, horizontal vs. vertical transmission, selection, and virulence can be used to inform approaches to inhibit tumorigenic progression, and, more generally, for cancer prevention and treatment.
Keywords: virulence, transmission, cancer, evolution, selection
Hypothesis
This paper proposes a conceptual similarity between the evolutionary trade-offs of virulence and transmission with respect to (a) microbial infection and (b) the behavior of neoplastic cells during carcinogenesis. The central hypothesis of this work is: Characteristics of the host-pathogen relationship observed in microbial infection can be utilized to describe patient-cancer interactions during carcinogenesis. Therefore, the human body is an ecosystem in which normal and neoplastic cells interact under selective pressures, increasing or decreasing cancer virulence and tumor cell transmissibility, in an evolutionarily stable manner, to maximize the adaptive fitness of neoplastic cells. The evolutionary imperative of living organisms, including pathogens and cancer cells, is genetic continuity. Under optimal conditions genetic expansion is adaptive, but most fundamentally it is adaptive to avoid genetic extinction and ensure continuity, even under adverse conditions. Pathogens will evolve toward lesser virulence under conditions of decreased horizontal transmission and/or conditions in which vertical transmission is favored over horizontal; we posit that the same tendencies apply for neoplastic cells within the host patient. While the optimal outcome of anti-cancer therapy is the complete elimination of all neoplastic cells, this is not always possible. Under those circumstances, we hypothesize that neoplastic cells can be forced to evolve toward decreased virulence by establishing conditions that favor vertical transmission of the neoplastic genome and impair horizontal transmission of the same. Thus, the objective of this paper is to understand how concepts of virulence and transmissibility apply to cancer, how different selective regimens can influence those phenomena, and how these processes can be leveraged for therapeutics.
General Considerations
What is the fundamental theoretical basis for our hypothesis? And how do we define virulence, particularly with respect to cancer?
Virulence is generally defined as the ability of a pathogen to cause morbidity and mortality in infected hosts [1]. We can consider the relationship between virulence and transmissibility. On the one hand, natural selection may promote decreased virulence that would enhance the mobility of the host and increase the probability for pathogen transmission. On the other hand, transmissibility may be enhanced by increased virulence, which would lead to higher pathogen load and, in some cases, superior evasion from host defenses. Thus, Lipistch and Moxon [1] postulated an “evolutionary trade-off” between (a) the ability of the pathogen to maintain opportunities for long-term transmission (through decreased virulence and increased host viability) and (b) increased short-term transmission via enhanced virulence resulting in higher rates of pathogen reproduction and/or repressed host defenses. Pathogens evolve toward a level of virulence that maximizes their reproductive potential in specific environmental contexts. There is a positive correlation between horizontal transmissibility and virulence, particularly when virulence affects rates of pathogen replication and transmission. Circumstances that facilitate transmissibility between unrelated hosts (horizontal transmission) would encourage greater virulence, with the standard example being that of the 1918 influenza pandemic, with virulence presumably enhanced given easier transmission due to war conditions. However, a contrarian view (see below, “An Alternative Viewpoint on Virulence and Transmission”) on the association between transmissibility and virulence [2] raises the possibility that the extreme virulence of the 1918 influenza strain may have been due to it being the first H2N1 serotype, infecting an immunologically naïve host population.
We propose an analogy between the association between transmissibility and virulence in (microbial) pathogens and that observed in cancer; in the latter, the outcome of invasive growth and metastasis is analogous to transmissibility and virulence is defined by the overall aggressiveness of the cancer, its growth rate and invasiveness, its potential for future possible growth and metastasis, its current state of invasion and metastasis, its degree of differentiation, other characteristics that contribute to its staging, and the extent to which the cancer contributes to patient morbidity and mortality. This analysis may contribute to a better understanding of carcinogenesis and cancer therapy from an evolutionary perspective. Further, there is advantage in exposing the cancer research community to ideas and studies of which they may have previously been unaware; including virulence, pathogen selection, and horizontal vs. vertical transmission. This cross-disciplinary approach to studying cancer from an evolutionary perspective may stimulate a better understanding of carcinogenesis and the development of innovative preventive and therapeutic approaches against cancer.
Horizontal vs. Vertical Transmission
At this point, it is important to define what we mean by “horizontal transmission” and “vertical transmission” with respect to cancer. Horizontal transmission of a pathogen, defined as any transmission between conspecifics that is not hereditary, or in utero, transmission from parent to child, is generally associated with enhanced virulence. In contrast, vertical transmission, intergenerational transmission between parent-offspring, is associated with decreased virulence. Can this paradigm be applied to disease processes taking place within a single patient (i.e., cancer)?
In cancer, metastasis and the potential for metastases, and the mechanisms whereby it occurs, is analogous to horizontal transmission while a (optimally slow-growing) stable primary tumor (including benign neoplasms) is analogous to vertical transmission of the “pathogenic” genetic information. In the former case, the neoplastic cells are horizontally (spatially) transmitted to different sites throughout the body; in the latter case, the neoplastic cells are vertically (temporally) transmitted by maintenance over time at one given location (e.g., the site of the original, primary neoplasm).
Of relevance, a study [3] has shown that genetic hybridization of cancer and normal cells can transmit malignancy from the former to the latter; furthermore, cell-free human cancer DNA exhibits the potential to transfer genetic information to, and induce malignancy in, normal cells [3]. Hence, gene information transfer from cancer cells to normal stromal cells [3], a physical manifestation of horizontal transmission, can promote neoplastic progression, including metastasis.
Figure 1
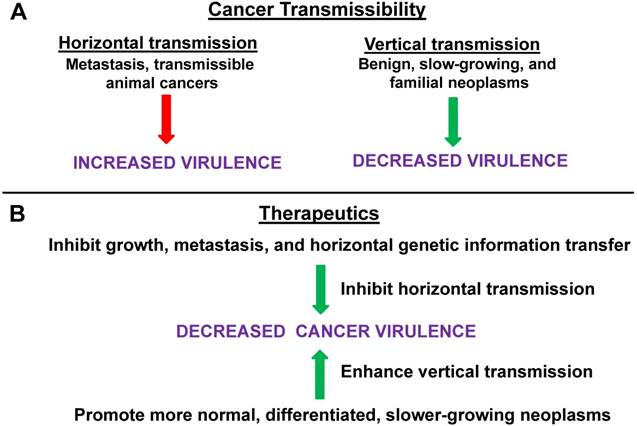
Horizontal vs. vertical transmission in cancer virulence. (A) Horizontal transmission in cancer can be represented by metastasis or by the rare cases of directly transmissible animal cancers; this form of transmission tends to lead to increased virulence. Vertical transmission, which tends to suppress virulence, can be modeled by benign neoplasms or (slow growing) primary tumors, and, possibly, by familial forms of cancer. (B) Therapeutics that would inhibit growth, metastasis and, in general, horizontal genetic information transfer, would tend to suppress virulence, while therapies that promote more normal, differentiated, and slower-growing (primary) neoplasms, as opposed to metastases, are more akin to vertical transmission and would likely suppress virulence. Thus, inhibiting horizontal mechanisms or (relatively speaking) promoting more vertical mechanisms, would tend to reduce cancer virulence. For both (A) and (B), green arrows represent processes leading to deceased virulence and red arrows represent processes leading to increased virulence.
Therefore, the language of transmissibility, horizontal vs. vertical transmission, and virulence can be used to describe the neoplastic lifecycle (Fig. 1A) and may inform approaches to inhibit tumorigenic progression (Fig. 1B). Hence, strategies that would tend to inhibit the growth, metastasis, and gene information transfer ability of neoplastic cells and/or reduce the fitness of neoplastic cells compared to their normal counterparts (in their original and/or metastatic location) would reduce “transmissibility” and promote less “virulent” cancer phenotypes, with positive outcomes for patient morbidity and mortality. For example, signaling pathways at the potential site of metastatic seeding can enhance neoplastic transmissibility; thus, Hedgehog signaling in bone promotes breast cancer metastasis at that site [4]. Specific characteristics of the potential metastatic tumor cell as well as the potential site of metastasis can both influence whether or not a given metastasis “takes” at a given site and grows; different types of cancer seem to have preferred sites of metastasis [5 and refs therein]. Repressing cell signaling at the potential recipient site [4], and/or blocking the specific combinations of integrins that mediate targeting of tumor cell endosomes to tumor-specific recipient sites and that “prepare” that site for metastasis [5], can inhibit transmissibility.
According to our hypothesis, reduced transmissibility (ease of metastasis) will also reduce long-term cancer virulence (growth, invasiveness, potential for future metastasis, interference with host metabolism) overall, particularly at the primary tumor site. Eliminating the possibility of horizontal transmission, and limiting the neoplasm to localized vertical transmission only, would select for decreased virulence. This would occur since we predict that the fitness of the neoplastic cells in a spatially restricted environment (enforced localization) would be optimized via the adoption of strategies (e.g., moderate proliferation and minimized invasiveness) that did not destroy the local cellular ecology. Thus, selective pressure would favor the neoplastic cells minimizing, to the extent possible, damage to the microenvironment that is required for their survival. Such cells would be expected to be more “normal-like” than those with unrestricted potential for invasion and metastasis. This selection for minimized virulence would be important for inoperable primary tumors, cancers in early stages of metastasis, as well as to prevent overall progression and recurrence. The meme of cancer becoming a “managed, chronic disease” may very well depend on controlling cancer virulence by reducing those factors that would facilitate transmissibility of the neoplasm within the patient.
To summarize: if the only way for the cancer cell to transmit its altered genome is via “vertical transmission” (defined as genetic continuity and potential limited expansion at the primary tumor site) rather by “horizontal transmission” (defined by invasion and metastasis) then this may favor selection of less “virulent” tumor cells. These less virulent neoplastic cells would be characterized, e.g., by slower growth rates, more differentiated phenotypes, more epithelial phenotypes (in epithelial tissue cancers), slower metabolic rates, and a lowered potential for future metastasis. Thus, we posit that there is a tight association between cancer transmissibility and cancer virulence. Metastatic transmissibility is itself a form of virulence as well as being an important potential contributing factor for subsequent further metastasis and for promoting other forms of virulence (e.g., dedifferentiation, proliferation, altered metabolism, etc.). In this context, one can consider the differences in intra-host transmissibility, aggressiveness, and potential for host mortality, comparing a more “vertical” form of skin cancer, basal cell carcinoma, to a more “horizontal” form, melanoma.
We further speculate that initiation and early progression could be inhibited through similar application of the transmissibility/virulence meme; thus, an environment that inhibits transmission of the altered genome and which favors a more normal, differentiated, slower growing phenotype, can retard neoplastic progression. For example, an increase of neoplastic virulence from, e.g., adenoma to carcinoma may be inhibited through approaches that favor a reduced transmissibility of the altered cancer genome. Thus, the difference between horizontal and vertical transmission in cancer is context-dependent. For an established cancer, as stated above, horizontal transmission is invasion/metastasis and vertical transmission is growth in situ. For a benign neoplastic cell/lesion, horizontal transmission can be considered transmission of the altered genome to many rapidly expanding cells at the primary site, while vertical transmission at this early stage of neoplasia can be considered as immortalization of the transformed cell, and low level, highly localized reproduction as part of a benign, non-invasive lesion (e.g., an adenoma). In both cases (early and advanced neoplasms) erecting barriers against horizontal transmission, so defined, will favor less virulent and more benign neoplastic cells. In contrast, anything that facilities easier (horizontal) transmission of the cancer genotype will select for more virulent and aggressive neoplasms consisting of cells with a clear selective advantage over normal cells in this cancer-promoting permissive environment. Thus, our hypothesis suggests that the modeling and therapeutic approaches heretofore utilized in the analysis of infectious pathogen virulence, as a function of transmission mode, will also apply to cancer.
Familial Cancer and Directly Transmissible Animal Cancers
While our hypothesis focuses on vertical vs. horizontal transmission of cancer in a single human host, modeling that host and its cells and tissues as an integrated ecosystem, are there examples of cancer that are transmitted between different conspecific individuals, either vertically or horizontally, akin to infections pathogens and parasites? What can these examples inform us about our hypothesis?
Human cancer in the individual patient is typically counter-selective for its own long-term survival. However, one can also consider horizontal vs. vertical cancer transmission in terms more precisely analogous to that of microbial pathogens. Here, vertical transmission describes familial cancer, including hereditary cancer syndromes [6]. While familial cancers typically occur at younger ages than purely sporadic forms, many (albeit not all) of these cancers typically manifest past the age range where most human reproduction takes place [6]. If these forms of cancer were to instead inflict severe morbidity and mortality before and during prime human reproductive ages, then the relevant oncogenic gene variants would have been selected against. A more precise determination of virulence of the familial vs. sporadic forms of the same cancer type remains to be performed; our hypothesis would suggest decreased virulence for familial cancers, at least in those cases in which a credible association between virulence (including age of onset) and host reproduction can be established. Of course, a proper comparison of transmission of cancer between individuals requires examples of direct horizontal transfer of cancer (as opposed to indirect transmission via viral infection) which superimposes issues of viral transmissibility and virulence onto the cancer ecology. Direct horizontal transfer in this context would be defined as transmission of viable cancer cells between individual hosts other than parent-offspring, resulting in an established tumor in the new host (and, optimally, one capable of being transmitted to subsequent hosts).
In the laboratory, horizontal transmission of cancer between hamsters has occurred via cannibalism and insect vectors (mosquitoes); a number of extremely rare accidents and other occurrences have transmitted neoplasms between two humans, albeit with no further dissemination [7 and refs therein]. But there has been no sustained transmissible cancer in those two species in nature. Thus, direct horizontal transmission of cancer does not naturally occur, insofar as we know, in humans, but does occur in Tasmanian devils (Tasmanian devil facial tumor disease, DFTD), in dogs (canine transmissible venereal tumor, CTVT), and in soft-shell clams (leukemia) [8,9]. Some characteristics of these horizontally, clonally transmitted, animal cancers in the mammalian species (DFTD and CTVT) are instructive.
DFTD, most likely transmitted through biting, is more aggressive and virulent compared to the sexually transmitted CTVT, which typically undergoes spontaneous regression after several months, leaving the dog immune to reinfection [7,8]. The ability to undergo regression may be an adaption of CTVT to maintain host viability; for example, tumors that grow too large can interfere with male sexual function, hence interfering with transmission [7,8 and refs therein]. Hence, decreased horizontal transmission of CTVT for such mechanical reasons facilitated the evolution of lesser virulence.
While DFTD is a relatively recent disease, CTVT has apparently been extant for thousands of years, indicative of depressed virulence that allows for transmissibility over an evolutionarily significant timeframe [7,8]. While the more virulent DFTD is currently driving the Tasmanian devil population to possible extinction, it is possible that selective pressures on both cancer and host may result in a less virulent, more-CTVT form of DFTD over time [8]. While such a decrease in virulence may be the long-term outcome, as it would enhance pathogen fitness by promoting long-term transmissibility, in the short-term, the opposite has been observed. Thus, in a Tasmanian devil population, a less virulent tetraploid form of DFTD was rapidly replaced by a more virulent diploid form [10]. This finding, observed in the early stages of the epidemic, may be due to an initial surplus of potential hosts (easy transmissibility) and a lack of antigenic response to the tumor cells (unlike the case of CTVT in dogs) [10]. Thus, there is a clear correlation between highly efficient horizontal transmission and greater virulence in DFTD.
The transmissible cancers DFTD and CTVT apparently evade host immune responses through the downregulation of MHC expression as well as repressed response from NK cells [11]. The inter-host transmission of these cancers, including immune escape, is analogous to intra-host metastasis as occurs in human cancer [11]. In addition, much is known about the mechanisms of CTVT transmission [7]. Besides immune evasion, CTVT had two other major hurdles to overcome for effective dissemination into new hosts. First, to enhance transmission, the tumor typically grows in an external region, is fragile and prone to ulceration, exploits the lengthy and typically aggressive coitus characteristic of dogs, and may influence host behavior [7]. Canine coitus typically results in superficial tissue damage that facilitates transmission of the neoplastic cells [7,8] analogous to how sexually transmitted pathogens in humans can more efficiently enter the body if epidermal integrity is compromised.
Second, the cancer must maintain a reasonably stable genome over evolutionary significant time scales; this may be achieved by various DNA maintenance and repair mechanisms, as well as by capturing host mitochondria to replace those of the tumor [7]. Further, differences between DFTD and CTVT in their interactions with host defenses may in part account for the high virulence of DFTD compared to CTVT (i.e., CTVT is evolutionarily stable while DFTD seems to be driving both its host and itself to extinction) [11]. Another question is why these two cancers were able to develop direct inter-host transmissibility while “contagious cancers” in general are so very rare. One possibility is the cell type from which these two neoplasms developed, likely from a Schwann cell in the case of DFTD and from a macrophage cell in the case of CTVT, were well suited to allow for inter-host transmissibility [11 and refs. therein]. Would similar-derived human cancers also possess potential for inter-host transmissibility?
While indirect and low-penetrant “human cancer transmission” can be considered to occur today via viruses (also see section Viral-Induced Tumorigenesis, below) such as HPV, apart from very rare occurrences [12], clonal (direct) horizontal transmission of human cancer does not occur in nature. However, the theoretical possibility exists that a novel form of cancer may develop utilizing such a mechanism, possibly via a sexually transmitted CTVT-like disease. It is plausible that virulent metastases [13,14], already pre-adapted for high transmissibility, could produce cells capable of horizontal transmission between individual human hosts. Further, it has been theorized that neoplasms derived from nervous system or immune cells may be particularly prone to the type of changes that would make direct transmission possible [11]; a future transmissible human cancer may derive from similar cell types. In addition, such transmissible human cancers may initially occur in relatively inbred human populations containing less person-to-person immunogenic divergence. DFTD transmission and virulence is enhanced by the inbred nature of the Tasmanian devil population that exhibits relatively little genetic diversity, and therefore demonstrates a muted immunological response to cancer cells derived from “unrelated” conspecifics [11]. Similarly, more inbred human populations may exhibit a similar immunological vulnerability to a cancer arising in “unrelated” members of that same population. In this scenario, such a cancer may or may not be able to make the jump into other, more genetically and immunologically divergent, human populations.
Therefore, studying diseases such as DFTD and CTVT may inform about possible future human health threats, and one can use these diseases to model different transmission modes (i.e., vertical vs. horizontal) that can occur within a single host, rather than between conspecifics at the population level.
An Alternative Viewpoint on Virulence and Transmission
Given the importance our hypothesis places on the association between type of transmission and virulence, it is worthwhile to consider an important contrarian viewpoint on the subject before proceeding. Ebert and Bull [2] offered what they referred to as a “devil's advocate viewpoint,” asserting a view that selection based on the trade-off between transmissibility and virulence would be too weak to result in rapid changes in virulence; they instead suggest direct selection against virulence itself. While the link between transmissibility and virulence is strong in vertical transmission, it is seemingly weaker for horizontal transmission, and the authors provide several examples where altered transmission did not result in the expected modification of virulence. Thus, Ebert and Bull identify what they see as fundamental problems with the “trade-off” paradigm. First, for a subset of pathogens, virulence has little or no selective consequence. Related to this is the understanding that virulence is not only a fixed characteristic of the pathogen, but is strongly influenced by variation in the host and in host-pathogen interactions. Then, and derived from the above, to the extent that a “trade-off” exists between transmissibility and virulence, it would involve more than two dimensions of influence, and attempts to evaluate findings in the context of only transmissibility vs. pathogen-mediated virulence would yield vague and/or misleading findings. Thus, Ebert and Bull are skeptical that modifying transmissibility will reliably result in expected and desired rapid and significant changes in virulence. Citing examples such as diphtheria, they instead advise direct selection against virulence itself (rather than indirect selection on correlated factors such as transmissibility).
On the other hand, the experience with avian vaccination against Marek's disease seems to legitimize that, for at least some pathogens, the transmissibility-virulence model has merit [15]. Thus, vaccination that does not prevent transmission increases virulence by increasing host survival and facilitating transmission to new hosts [15]. Therefore, it may well be that both strategies, modulating transmissibility and direct selection against virulence, are useful. Consistent with this, the original Ebert and Bull paper had comments [16,17] and an author response [17,18] on the relationship of transmissibility, virulence, and virulence management. Thus, Eliot [16] agrees with some of Ebert's and Bull's assertions, but suggests that they were too rigid and doctrinaire, and suggests that further study can reveal approaches to productively manage virulence through both direct selection and indirect selection, even in the case of horizontal transmission. Gandon and Day [17] consider the contrarian view of Ebert and Bull to be too narrow and that direct selection against virulence may not be possible (Eliot made a similar point). Gandon and Day also raise the possibility of directly targeting transmissibility as an approach. In general, both Eliot and Gandon and Day do not believe that Ebert's and Bull's argument was a convincing refutation of the “trade-off” model; in their riposte [18], Ebert and Bull disagree with these criticisms, and urge a broader analysis of these issues, going beyond the “trade-off” paradigm. Resolution of these controversies would inform the relevance of issues of virulence and transmissibility to cancer within the human patient, which is the overarching theme of our present analysis.
This manuscript's fundamental thesis accepts the validity of the basic trade-off model of virulence vs. transmission; however, as is standard with the scientific method, that model will need to be modified or rejected dependent upon future observations and experimental data. That said, the Ebert-Bull idea of direct selection against virulence is also consistent with our fundamental hypothesis; thus, the dual-strategy approach of targeting both virulence (directly) and transmission (to indirectly affect virulence) may be optimal.
Stem Cells, Clonal Dominance, and Somatic Evolution
We will now consider issues related to stem cells, clonal dominance, and somatic evolution [19-22]. Nowell [21] proposed an evolutionary view of cancer as a disease resulting from the interplay of genetic instability and natural selection. Therefore, as is usually the case with mutations, most of the genetic alterations that occur (at low frequency) are harmful, leading to clonal extinction. However, some fraction of these alterations are beneficial, leading to clonal expansion. Nowell's theory therefore predicts neoplastic uniqueness; in other words, every tumor will have its own genetic signature, resulting from (random) mutation and selection (channeled in specific directions by the tumor microenvironment), as well as by human genetic polymorphism. These predictions have been confirmed by subsequent experiment and observation. In addition, this hypothesis predicts that cancer therapeutics further shape the tumor mutational landscape by exerting additional selective pressures, and this too has been confirmed by experiment and observation. Of relevance to the cancer stem cell paradigm, clonal dominance of stem cells that occurs with age can result in accumulation of deleterious mutations that can lead to cancer [19]. Altering the environment can in theory either select for the preservation of polyclonality or selection of clonal dominance of cells relatively lacking in deleterious mutations [19].
Why do multicellular organisms exhibit continued cycles of cell differentiation even after the period in which production of new types of cells is required? One analysis [20] associates “serial differentiation,” defined as a sequence of differentiating stages (e.g., stem cells, transient amplifying cells, terminally differentiated cells, and cell turnover), with repression of somatic evolution [21]. Thus, within the population of cells making up a multicellular organism, selective pressure will favor the expansion of cells with superior viability and reproduction (“somatic evolution”) [20], which can be conferred by somatic mutation, epigenetic changes, altered environment, etc. This somatic evolution often leads to cancer; however, tissues that exhibit a serial differentiation pattern of organization are more resistant to somatic evolution than are more basic cellular organization patterns. Further, self-renewing cell populations (e.g., stem cells) are particularly vulnerable to somatic evolution, and mutations that interrupt or disorder the normal differentiation process can generate new populations of self-renewing cells prone to somatic evolution, leading to cancer. In this context, it is important to distinguish between (a) neoplasms that originate from normal stem cells that acquire oncogenic mutations, and (b) non-stem cells that first undergo a dedifferentiation step to transition to self-renewing cells capable of subsequent somatic evolution [20]. Although the eventual outcome is the same, it has been proposed that neoplasms derived from these two alternative mechanisms may retain genetic/epigenetic differences that could be exploited for medically relevant diagnostic and/or therapeutic approaches [20]. Dedifferentiation and disrupted differentiation are likely key steps in somatic evolution, contributing to various forms of cancer [20]. Another related factor to be considered is that of feedback control of tissue homeostasis [20]. Thus, for example, given scenario (b) above, further downstream differentiation is blocked and this can deplete the numbers of differentiated cells, stimulating a compensatory proliferation of stem cells and transient amplifying cells [20]. This stimulatory signal can also increase growth or the neoplastic cells responsible for this derangement of differentiation, thus promoting carcinogenesis. Of relevance to our present analysis, somatic evolution in neoplasia is correlated to enhanced horizontal transmissibility and virulence, as the default pathway is for enhanced growth and metastatic potential as part of the evolution of the neoplastic somatic cell [20-22]. Somatic evolution, based on selective pressures derived from specific environmental challenges, drives changes in transmission mode (vertical to horizontal and vice versa) that affect cancer virulence; therefore, the challenge is to channel somatic evolution in the direction of vertical transmission and decreased virulence. These therapeutic implications of our hypothesis are discussed in the last sections of our paper, below.
Kin Selection, Soft vs. Hard Selection, Genetic Variation, and Virulence
Interactions between pathogenic organisms within a host can affect individual and group virulence; the same principle, according to our hypothesis, applies to cancer cells that are being transmitted within a host vertically or horizontally. According to basic evolutionary theory, fitness outcomes for interacting organisms will depend on their relative genetic relatedness; thus, the genetic relatedness of interacting parasites/pathogens can affect virulence (Fig. 2A).
Although experimental confirmation is currently weak, the following has been proposed [23]. In general, the “social behavior” of the pathogen population influences the relationship between kin selection (selection that may decrease the actor's probability of survival but enhances that probability for kin who carry some fraction of the actor's genes, thus in this manner enhancing actor's fitness), and virulence. Hence, in situations in which host resources are limited and cooperation to efficiently utilize those limited resources would maximize reproduction, decreased relatedness would lead to increased virulence. This would obtain due to decreased kinship resulting in less cooperation in managing limited resources and maximal competition to exploit those host resources. On the other hand, in a situation in which cooperation between pathogens within the host would increase the pathogen growth rate, then increased relatedness would lead to greater virulence. “Spiteful” behaviors (behavior that imposes fitness costs on both actor and recipient, but imposes greater costs on the recipient) between pathogens would be maximal at intermediate levels of relatedness and this would minimize virulence as pathogens interfere with each other's growth rate within the host. When resources are limited, greater genetic relatedness between pathogens would tend to decrease virulence, since adaptive fitness would necessitate a “prudent sharing” of limited resources among the related pathogens.
Figure 2
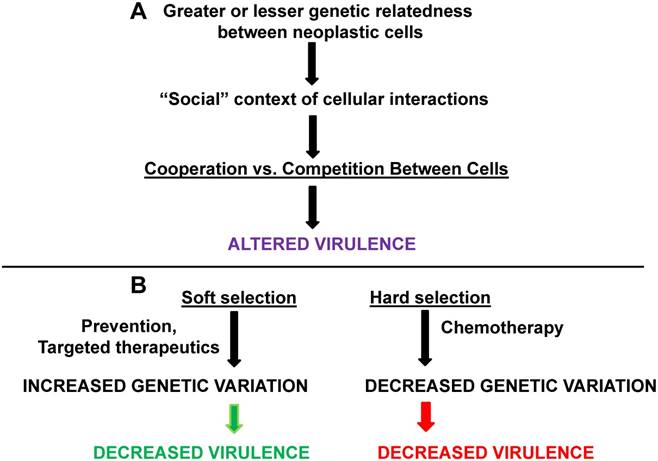
Kinship, soft vs. hard selection, and cancer virulence. Greater or lesser degrees of genetic relatedness (influenced by mutation, aneuploidy, etc.) between normal and neoplastic cells and between different neoplastic cells can affect cooperation vs. competition in different “social” contexts. The result is changes in the degree of virulence. (B) Soft selection in cancer may be represented by prevention or by highly targeted therapeutics; these approaches may maintain genetic variation but decrease virulence. Hard selection in cancer, as best represented by whole-body chemotherapy, may decrease genetic variation in neoplastic cells but increase virulence (assuming that the neoplastic cells are not eliminated by the treatment). The green arrow represents processes leading to deceased virulence and the red arrow represents processes leading to increased virulence.
Of direct relevance for our hypothesis, the language of kin selection and pathogen cooperation and competition can be applied to interactions between normal and neoplastic cells that differ genetically due to mutation and aneuploidy, as well as interactions between neoplastic cells themselves, reflecting the well-known genetic heterogeneity found in most cancers.
Can “spiteful” behaviors in part account for the relative rarity of synchronous primary tumors [24], despite the existence of “field effects” [25] that would tend to increase the frequency of such cancers? Further, with respect to the issue of cell-to-cell competition and carcinogenesis [26], relative genetic relatedness needs to be addressed. While the normal and neoplastic cells in an individual all have the same basal genetic background, mutation, aneuploidy, etc. in neoplastic cells results in varying degrees of genetic divergence from normal cells (and from other neoplastic cells). Thus, in cases where host resources are limited (e.g., caloric restriction, suppressed angiogenesis), and where neoplastic cells would be competing for these limited resources, greater cancer genetic heterogeneity may be expected to result in greater virulence, while more genetically homogenous tumors would be expected to exhibit less virulence. On the other hand, in situations in which cooperation between neoplastic cells can enhance their fitness, greater genetic homogeneity could be associated with higher degrees of virulence and vice versa. Is there any association between the degree of genetic relatedness of neoplastic and surrounding normal cells and the degree to which normal cells can support neoplasia? These questions require further analysis and experimental evaluation, and are of direct interest for therapeutic approaches, such as “benign cell boosters” (see below). We deal with the related phenomenon of “competitive release” below.
Greater genetic variation in pathogen/parasite populations can fuel resistance to therapy due to increased probability that gene variants encoding resistant phenotypes exist in the population [27]. Thus, on the one hand, one approach to rational therapy would be to minimize genetic variation; on the other hand, decreased variation may select for the most virulent and/or resistant strains, and greater genetic variation may result in the continuity of more maladaptive and less virulent strains. In this case, approaches that allow for maintained genetic polymorphism may be preferable. Further, there may be “trade-offs” between relative genetic variation and virulence, as stated [27].
An important concept related to genetic variation and virulence among pathogens that exist, or can exist, in spatially separated environments, is the distinction between soft vs. hard selection (Fig. 2B). This distinction between these forms of selection can assist in clarifying the association between genetic variation and virulence in different environmental contexts. For soft vs. hard selection, the condition of initial spatial separation is important, as different pathogen genotypes would be expected to have differential fitness in varying environments, thus resulting in a degree of genetic polymorphism in the total pathogen metapopulation (defined as all pathogens of the same type in all relevant environments) [27]. In soft selection, regulation against pathogen types takes place within spatially separated environments, which reduces pathogen numbers before the pathogens mix in a common environment and then disperse to new hosts (transmission). In soft selection, similar representation of the different pathogen types in the mixed population would be observed, thus maximizing pathogen diversity and genetic variation [27]. In hard selection, regulation of pathogen numbers occurs after the different pathogen types have already mixed, selecting in favor of those pathogen types best suited for their environment (and, hence, represented at higher levels in the mixed population). Hard selection would therefore minimize genetic variation as selective pressure in the mixed population favors the more abundant type (typically with high growth rates) and/or the type most resistant to the treatment being simultaneously applied to the entire metapopulation [27].
Soft selection would tend to favor decreased virulence, while the hard selection regimen would tend to favor fast-growing (potentially resistant) strains, leading to increased virulence. It is likely that “spiteful” interfering behavior would minimize virulence if genetic relatedness between strains is intermediate; further, if cooperation among pathogens is required for growth, the decreased relatedness (greater generic variation) inherent in the sort selection scenario would lead to less virulence as cooperation is minimized [23]. Therefore, soft selection offers more possibilities for lower virulence than does hard selection [27]. However, it is also possible that soft selection could lead to increased virulence if kinship between pathogen strains is low enough to favor competition for limited host resources [23].
Cancer cannot be described with soft selection and hard selection as precisely as can most exogenously-originating pathogens; nevertheless, this paradigm can contribute to a better understanding of the interactions between neoplasms and various preventive and/or therapeutic approaches. Current cancer therapy is most analogous to hard selection, the type that decreases genetic variation but can enhance virulence. Therapy is focused on (a) primary tumors of established cancers, which already are a mixed population of genetic variants, some being more numerically dominant; and/or (b) metastases, which are post-dispersal and which are typical treated by body-wide chemotherapy, an approach in which all neoplastic cells are affected simultaneously as one metapopulation. Therefore, typical cancer therapeutics would be expected to reduce genetic variation among the neoplastic cell metapopulation; if the cancer is not fully eliminated, it is likely to recur in a more virulent form.
Soft selection in cancer is more problematical to describe, but is most likely best modeled by preventive strategies. Such strategies, for epithelial cancers such as colorectal (CRC), which would tend to target (a) isolated preneoplastic or very early stage neoplastic isolated cells; (b) small genetically homogenous lesions in spatially separated microenvironments, or, perhaps most likely; (c) a mixture of genetically variable cell types within a single lesion, with the cells constituting this lesion being affected differently by their respective neighboring microenvironments (which could be viewed as “spatially separated” with respect to their activity if not with respect to the proximity). If these approaches do not eliminate the neoplastic cells, the result, particularly from scenario (c), from this soft selection regimen is likely to be a genetically heterogeneous neoplastic mass of relatively low virulence. In other words, a failed preventive (e.g., anti-CRC) regimen would result in a typical pre-cancerous benign neoplasm (e.g., adenoma) or less aggressive very early stage carcinoma, consistent with actual observation. Soft-selection in therapy for established cancer might involve (a) highly specific targeting of particular cell types within the neoplasm and their associated microenvironments (cancer stem cells?) and/or (b) techniques that would specifically target individual metastatic cells and their associated microenvironments rather than broad spectrum chemotherapy (in both cases a form of functional “spatial separation”). Further development of soft selection approaches for cancer treatment will require a better understanding of the complex interactions between types of selection, neoplastic genetic variation, and cancer virulence. Soft selection approaches can be (as described above) expected to decrease virulence, but by maintaining greater genetic heterogeneity, soft selection runs the risk of maintaining variants capable of escaping local competitive controls on neoplastic growth, via metastatic colonization.
Metastatic Colonization Genes and Horizontal Transmission
Considering metastasis as a form of internal horizontal transmission that facilitates escape from local competition, we can consider genetic changes facilitating this process. “Metastasis virulence” genes are those whose expression confers a selective advantage to the neoplastic cells in their secondary (metastasis) sites, but not in the primary tumor [13]. Therefore, these are “metastatic colonization” genes, not genes involved in primary tumor development. Expression of these genes integrate the horizontal transmission of the neoplastic “pathogen” to different body sites with the increased cancer virulence associated with metastasis, negatively affecting patient morbidity and mortality. Tumor aggressiveness and metastasis are likely associated with an “epigenetic programming” that results in the expression of “off-context” genes, such as that of the germline and placenta [14]. Thus, expression of normally silenced genes in somatic (cancer) cells can be used an as an index of aggressive behavior and poor prognosis. These changes in gene expression may be linked, directly or indirectly, to the alteration in cancer virulence consequent of modified transmissibility of the initial primary tumor. The specific changes in gene expression, coupled to the effects of the tumor microenvironment, can influence whether the metastasis is relatively “dormant” (i.e., less virulent) or demonstrates more aggressive proliferative and invasive properties (i.e., more virulent) [28,29]. This is explored in more detail in the Molecular and Cellular Profiles section, below, using colorectal cancer as an example.
Further, consistent with our general hypothesis, a case of a “tertiary” metastasis (i.e., a metastasis derived from another metastasis, rather than from the original primary tumor) has been observed, and this tertiary neoplasm exhibited a more aggressively proliferating phenotype compared to the relatively dormant original metastasis [30]. This finding is consistent with the idea that increased horizontal transmission (e.g., further metastatic dissemination of the neoplastic cells in the patient) leads to a more virulent phenotype (e.g., very rapidly proliferating neoplasm). As we shall see (below), the tumor's use of metastatic colonization (via horizontal transmission) can result from selective pressures, which can be exerted by therapies that enhance the fitness of (more) normal cells over those that are neoplastic.
Game Theory, Cancer Heterogeneity, and Virulence
The basic processes described above depend upon tumor heterogeneity. For example, kin selection and spiteful behavior in cancer presume genetic variation between cancer cells (and of course between normal cells and cancer cells). The differences in gene expression and cell phenotype driving differences in transmission and virulence, all presuppose the existence of different cell types in the tumor, i.e., heterogeneity. From an evolutionary standpoint, why is such heterogeneity maintained? Why doesn't one clone always outcompete the others, forming homogenous tumors?
Cooperation between pathogen types in a host can enhance or suppress virulence, in part dependent upon the relatedness of those types to each other. In a human tumor, cooperating types imply tumor heterogeneity, a well-known attribute of many cancers. Thus, tumor heterogeneity is, as suggested above, a key contributor to cancer metastasis [31 and refs therein] and, hence, to cancer virulence. Maintenance of cancer heterogeneity is an important question for evolutionary oncology and for tradeoffs inherent in cancer virulence, since more rapidly reproducing cells (i.e., more virulent) should outcompete others [32]. That this does not always occur implies cooperation between tumor subtypes that, in specific environmental contexts, leads to maintenance of heterogeneity and the potential for altered virulence, as well as the maintenance of sufficient genetic diversity and phenotypic plasticity to adaptively respond to different environmental challenges. The type of association (i.e., positive or negative) between cancer heterogeneity (genetic variation) and virulence would depend upon the relative genetic similarity of the cells, cooperation (or the lack thereof) between cell types, and the environmental context, including both natural and therapeutic selective pressures.
One possibility for the maintenance of heterogeneity is for cell types to be mutually interdependent; thus, one type produces factor “A” and the other type produces factor “B” and both factors are required for survival of both cell types. However, more typically, such symbiotic interdependence does not occur and cells can develop mutations that suppress production of the necessary factors; these non-producer cells are dependent upon the remaining producing cells in the tumor population [32]. Thus, cells (“free-riders” or “defectors”) that use but do not contribute to “public goods” (e.g., growth factors) can develop in the population and such free-riders would in theory have a growth advantage over the producers. After all, the non-producers do not need to invest energy and resources to produce factors that are provided exogenously, and therefore these non-producers can instead invest more into their own reproduction. This being so, how are producers maintained, avoiding a “tragedy of the commons” scenario in which free-riding cancer cells outcompete producers, thus dooming both cell types to extinction [32]?
Archetti et al. [32] present a model of producer/non-producer cell heterogeneity utilizing a neuroendocrine pancreatic cancer (insulinoma) cell system cooperating by sharing insulin-like growth factor II (IGF-II). Cells that do not produce IGF-II will grow very slowly in the absence of exogenously provided factor, but would have a growth advantage in a mixed cell population, as these non-producers utilize the IGF-II produced by others without incurring the costs of production themselves. In this context, the IGF-II, which diffuses and optimizes growth of all cells including those that are non-producers, can be labeled a non-linear public good [33-35].
Maintenance of a mixed population (see comments on “intratumor heterogeneity” below) in this model is dependent upon the starting numbers of each cell type as well as concentrations of serum/IGF-II in the cell medium [32]. The first variable to be considered derives from stochastic processes and cell competition; thus, too few producer cells in the initial cell seeding would not allow their establishment as a stable population. Of greater interest is how the evolutionary dynamics play out when both cell types are seeded in numbers appropriate for maintenance, yet the environmental contexts differ. At high concentrations of serum/IGF-II, the producer cell requirement is relaxed; given that non-producers can survive from the factors present in the medium, they will outcompete producer cells [32]. Here, the cost of production is much higher than the benefit, resulting in reduced relative producer cell fitness; in contrast, very low serum/IGF-II concentrations result in enhanced producer cell growth, as the benefits of production outweigh costs [32]. Intermediate levels of serum/IGF-II result in the stable maintenance of both cell populations, an established heterogeneity [32]. When evaluating this model from the standpoint of proportion of cell types, optimal tumor growth would occur with an intermediate level of producer cells [31], again maintaining heterogeneity of the neoplastic cell population.
Thus, increased virulence deriving from heterogeneity can be modeled by evolutionary game theory, in which environmental contexts affect the cooperative and competitive interplay between cell types, including “altruistic” producers and “free-riding” non-producing “defectors.” The heterogeneity thus maintained sets the stage for further differentiation and specialization of cell types and functions, leading to further enhanced virulence including metastasis. Of course, the IGF-II model can be applied to many other scenarios [32] in which cancer cell types cooperate and compete to optimize survival and growth and spread of the cancer genome, both “vertically” and “horizontally” (virulence, metastasis). This modeling, based on evolutionary game theory, has implications for therapeutics (see below), and directly influences selective pressures affected transmission modes and consequent cancer virulence.
Viral-Induced Tumorigenesis
Considering analogies between cancer and infections pathogens inevitably leads to an evaluation of cancers that are initiated by pathogen infection. The genetic diversity underlying decisions affecting cancer virulence and transmissibility are typically caused by mutation or epigenetic modification, although other mechanisms have been proposed [36-38]. One source of carcinogenesis in humans, briefly mentioned in the section on transmissible animal cancers, is viral-induced tumorigenesis. The pioneering work of Rapp [39-43] demonstrated that a significant number of human cancers are ultimately of viral origin. Importantly, the “hit and run” hypothesis [44] suggests that the fraction of human cancers of viral etiology is underestimated. This hypothesis suggests that a given tumorigenic initiation (“hit”) can be virus-dependent, however the subsequent maintenance and progression of the neoplastic state can be independent of the virus and is thus fully compatible with viral loss (“run”) from tumor cells [44]. This ties into the clonal expansion paradigm of Nowell [21, 45,46], since a viral-initiated cell can clonally expand after viral loss, resulting in a tumor whose viral origin is not observable.
Rapp's work raises two important questions with respect to the fundamental hypothesis under consideration. First, is the “run” stage associated with a change from vertical transmission of the cancer (e.g., in situ growth initiated by the viral “hit”) to horizonal transmission (e.g., invasion and metastasis)? This is unknown, but amenable to study. Second, is there any clear association between the virulence and transmissibility of a virus and the consequent virulence and transmissibility (as defined in this paper) of the cancer resulting from infection with that virus? It would be tempting to speculate that enhanced horizontal transmissibility of the viral agent would result in greater cancer virulence (e.g., shortened timeframe for tumorigenesis, increased relative morbidity and mortality), if an increased cancer burden would reduce the likelihood of viral transmission. If so, this would in turn raise the possibility that the virulence of viral-induced cancer can be decreased by making viral transmission more difficult (e.g., vaccination). Thus, inhibited horizonal transmission of the virus would result in decreased cancer virulence as an adaptive response, so that the afflicted patient would have more opportunities to transmit the viral pathogen.
This scenario assumes a relatively strong link between viral infection, carcinogenesis, and the ability of the patient to pass on that viral infection. However, if the period between viral infection and cancer development is long, and if the patient can pass on the virus during this “incubation” period, (as is typically the case) then one could reasonably expect cancer virulence to be uncoupled from viral transmissibility. If on the other hand the “incubation” period between viral infection and cancer is short, a stronger (inverse) link between cancer burden and viral transmissibility can be expected; e.g., a very ill cancer patient would have fewer opportunities to transmit the viral pathogen. Further, the degree of penetrance is crucial; the potential link between viral transmissibility and cancer virulence would be strengthened in proportion to the probability that viral infection leads to cancer development. This scenario is also complicated by the effects of viral virulence itself. If this virulence would decrease rates of horizonal transmission of the virus, then cancer virulence may be decreased for the reasons discussed above; on the other hand, higher degrees of viral virulence may result in more efficient tumor initiation in any given infected patient.
Molecular and Cellular Profiles: Colorectal Cancer, Influenza, and Pathogen Subversion of Host Pathways
It is important to note that the fundamental hypothesis of this manuscript is a conceptual analogy of virulence and transmissibility comparing microbial infections and cancer. We do not necessarily suggest that the molecular and cellular profiles and pathways controlling virulence vs. transmissibility are always the same or similar comparing cancers and microbes, only that the evolutionary trade-offs and selective pressures may be similar. Having said that, in some cases, there are in fact similarities between the molecular mechanisms influencing virulence and transmissibility of cancers and microbes. Thus, in this section, we will examine colorectal cancer (CRC) as an example, make a brief comparison to influenza virulence, and then note examples where microbial pathogens subvert host pathways that can have significance to cancer.
CRC is typically characterized by a series of mutations in driver genes, particularly tumor suppressors and proto-oncogenes, promoting neoplastic progression from adenoma to carcinoma to metastasis. The early stages of this process can be considered akin to vertical transmission with lesser neoplastic virulence, while the latter stages of CRC are akin to horizontal transmission with greater virulence. Mutations in the Wnt signaling pathway initiate most cases of CRC; mutation in the APC tumor suppressor is the most frequent initiating genetic alteration, resulting in deregulated Wnt activity, excessive cell growth, and adenoma formation. Interestingly, the two hits of APC required to fully initiate the neoplastic process are balanced to retain a degree of APC function; this results in an intermediate (“just right”) level of Wnt signaling conducive to cell proliferation at the early stages of the neoplastic process [47]. Thus, vertical transmission and reduced neoplastic virulence in early stage CRC requires careful control of Wnt signaling levels. Later, during progression, analogous to transition to greater virulence accompanying horizontal transmission (i.e. invasion and metastasis), a third hit may occur in an APC allele, further increasing Wnt signaling to drive advanced tumorigenesis (increased proliferation, invasion, and metastasis); this is known as the “three hit hypothesis” [48]. Superimposed on this scenario is that cells with deregulated Wnt signaling due to mutation can still be responsive to exogenous Wnt signaling, further fine-tuning Wnt activity to meet the adaptive requirements of the neoplastic cells. As the CRC cells transition to a more virulent profile associated with horizontal transmission, the ZEB expression axis driving epithelial to mesenchymal transition is activated [49], promoting expression of vimentin [50] and eventual metastasis (maximized horizontal transmission leading to increased virulence).
Underscoring the importance of exogenous environmental factors in this process, it has been noted that obese CRC patients exhibit fewer driver mutations than their normal weight counterparts, likely due to obesity-related cytokine signaling substituting for gene mutation [51] in the transition from vertical transmission (and lesser virulence) to horizontal transmission (and greater virulence). Thus, the careful balance between virulence and transmissibility in CRC is tightly controlled by molecular and cellular profiles altering signaling pathways to affect changes in cell proliferation, invasion, and metastasis. In addition, human tumors, as they progress (increase virulence), suppress immune function to evade tumor surveillance mechanisms.
How do the basic molecular profiles and pathways typical of CRC compare to that of a well-studied infectious pathogen? Influenza is a classic example of a pathogen demonstrating a balance between transmissibility and virulence. Transmissibility for the most part is influenced by external factors (social structures, war, vaccination, human mobility and global travel, etc.) leading to altered rates of horizontal transmission and consequent changes in the degree of viral virulence. An examination of gene expression related to influenza virulence [52] provides some analogies to CRC, and to cancer in general. In most cases of CRC, APC mutation drives cell proliferation and a third hit is associated with increased cancer virulence; in influenza, mutations in the viral RNA polymerase complex drives enhanced viral RNA production and viral proliferation to enhance viral virulence. Changes in the HA protein, which is responsible for host cell infection (horizonal transmission) and viral virulence, are analogous to cancer genetic profiles (e.g., ZEB in CRC) that promote invasion and metastasis. The NS1 gene product suppresses host immune response to influenza, and mutation of the NS1 gene can increase virulence similar to the suppression of immune tumor surveillance by cancer cells. Therefore, cancer and infectious pathogens may share certain basic patterns of molecular control of virulence and transmission that can be similarly influenced by selective pressures. Further, the virulence of both cancer (e.g., CRC) and of a microbial pathogen (e.g., influenza) are both also significantly affected by the characteristics of the host; e.g., host gene expression, immune surveillance, etc. The same host pathways affecting the one may affect the other, and this is clearly demonstrated by the fact that microbial pathogens can influence host signaling pathways that can be deregulated in cancer.
Thus, we note that, besides the well-known phenomenon of viral oncogenes and cancer initiation [39-43], microbial pathogens can subvert host signaling pathways in ways that enhance pathogen virulence [53]; some of these pathways are associated with carcinogenesis. Thus, various pathogenic bacterial species can affect MAPK signaling as well as G protein signaling (including Rac1), and various pathogens, including viruses, can affect actin and ubiquitin-mediated signaling [53]. In this manner, the molecular and cellular profiles of an infectious pathogen that influence virulence can directly modulate molecular and cellular profiles in the infected host to alter signaling pathways that potentially can affect cancer virulence and transmissibility.
While this section is not meant as a comprehensive comparison of the molecular and cellular profiles that exhibit similarity between the pathogenesis of cancer vs. infectious disease, the examples provided do demonstrate certain fundamental conceptual similarities. More in-depth comparative analyses of the molecular mechanisms underlying cancer vs. microbial pathogen virulence is a fruitful area of future inquiry.
Transmission Modes, Evolutionary Trade-offs, Tolerance, and Initial Therapeutic Approaches
Advances in the theoretical consideration of virulence and transmissibility [54-56], in some cases supported by experimental observation, can provide insights leading to therapeutic approaches.
Antonovics et al. [54] discuss the evolution of “transmission modes” and note that vertical transmission of a pathogen is maximized when the host is below its carrying capacity, allowing for greater expansion of the host and thus greater opportunity for pathogen vertical transmission. By analogy, we speculate that vertical transmission of neoplastic cells will be favored in those environments that retain, spatially and molecularly, open niches for local neoplastic expansion. Many pathogens have “mixed” (i.e., simultaneous horizontal and vertical) transmission modes, and evolutionary trade-offs affecting pathogen fitness determine which of these modes are favored [54]. The same likely holds true for cancer.
Cressler et al. [55] analyze the evolutionary trade-offs involved in virulence, providing theoretical support for the themes discussed above. In addition, they consider experimental evidence supporting or refuting various relevant theories. There is strong experimental support for the link between vertical transmission and reduced virulence; the authors also point out that since almost all parasites that can be transmitted vertically can also be transmitted horizontally, in practice what we are evaluating is the “mixed” rather than purely “vertical” transmission mode. Mixed transmission is predicted to reduce virulence and cause an increased dependence on vertical transmission over time, reinforcing decreased virulence [55]. In cancer, this is supported by the finding of “spiteful behavior” between primary tumors and metastases (which are not genetically identical due to mutational divergence), in which the metastases exhibit enhanced growth after the primary tumor is removed [57]. One possibility for this observation is that the primary tumor secrets anti-angiogenesis factors that suppress metastatic growth at distant sites [57], and removing this inhibition allows for outgrowth of metastases after resection of the primary tumor. Perhaps, suppression of metastatic growth allows the primary tumor to optimize limited host resources, a clear demonstration of vertical cancer transmission (continuity of a localized primary tumor) leading to decreased cancer virulence (e.g., cancer mortality, which is typically due to metastasis). Another point mentioned by these authors that is worthy of further study is the hypothesis that resistance to immune clearance, rather than the mode of transmission, is the major driver of pathogen virulence [55 and refs. therein]. However, there is, as stated above, a large degree of experimental evidence specifically linking virulence with horizontal vs. vertical (or “mixed”) transmission [55].
Another paper [56] extends the analysis of cancer, virulence, and transmissibility to the concepts of “resistance” and “tolerance,” both of which have obvious therapeutic implications. Resistance is here defined as the ability of the host to eliminate (or at least reduce) pathogen load; tolerance is defined as the ability of the host to adapt to, or accommodate, whatever pathogen load exists at a given time. Just as different hosts can tolerate microbial virulence to varying degrees (as the phenomenon of the “asymptomatic carrier“ makes clear), so do hosts differ in their tolerance of cancer. Also of interest is the point made that negative effects of a tumor on the host (what we call virulence and they call pathogenicity) can be due to tumor load (quantitative) or due to the unique characteristics of the tumor itself (qualitative), and these are both modifiable by host responses.
We also note differences in terminology of interest to the reader. Our manuscript defined what we mean by cancer transmissibility and virulence in the General Consideration section; briefly, transmissibility is defined by the spread of cancer cells within the patient (e.g., invasion and metastasis), while virulence encompasses all the factors that affect patient morbidity and mortality to a greater or lesser degree. However, van Niekerk et al. [56] define cancer virulence in terms of the tumor's “spread and dissemination within the host” (i.e., transmissibility), and use the term “pathogenicity” (instead of virulence) to describe the costs imposed on the host. Further, they do not distinguish between horizontal and vertical transmissibility of a cancer within its host. Readers need to be mindful of these semantic distinctions, although they do not affect the relevance of resistance vs. tolerance for cancer therapeutics (see below).
In addition to what has been discussed above (Figs. 1B, 2B), one can consider possible approaches based on an evolutionary appreciation of cancer and on a cancer virulence paradigm. These approaches could be utilized at all steps of the neoplastic process, but would be most required for cancers that would need to be treated as “maintained chronic diseases;” for example, inoperable tumors, intractable metastases, recurrent cancer, or cases that have a high probability of recurrence. One could in theory target selective pressures against fast-growing cells, epithelial to mesenchymal transition and metastasis, ability to proliferate independent of exogenous growth factors, abnormal cellular metabolism, and other indices of virulence.
Benign cell boosters have been proposed [22 and refs therein]. Thus, these agents would enhance the relative fitness of normal cells vs. their neoplastic counterparts, allowing the normal cells to outcompete neoplastic cells in given ecological niches. This approach can be applied at various steps in the neoplastic process, at each point favoring less virulent cells over their more virulent counterparts. For example, even within the neoplastic cell population, one can favor benign adenoma over carcinoma, or favor slow-growing, less invasive carcinoma over more aggressive counterparts, or favor primary tumor cells over metastatic ones. In theory, since these agents increase cellular fitness, selective pressure would favor wild-type phenotypes and not necessarily select for enhanced cancer virulence [21, 22 and refs therein]; however, our hypothesis suggests this may not necessarily be the case if increased niche competition from boosted benign cells exerts selective pressure on neoplastic cells to metastasize to more promising niches, in a horizontal transmission mode that would, eventually and inevitably, increase cancer virulence.
Enhancing host tolerance to a cancer [56] that cannot be eliminated not only will improve patient quality of life but would not result in a virulence-enhancing adaptive response by the cancer. In this case, the therapeutic approach is targeted to host mechanisms that allow better accommodation to, and adaption to, an existing cancer load, rather than an attempt to target the cancer load itself [56]. Therefore, therapeutic approaches aimed at improving tolerance rather than resistance should not result in a counter-productive increase in the tumor's virulence and horizontal transmission.
Of importance with respect to evolutionary optimization of therapeutics is the concept of “competitive release” [58-60]. Neoplastic cells that are highly resistant to therapy often exhibit sub-optimal fitness compared to normal and low-resistant cells, due to costs incurred by the more resistant cells to maintain their phenotype. More highly resistant cells are held in check by the presence of those less resistant (these less resistant cells are more fit in the absence of the therapeutic selective pressure), in a form of niche and resource competition. However, elimination of normal and low-resistant competitors (e.g., by therapy) releases the more resistant cancer cells from this competition, allowing for the outgrowth of a resistant and virulent tumor.
An example using anti-microbial chemotherapy is illustrative [60]. High dose therapy has trade-offs. On the one hand, it attempts to select against mutational development of resistance; on the other hand; it more effectively eliminates less resistant pathogens, facilitating competitive release and expansion of highly resistant strains [60]. Low dose therapy would have the opposite effect; it would preserve less resistant competitors (good) but provide greater opportunity for the development of resistance (bad). Intermediate dosing, which is the most common clinical choice (as the “middle ground” between potential toxicity of higher doses and decreased effectiveness of lower doses), is the worst choice, as it both facilitates mutational resistance and facilitates competitive release by eliminating more benign microbial competitors [60]. Thus, optimal dosing typically lies at the extremes of the continuum (high or low), with the choice dependent on context [60]. This principle applies to cancer, and has been demonstrated experimentally [58,59].
The underlying evolutionary rationale is that highly chemoresistant cancer cells incur costs (resources, energy, etc.) to maintain resistance and as such are less fit than less resistant cells in the absence of the chemotherapeutic selection. Similarly, by analogy, researchers who perform plasmid preps understand that the bacteria must be grown under antibiotic selection to retain the plasmid; otherwise, bacteria that have lost the plasmid will overgrow the others, since they no longer incur the (unnecessary in the absence of antibiotic selection) cost of reproducing the plasmid along with their own genome. For cancer therapy, it is therefore optimal to maintain a population of therapy-sensitive cells to restrain resistant outgrowth via intratumor competition [58]. Thus, maximal dose therapy, which would eliminate the less-resistant and non-resistant populations, is to be avoided, and replaced by flexible, minimal regimens that restrain tumor growth without facilitating competitive release of more resistant strains from the intratumor competition. This approach was experimentally verified in a orthotopic mouse model of breast cancer [59].
Obviously, therapies, such as benign boosters, that enhance the survival of normal cells, will constrain competitive release of more virulent and resistant neoplastic strains. One caveat, based on the hypothesis outlined in the current manuscript, is whether enhanced niche competition of normal vs. neoplastic cells would select against vertical transmission of neoplastic cells (e.g., primary growth in situ) and select for horizontal transmission (i.e., metastasis), as the cancer cells attempt to escape local competition by spreading elsewhere.
One could invoke a type of “sink-source” dynamics, in which overpopulation of the higher quality niche space (the “source” that is the location of the primary tumor) due to local competition (normal cell-to-cancer cell, as well as intratumor, competition) leads to migration (metastasis) of cancer cells to lower quality habitats (the “sink” that is the sites of metastasis to which the cells need to become adapted). One can suggest that benign boosting is therefore required not only at the site of the primary tumor but at the likely sites of metastasis. One could also speculate that the concept of benign boosting can not only include boosting normal over neoplastic cells, but, in those cases where it is not feasible to eliminate tumor cells, to boost less virulent neoplastic cells over those more virulent, and to, in a related sense, boost vertical over horizontal transmission. Another therapeutic possibility is a variation of the “ecological trap” idea, in which rapid environmental change “tricks” organisms to prefer sub-optimal “sink” habitats. Thus, if horizontal transmission is unavoidable in some cases (or at least potentially unavoidable), environmental change, in this case specific therapeutic applications, could be used to “trick” metastatic cells to migrate to suboptimal locations at which they would be less efficient in establishing a metastasis and/or be more vulnerable to follow-up therapeutic targeting.
Therefore, the hypothesis presented in our work adds another layer of complexity to the question of optimal therapeutics, given that strategies that affect relative fitness of cells at the primary site (vertical) could in theory inadvertently select for cells (horizontal) that metastasize to escape enforced intratumor competition.
However, when considering intratumor competition, we must understand that we are, in essence, “aiming at a moving target.” For example, a study on the evolutionary dynamics of non-small-cell lung cancer [61] demonstrated that ongoing chromosomal instability, as well as genome doubling, was positively associated with an increased risk of cancer recurrence or death [61]. Note that this analysis was done on cells isolated before systemic therapy, with the finding that mechanisms generating intratumor heterogeneity was “almost universal” [61]. These processes do not end upon initiation of therapy, and can be a driver of adaption of neoplastic cells, thus initiating horizontal transmission modes to colonize host areas of lesser initial competition for resources. Generation of further intratumor heterogeneity will furnish the tumor population with those types that can evade local intratumor competition, as well as competition from normal cells.
In addition, therapeutics that attempt to contain virulence by altering environmental factors influencing cell behavior may run into the problems inherent in the game theory model discussed above. For example, consider a mixed population of cell types, some of which are dependent on some factor produced by others. Reducing serum levels of that factor may result in short-term benefit (e.g., decreased tumor volume via impaired growth) but may fail long-term (recurrence) because the treatment simply shifts the equilibrium in favor of the producer cells [35].
Oncolytic viruses are another approach with promise, since the viruses are evolved entities capable of adaption in response to selective pressures [22 and refs therein]. Thus, if neoplastic cells develop resistance to the viruses, the viruses may adapt to restore efficacy; further, it would be possible to utilize exogenously-applied selective pressures to develop viruses with traits more beneficial for therapeutic utility [22]. One potential problem is adaption of the viruses to infect normal cells, but this has so far not been identified as a problem in model systems [22 and refs. therein]. Many of these approaches would be complicated if adaptive mutation, particularly in its quantum mechanical form [28], affected the adaptive landscape (see above).
In addition, control of gut microbiota may be another therapeutic or even preventive option to modulate gastrointestinal cancer virulence. In addition to the well-known role of H. pylori in gastric cancer, several bacterial species are associated with colorectal cancer, including but not limited to S. bovis and F. nucleatum [62]. With respect to the association between microbiota and tumorigenesis, microbial virulence may be mechanistically and causally connected to cancer virulence. Thus, factors expressed by pathogenic microbes that enhance their virulence can stimulate inflammation, activate signaling pathways in colonic cells, induce DNA damage in colonic cells, alter colonic cell proliferation and apoptosis, etc., which all can contribute to enhanced neoplastic virulence [62 and refs. therein]. Modification of the gut microbiota therefore can constitute a powerful approach to alter the cellular microenvironment to select for decreased tumor virulence. Further, if primary tumor removal, and the elimination of vertical transmission, releases the brakes on metastatic horizontal transmission [57], then perhaps treatments to prevent metastatic growth should be more routine even in cases (e.g., very early stage primary tumors) where metastasis is thought to be highly unlikely. This requires further study and careful evaluation of costs vs. benefits.
The suggested approaches are presented to initiate evaluation of the utility of modeling carcinogenesis from the perspective of host-pathogen interaction. As understanding of these principles, and their practical applications increases, it may be possible to optimize more powerful approaches for the prevention and treatment of cancer. Further, an understanding of the causes and prevention of cancer virulence, with an emphasis on suppressing horizontal transmission, can inform strategies to adjust perioperative treatment approaches to prevent metastasis secondary to surgical intervention [63]. Blocking metastasis, e.g., via various treatments aimed at downregulating metastatic gene expression and signaling pathways [4,5], thus preventing horizontal transmission of the cancer, would possibly promote its vertical transmission instead. According to our fundamental hypothesis, this would select for decreased cancer virulence.
In summary, the fundamental hypothesis under consideration in this manuscript can inform optimized treatment options for cancer.
Conclusion
Cancer can be viewed as the result of an evolutionary mismatch between the environments humans find themselves in today and the environments in which we evolved and for which our genomes are adapted [64]. Cancer is in another sense an atavistic phenomenon, in which neoplastic cells behave selfishly to promote their own fitness without the constraints of cooperative multicellularity; hence, they behave, in the adaptive sense, akin to single-celled independent organisms [64], or relevant to the current paper, akin to an infectious pathogen. Thus, the overarching hypothesis of this manuscript is that the host-pathogen relationship can be used to describe patient-cancer interactions; the human body is an ecosystem in which normal and neoplastic cells interact under selective pressures, increasing or decreasing cancer virulence. Therefore, host-pathogen modeling of transmissibility vs. virulence, horizontal vs. vertical transmission, soft vs. hard selection, kin selection and cooperation can be utilized to better understand carcinogenesis and to devise better preventive and therapeutic approaches for human cancer.
Abbreviations
DFTD: Tasmanian devil facial tumor disease; CTVT: canine transmissible venereal tumor; CRC: colorectal cancer.
Acknowledgements
MB is supported by funding from Geisinger Commonwealth School of Medicine.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lipsitch M, Moxon ER. Virulence and transmissibility of pathogens: what is the relationship? Trends Microbiol. 1997;5:31-7
2. Ebert D, Bull JJ. Challenging the trade-off model for the evolution of virulence: is virulence management feasible? Trends Microbiol. 2003;11:15-20
3. Goldenberg DM, Zagzag D, Heselmeyer-Haddad KM. et al. Horizontal transmission and retention of malignancy, as well as functional human genes, after spontaneous fusion of human glioblastoma and hamster host cells in vivo. Int J Cancer. 2012;131:49-58
4. Das S, Samant RS, Shevde LA. The Hedgehog Pathway Conditions the Bone Microenvironment for Osteolytic Metastasis of Breast Cancer. Int J Breast Cancer. 2012;2012:298623
5. Hoshino A, Costa-Silva B, Shen TL. et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329-35
6. Brandt A, Bermejo JL, Sundquist J. et al. Age of onset in familial cancer. Ann Oncol. 2008;19:2084-8
7. Strakova A, Murchison EP. The cancer which survived: insights from the genome of an 11000 year-old cancer. Curr Opin Genet Dev. 2015;30:49-55
8. Murchison EP. Clonally transmissible cancers in dogs and Tasmanian devils. Oncogene. 2008;27(Suppl 2):S19-30
9. Metzger MJ, Reinisch C, Sherry J. et al. Horizontal transmission of clonal cancer cells causes leukemia in soft-shell clams. Cell. 2015;161:255-63
10. Hamede RK, Pearse AM, Swift K. et al. Transmissible cancer in Tasmanian devils: localized lineage replacement and host population response. Proc Biol Sci. 2015; 282[1814]. pii. 2015 1468
11. Siddle HV, Kaufman J. A tale of two tumours: Comparison of the immune escape strategies of contagious cancers. Mol Immunol. 2013;55:190-3
12. Gärtner HV, Seidl C, Luckenbach C. et al. Genetic analysis of a sarcoma accidentally transplanted from a patient to a surgeon. N Engl J Med. 1996;335:1494-6
13. Nguyen DX, Massague J. Genetic determinants of cancer metastasis. Nat Rev Genet. 2007;8:341-52
14. Rousseaux S, Debernardi A, Jacquiau B. et al. Ectopic activation of germline and placental genes identifies aggressive metastasis-prone lung cancers. Sci Transl Med. 2013;5:186ra66. doi: 10.1126/scitranslmed.3005723
15. Read AF, Baigent SJ, Powers C. et al. Imperfect vaccination can enhance the transmission of highly virulent pathogens. PLoS Biol. 2015;13:e1002198
16. Elliot SL. The virulence-transmission trade-off and virulence management. Trends Microbiol. 2003;11:205-6 author reply 208-9
17. Gandon S, Day T. Understanding and managing pathogen evolution: a way forward. Trends Microbiol. 2003;11:206-7 author reply 208-9
18. Ebert D, Bull JJ. Response to: Eliot and Gandon and Day: Revisiting virulence management. Trends Microbiol. 2003;11:208-9
19. Goodell MA, Rando TA. Stem cells and healthy aging. Science. 2015:350 1199-1204
20. Pepper JW, Sprouffske K, Maley CC. Animal cell differentiation patterns suppress somatic evolution. PLoS Comp Biol. 2007;3:e250
21. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23-8
22. Pepper JW, Findlay CS, Kassen R. et al. Cancer research meets evolutionary biology. Evol Applic. 2009;2:62-70
23. Buckling A, Brockhurst MA. Kin selection and the evolution of virulence. Heredity (Edinb). 2008;100:484-8
24. Peng YF, Gu J. Synchronous colorectal and lung cancer: Report of three cases. World J Gastroenterol. 2008;14:969-73
25. Lochhead P, Chan AT, Nishihara R. et al. Etiologic field effect: reappraisal of the field effect concept in cancer predisposition and progression. Mod Pathol. 2015;28:14-29
26. Moreno E. Is cell competition relevant to cancer? Nature Reviews Cancer. 2008;8:141-7
27. Vale PF. Killing them softly: managing pathogen polymorphism and virulence in spatially variable environments. Trends in Parasit. 2013;29:417-22
28. Aguirre-Ghiso J. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7:834-46
29. Kleffel S, Schatton T. Tumor dormancy and cancer stem cells: two sides of the same coin. Adv Exp Med Biol. 2013;734:145-79
30. Stanley GA, Balani JP, Miller DS. et al. Clinical evidence: metastases can metastasize. World J Oncol. 2012;3:138-41
31. Wagenblast E, Soto M, Gutiérrez-Ángel S. et al. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature. 2015;520:358-62
32. Archetti M, Ferraro DA, Christofori G. Heterogeneity for IGF-II production maintained by public goods dynamics in neuroendocrine pancreatic cancer. Proc Natl Acad Sci USA. 2015;112:1833-8
33. Nowak MA, Tarnita CE, Antal T. Evolutionary dynamics in structured populations. Phil Trans R Soc Lond B Biol Sci. 2010;365:19-30
34. Archetti M, Scheuring I. Review: Game theory of public goods in one-shot social dilemmas without assortment. J Theor Biol. 2012;299:9-20
35. Perc M, Gómez-Gardeñes J, Szolnoki A. et al. Evolutionary dynamics of group interactions on structured populations: A review. J R Soc Interface. 2013;10:20120997
36. Bordonaro M, Ogryzko V. Quantum biology at the cellular level-elements of the research program. Biosystems. 2013;112:11-30
37. Hara T, Kouno J, Nakamura K. et al. Possible role of adaptive mutation in resistance to antiandrogen in prostate cancer cells. Prostate. 2005;65:268-75
38. Mullighan CG, Phillips LA, Su X. et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322:1377-80
39. Rapp F. The role of the viral genome in oncogenesis. Cancer Res. 1968;28:1832-4
40. Rapp F. Westmoreland D. Do viruses cause cancer in man? CA Cancer J Clin. 1975;25:215-29
41. Rapp F. Viruses as an etiologic factor in cancer. Semin Oncol. 1976;3:49-63
42. Rapp F. Viral carcinogenesis. Int Rev Cytol Suppl. 1983;15:203-44
43. Rapp F. Current knowledge of mechanisms of viral carcinogenesis. Crit Rev Toxicol. 1984;13:197-204
44. Nevels M, Täuber B, Spruss T, Wolf H, Dobner T. "Hit-and-run" transformation by adenovirus oncogenes. J Virol. 2001;75:3089-94
45. Cole LJ, Nowell PC. Radiation carcinogenesis: the sequence of events. Science. 1965;150:1782-6
46. Nowell PC. Tumors as clonal proliferation. Virchows Arch B Cell Pathol. 1978;29:145-50
47. Albuquerque C, Breukel C, van der Luijt R, Fidalgo P, Lage P, Slors FJ, Leitão CN, Fodde R, Smits R. The 'just-right' signaling model: APC somatic mutations are selected based on a specific level of activation of the beta-catenin signaling cascade. Hum Mol Genet. 2002;11:1549-60
48. Segditsas S, Rowan AJ, Howarth K, Jones A, Leedham S, Wright NA, Gorman P, Chambers W, Domingo E, Roylance RR, Sawyer EJ, Sieber OM, Tomlinson IP. APC and the three-hit hypothesis. Oncogene. 2009;28:146-55
49. Lazarova D, Bordonaro M. ZEB1 Mediates Drug Resistance and EMT in p300-Deficient CRC. ZEB1 Mediates Drug Resistance and EMT in p300-Deficient CRC. J Cancer. 2017;8:1453-1459
50. Lazarova DL, Bordonaro M. Vimentin, colon cancer progression and resistance to butyrate and other HDACis. J Cell Mol Med. 2016;20:989-93
51. Bordonaro M, Lazarova D. Hypothesis: Obesity Is Associated with a Lower Mutation Threshold in Colon Cancer. J Cancer. 2015;6:825-31
52. Tscherne DM, García-Sastre A. Virulence determinants of pandemic influenza viruses. J Clin Invest. 2011;121:6-13
53. Alto NM, Orth K. Subversion of cell signaling by pathogens. Cold Spring Harb Perspect Biol. 2012;4:a006114. doi: 10.1101/cshperspect.a006114
54. Antonovics J, Wilson AJ, Forbes MR, Hauffe HC, Kallio ER, Leggett HC, Longdon B, Okamura B, Sait SM, Webster JP. The evolution of transmission mode. Philos Trans R Soc Lond B Biol Sci. 2017; 372(1719). pii. 2016 0083. doi: 10.1098/rstb.2016.0083
55. Cressler CE, McLeod DV, Rozins C, Van Den Hoogen J, Day T. The adaptive evolution of virulence: a review of theoretical predictions and empirical tests. Parasitology. 2016;143:915-30
56. van Niekerk G, Loos B, Nell T, Engelbrecht AM. Cancer tolerance, resistance, pathogenicity and virulence: deconstructing the disease state. Future Oncol. 2016;12:1369-80
57. Demicheli R, Retsky MW, Hrushesky WJ, Baum M, Gukas ID. The effects of surgery on tumor growth: a century of investigations. Ann Oncol. 2008;19:1821-8
58. Silva AS, Kam Y, Khin ZP, Minton SE, Gillies RJ, Gatenby RA. Evolutionary approaches to prolong progression-free survival in breast cancer. Cancer Res. 2012;72:6362-70
59. Enriquez-Navas PM, Kam Y, Das T, Hassan S, Silva A, Foroutan P, Ruiz E, Martinez G, Minton S, Gillies RJ, Gatenby RA. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci Transl Med. 2016;8:327ra24. doi: 10.1126/scitranslmed.aad7842
60. Day T, Read AF. Does High-Dose Antimicrobial Chemotherapy Prevent the Evolution of Resistance? PLoS Comput Biol. 2016;12:e1004689
61. Jamal-Hanjani M, Wilson GA, McGranahan N. et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med. 2017;376:2109-2121
62. Gagniere J, Raisch J, Veziant J. et al. Gut microbiota imbalance and colorectal cancer. World J Gastro. 2016;22:501-18
63. van der Bij GJ, Oosterling SJ, Beelen RH. et al. The perioperative period is an underutilized window of therapeutic opportunity in patients with colorectal cancer. Ann Surg. 2009;249:727-34
64. Greaves M. Evolutionary determinants of cancer. Cancer Discov. 2015;5:806-20
Author contact
![]() Corresponding author: Geisinger Commonwealth School of Medicine, 525 Pine Street, Scranton, PA 18509. Tel: 570-504-9646; Fax: 570-504-9636; Email: mbordonarogeisinger.edu
Corresponding author: Geisinger Commonwealth School of Medicine, 525 Pine Street, Scranton, PA 18509. Tel: 570-504-9646; Fax: 570-504-9636; Email: mbordonarogeisinger.edu
Citation styles
APA
Bordonaro, M. (2018). Hypothesis: Cancer Is a Disease of Evolved Trade-Offs Between Neoplastic Virulence and Transmission. Journal of Cancer, 9(10), 1707-1724. https://doi.org/10.7150/jca.24679.
ACS
Bordonaro, M. Hypothesis: Cancer Is a Disease of Evolved Trade-Offs Between Neoplastic Virulence and Transmission. J. Cancer 2018, 9 (10), 1707-1724. DOI: 10.7150/jca.24679.
NLM
Bordonaro M. Hypothesis: Cancer Is a Disease of Evolved Trade-Offs Between Neoplastic Virulence and Transmission. J Cancer 2018; 9(10):1707-1724. doi:10.7150/jca.24679. https://www.jcancer.org/v09p1707.htm
CSE
Bordonaro M. 2018. Hypothesis: Cancer Is a Disease of Evolved Trade-Offs Between Neoplastic Virulence and Transmission. J Cancer. 9(10):1707-1724.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.