Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(10):1846-1855. doi:10.7150/jca.21248 This issue Cite
Research Paper
Antagonistic role of GSK3 isoforms in glioma survival
Vidhi Vashishtha, Nupur Jinghan, Ajay K.Yadav ![]()
Cancer Genetics Laboratory, Dr. B.R. Ambedkar Center for Biomedical Research, University of Delhi (North Campus), Delhi- 110007, India
Received 2017-5-30; Accepted 2017-11-19; Published 2018-4-24
Citation:
Vashishtha V, Jinghan N, K.Yadav A. Antagonistic role of GSK3 isoforms in glioma survival. J Cancer 2018; 9(10):1846-1855. doi:10.7150/jca.21248. https://www.jcancer.org/v09p1846.htm
Other stylesAbstract
GSK3 (Glycogen Synthase Kinase-3) function in brain is contributed by two distinct gene GSK3 alpha and GSK3 beta. Present findings indicate that imbalance in between GSK3 alpha and beta isoform contributes oncogenesis. In gliomas, GSK3 isoform specific functions are different then as reported for melanoma, prostate cancer, lung cancer etc. Both the isoforms of GSK3 are inversely regulating hnRNPA1 (RNA binding protein) expression, subsequently affecting RNA alternative splicing (BIN1, RON, Mcl1, PKM) in gliomas. Elevated expression of c-Myc, hnRNPA1, Phospo-ERK1/2 and Cyclin D1 in GSK3 alpha knock down cells, resembles GSK3 beta isoform overexpressing glioma cells, promotes cell survival. HnRNPA1 dependent survival signaling pathway were elaborated using si RNA approach or by over expressing cloned hnRNPA1 gene in U87 glioma cells. Therefore, performed study empirically support GSK3β inhibition along with restoration of GSK3α would be a good strategy to target gliomas.
Keywords: Heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1), Alternative splicing, GSK3, Brain Tumor, ERK
Introduction
hnRNPA1, a member of the hnRNPA/B family, is aberrantly over expressed in various cancers. hnRNP family of proteins interact with newly derived mRNA transcript [1]. hnRNPA1 specifically bind to splicing silencer sequences and promote exon inclusion thus acting as splicing repressors [2]. hnRNPA1 play key steps in mRNA metabolism such as alternative splicing [3], mRNA export [4], translation [5], microRNA processing [6] and telomere maintenance [7]. Non- tissue specific gene de-arrangement by family of splice factors proteins is a leading cause for tumorogenesis [8]. EGFR dependent regulation of splice factor family [9] and hnRNA1 dependent Max splicing to develop therapeutic resistance in gliomas has been widely studied [10]. hnRNPA1 overexpression at the downstream level also interact preferentially with anti-apoptotic mRNA variants, for FGF dependent cell survival [11].
Two different GSK3 isoforms, where GSK3α is a resident of Chr19q13.2, and GSK3β from Chr 3q13.33, with 98% identical Kinase domain [12]. Both isoforms perform similar function and shows overlapping function in Alzheimer diseases progression, cell cycle and proliferation. Both isoform also play a key role in regulating APC dependent β-catenine phophorylation dependent degradation, is to regulate cell proliferation [13]. This redundancy or distinct function of GSK3 isoform could be varied depends entirely on cellular context, yet to be explore in transformed cells. Till date many reports, particularly in various cancer GSK3β is oncogenic in gliomas, whereas is tumor suppressor in certain epithelial cancer. However, recently few reports address GSK3α as therapeutic candidate in prostrate, melanoma, lung cancer.
Recently, we identified GSK3β to cooperate hnRNPA1 expression in U87 gliomas. hnRNPA1 overexpression modulates Pyruvate kinase (PKM) splicing, trigger Warburg effect in glycolytic tumors or glioblastomas. Here, we have reported that GSK3α and GSK3β isoforms are non-redundant, knocking down GSK3α transiently or stably using Lentiviral approach promote glioma survival, is functionally similar as observed in transiently transfected hnRNPA1.
Material & Method
Cell culture Material
U87, U373, LN229 and A172 cell lines (Generous support from Prof. Subrata Sinha, N.B.R.C, Manesar, Gurgaon (India)) were propagated in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum and antibiotic penicillin-streptomycin (PAN Biotech comp.) under 37˚C temperature and humidified chamber under 5% CO2 .
Antibodies
All western blots were developed using monoclonal antibodies such as anti-GSK3α/β and GSK3α/β-pTyr (Santacruz Biotechnology, Inc.), anti- GSK3 α (Abcam), hnRNPA1 (Santacruz Biotechnology, Inc. & Abcam), α-Tubulin (Santacruz Biotechnology, Inc.), phospho-ERK1/2 (Abcam), Cyclin D1 (Millipore), AKT1/2/3 (Santacruz Biotechnology, Inc.), c-MYC (Abcam)
Plasmid constructs
Total RNA was extracted from U87 cells; it was reverse transcribed using first strand cDNA (Bio-Rad Laboratories) as per the manufacturer's instructions. hnRNPA1 gene was PCR amplified, with annealing temperature(55°C) optimized for maximum product yield. The hemagglutinin (HA) tag was attached to the N terminus of hnRNPA1 by including the HA tag in the forward primer, carried out using specific primers: sense- 5'- AGGATCCATGACCatgtacccatacgatgttccagattacgctGGATCTAAGTCAGAGTCTCCTAAA-3'antisense- 5'- GGTCGACTTAAAATCTTCTGCCACTGCCAT -3' with BamH1 and Sal1 restriction endonuclease site (underlined), respectively, at their 5' and 3'end. The amplified products were then digested and cloned into BamH1 and Sal1 sites of pBABE mammalian expression vector.
Transient Transfections
U87 cells were grown to 80% confluence in 60mm dishes. Empty vector pBABE (100ng) and human hnRNPA1 - pBABE DNA (100ng & 200ng) was transfected into the cells using TransIT-LT1 transfection reagent (Mirus Bio LLC) as per the manufacturer's instructions. 48 h post- transfection, the lysates were prepared in cell lysis buffer containing phophatase inhibitor (Santa Cruz Biotechnology) and protease inhibitor cocktail (Abcam).
si RNA transfection and Lentiviral mediated knock down
U87 & A172 glioma cells were transfected next day after cell plating with specific siRNA against Gsk3α (10nM), Gsk3β (10nM), hnRNPA1 (10nM) (Santa Cruz Biotechnology, Inc.) as per protocol. In another experiment GSK3α specific Lentiviral (Santa Cruz Biotechnology, Inc.) for knock down studies were performed as per protocol. Lysates were made in lysis buffer (Cell Signaling Technology) containing protease inhibitor (Abcam) and phosphatase inhibitor (Santa Cruz Biotechnology, Inc.).
Cell Growth Assay
U87 MG CNT and GSK3α Lentiviral transfected stable cells were used to analyse the cell growth. Equally 1X104 cells (CNT & GSK3α RNA i) were plated in 4 dishes in duplicates. Cell were counted on 1st Day, 3rd Day, 5th Day, 7th Day in hemocytometer using Trypan Blue dye (0.1%), to exclude dead cell count[14].
Cyclohexamide treatment in Glioma cell
U87 cell were transfected after 17 hrs in freshly plated cells with si RNA against Gsk3α (10nM), Gsk3β (10nM), Akt (10nM) for 72 hrs, followed with cyclohexamide (100μg/ml). Lysates were made at different time scale (0hrs, 2hrs 4 hrs), in cell lysis buffer containing phosphatase inhibitors & protease inhibitor cocktail. All experiments were done three times (n=3), densitometry analysis were done using NIH ImageJ open access software, after normalizing each with representative loading control.
Over expression of GSK3β in U87 & U373 Glioma cells in comparison with empty vector
U87 & U373 cells were grown in DMEM medium (10% FCS, antibiotic) transfected after 17 hours of freshly plate cells with GSK3β (500ng) in pCDNA plasmid construct (Gifted Prof Justin V. McCarthy, Ireland) and empty vector (500ng) using TransIT-LT1 transfection reagent (Mirus Bio LLC). After 48 hours of transfection protein lysates were prepared in cell lysis buffer and protein expression were performed by western blotting.
qRT-PCR & RT-PCR
U87 cells transfected as mentioned above: scrambled siRNA (10nM), siGsk3α (10nM), siGsk3β (10nM) for 72hrs, followed with extraction of total RNA using TRIzol Reagent. 1μg of total RNA was reverse transcribed using 2-STEP RT PCR kit (Invitrogen comp.) following the manufacturer's protocol. Gene was amplified using specific forward and reverse primer [15] (Table S1). PCR cycling conditions were as follows: 98ºC initial denaturation for 30 sec. 35 cycles of 98 ºC denaturation for 10 sec, different annealing temperature for different genes (Table S1) for 30 sec, and 72 ºC extension for 30 sec. 72ºC Final extension for 7 mins and final hold at 4ºC.
Similarly, hnRNPA1 transcript in U87 & LN229 cells were analysed using Taqman primer probe (Applied Biosystem) with specific control 18s rRNA (Invitrogen) and mRNA expression of genes (Bcl-xL, c-Myc, surviving & Cyclins) were analyzed with the help of SYBR green (Eurogentec) as followed with Real time PCR (Applied Biosystems) n- fold analysis, after calculating the ct value. Each experiment was done and analyzed in triplicate.
Caspase-3/CPP32 Colorimetric Assay and Cell Viability Assay in glioma cells
U87 & LN229 cells were transfected after 17 hours of freshly plated cells with specific siRNA's (CNT (10nM), Gsk3α (10nM), Gsk3β (10nM) and hnRNPA1 (10nM)). After 72 hours of transfection cell lystaes were made using cell lysis buffer. Protein quantification was performed using BCA reagent (Bicinchoninic Acid) (GeNei). 100μg of protein was taken for Caspase-3 colorimetric assay (BioVision) as per manufacture's protocol, measure at 400nm. In another set of experiment, cell viability was assessed using MTT colorimetric assay, based on the ability of viable cells to reduce yellow MTT (3-(4-5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide, a tetrazole) to purple formazan product, absorbance measures at 570 nm. Experiment was performed in triplicate.
Statistical Analysis
Numerical values obtained from individual experiments were expressed here, as mean ± standard error of the mean, student T-test.
Results
Analysis of GSK3α, AKT and hnRNPA1 in glioma cells
Protein lysates from U87, U373, LN229 & A172 glioma cells were taken to study the comparative expression of GSK3, AKT, and hnRNPA1 protein. A172 comparatively expresses higher GSK3α and low hnRNPA1, where as U373 and U87 expresses less GSK3α (Fig.1). LN229 and U87 expresses high level of AKT kinase, were used in subsequent studies, for hnRNPA1 stability.
GSK3α knock down promotes survival signaling pathway
Knocking down of GSK3α with two doses of siRNA viz. 6nM & 10nM in U87 & A172 Glioma cells augments phosphorylation at GSK3β (Tyr216) activation and phosphorylation at GSK3 alpha (Ser 9) is inactivation, resulted increase in expression of hnRNPA1, c-Myc, p-ERK1/2 & Cyclin D1 (Fig.2A). Similarly in A172 glioma cells expression of hnRNPA1, c-Myc, p-ERK1/2 & Cyclin D1 were prominently higher in GSK3α knock down cells at high dose (Fig.2B). We also examined and compared proliferation rate of U87MG control and U87-GSK3α RNAi (lentiviral transduced) cells. From here our data indicate that U87-GSK3α RNAi stable cells proliferated many fold higher than U87MG control cells, additionally compared proliferation rate of cell after incubating cells with Temozolomide -alkylating drug (100µM) (Fig. 2C). Under TMZ stress, GSK3 alpha knock down cells delayed Temozolomide dependent inhibition in growth of cells. Cell counting was performed using Hemocytometer at different days (0, 1, 5 7). Then similar experiment was performed where cytoplasmic and nuclear extracts were made for normal cells as well as after treating with Temozolomide for 48 hrs. Interestingly, translocation of phosphorylated form of ERK1/2 kinase from cytoplasm to nucleus is relatively very high. along with nucleus translocation effect on cMYC and beta catenine was observed in Temozolomide treated cells (Fig. 2D). This all making sense as reported previously, that MAPKinase signaling and β-catenin proto-oncogene in case of Glioma promote proliferation and cancer stemness [47, 48].
Figure 1
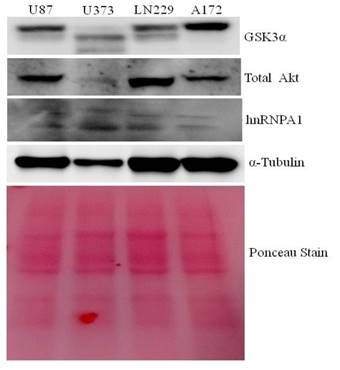
Comparative expression of GSK3, Akt kinase and hnRNPA1 in glioma cell lines: Glioma cell lines (U87, U373, LN229 and A172) protein Lysate were evaluated by western blotting for GSK3, Akt & hnRNPA1. Ponceau staining was performed for equal loading control.
Over-expression of GSK3β or siRNA against Gsk3α and Gsk3β kinase in glioma cell
hnRNPA1 expression was assessed in transiently transfected GSK3β plasmid and Empty vector in U87 & U373 cells, where we have observed GSK3β dependent increase in hnRNPA1 expression, with decrease in GSK3α expression (Fig.3A), reveals auto regulatory effect of hnRNPA1 between GSK3(α/β) isoforms. Further-more validated by measuring the stability of hnRNPA1 in Gsk3α and Gsk3β knock down cells at protein level, using cycloheximide (100μg/ml) treatment at different time points (0, 2 & 4hr). Stability of HnRNPA1 is very much reduced in Gsk3β knock down as well as in Akt knock down cells, whereas hnRNPA1 is comparatively stable in Gsk3α knock down cells (Fig.3B). Gene knock down effect was phenotypically observed and pictures were taken under phase contrast microscope, with or without cyclohexamide (100μg/ml) treatment before making lysates. More number of stressed cells or loss of spindle shape phenotype were visualized in Gsk3beta knock down cells compared to Gsk3 alpha knock down U87 cells (Fig S3). Whereas, hnRNPA1 dependent signalling cascade after silencing Gsk3alpha or Gsk3 beta in MCF7 cells (Breast Cancer cells), is not significantly antagonistic in nature, as we observed in glioma cells (Fig S2).
In another set of experiment using qRT-PCR approach hnRNPA1 mRNA transcript is relatively much higher in Gsk3α knock down cells however is significantly reduced in Gsk3 β knock down cells (Fig.4A). Therefore, hnRNPA1 expression dependent alternative splicing in BIN1, RON, Mcl-1 & PKM (Pyruvate Kinase) were evaluated in Gsk3α and Gsk3 β knock down U87 glioma cells. Expression of anti apoptotic variants of BIN1 and MCL-1 along with apoptotic splice variants of RON are therefore decreased in Gsk3α knock down cells, whereas is oppositely regulated in GSK3β knock down cells. Moreover, ratio PKM2/PKM1 (Pyruvate kinase) is also getting higher in Gsk3α knock down cells, marker for glycolytic pathway (Fig.3C).
Expression of survival genes in Gsk3α & Gsk3β knock down glioma cells
Proto-oncogene c-Myc co-operates BCL-XL in gliomagenesis. Moreover, Survivin is a potential cancer biomarker of IAP family member, inhibits apoptosis. These all three genes were significantly much higher in Gsk3 alpha knock down cells and are less in Gsk3β knock down cells (Fig 4B). Also observed high mRNA transcript of differentially expressed Cyclin's (B1, D1, E1, E2) in Gsk3α knock down & Gsk3β over expressing cells, however is much lesser in Gsk3β knock down or hnRNPA1 knock down U87 cells (Fig 4C). Satisfactory knock down of Gsk3α and Gsk3β, hnRNPA1 and over expression of Gsk3β at mRNA level expression were evaluated by real time- qRT-PCR (Fig.S1).
HnRNPA1 gene cloned in expression vector was transfected in U87 cells to study survival signaling cascade. Our results demonstrated increase in phospho-ERK1/2, phospho-AKT and c-Myc (Fig.4D), thus felicitate survival signaling cascade. Furthermore, expression of GSK3α decreases in hnRNPA1 over-expressing cells, indicating autoregulation between GSK3α/β and hnRNPA1.
Comparison of survival signaling and apoptosis in GSK3α and hnRNPA1 knock down cells
p70S6K acts downstream of the mammalian target of rapamycin (mTOR). Activation of p70S6K and its downstream target ribosomal protein S6Kinase mediates nutrient and mitogen-stimulated translation, and is essential for cell growth and proliferation [16]. From our study, we observed activation of p70S6Kinase in GSK3α knock down cells where as was downregulated in hnRNPA1 knockdown cells, along with apparent increases GSK3α (Fig 5A), thereby affecting cell viability (Fig 5B) and apoptosis in Gsk3α and hnRNPA1 knock down cells (Fig.5 C).
Consistent with our previous results, our study indicates oncogenic hnRNPA1 expression is depend upon the absence of GSK3 alpha and over expressed GSK3 beta in glioma cell.
Discussion
GSK3, a regulatory Ser/Thr protein kinase, exists in two distinct isoforms, GSK3α and GSK3β [17]. AKT kinase dependent GSK3α/β signaling network in various cancers is interpreted in varied outcomes, more likely to be a survival, differentiation, migration, apoptosis etc [18, 19]. Interfering directly GSK3α/β in lung [20-22] or prostate cancer [23, 24] promotes apoptosis. Under wide scale study, where selectively inhibiting GSK3 kinase in gliomas intended to be a promising candidate[25-28] promotes expression of c-Myc dependent apoptotic proteins, and induces cytotoxicity[29]. Majority of inhibitors used selectively against GSK3β kinase also have many off targets, includes GSK3α isoform, which is been reported with distinct apoptotic function as reported in various other cancers. Inhibition of GSK3β using AR-A014418 has been demonstrated to be a glioma inhibitory in xenograft mouse model [30, 31], where also found inactivation of GSK3α phospho-Tyrosine in U87 MG. From our previous study, inactivation of both GSK3β along with GSK3α kinase, enhances apoptosis could be because of dominant effect of GSK3β kinase in gliomas [32]. Initiating apoptosis with challenge of reoccurring event attempted us to study individual GSK3 isoform function. One of the most prominent observations of the present study is counteracting effect of GSK3α and β isofrom, where specific knock down GSK3α promote ERK1/2 activation & cell survival where as knocking down of GSK3β promotes apoptosis.
Figure 2
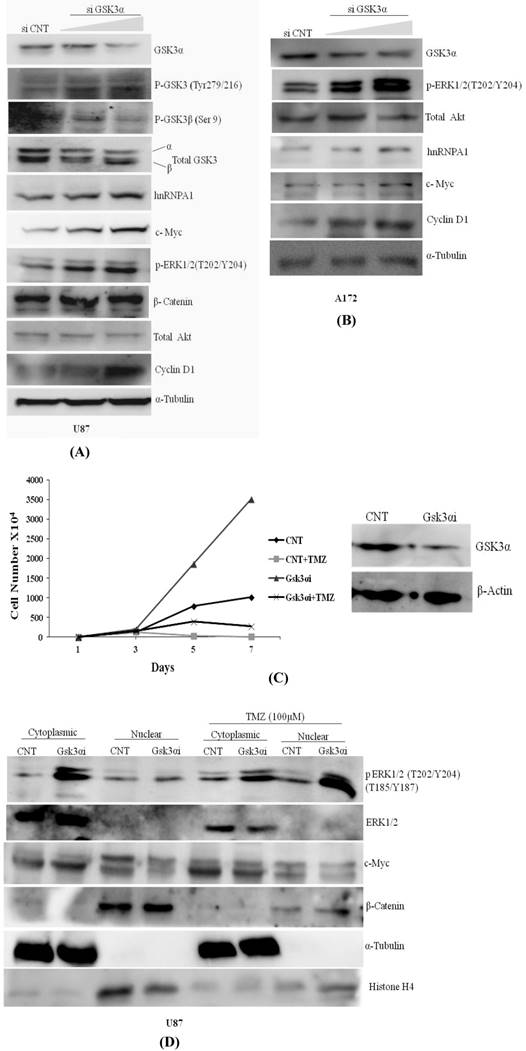
Knock down of GSK3α (6nM &10nM) elevates the expression of hnRNPA1, c-Myc, Cyclin D1, pTyr-GSK3: A) U87 MG, B) A172 gliomas, C) U87 MG CNT and GSK3 alpha Lentiviral transfected stable cells were plated in dishes (1X104/plate). Next day TMZ (100µM) were added in the cells. Cells were harvested on the indicated days (days 1-7) and counted using hemocytometer. Proliferation curves were generated by plotting the number of cells against number of days. D) U87 MG CNT and GSK3 alpha Lentiviral transfected stable cells were plated in dishes (1X104/plate). Next day TMZ (100µM) were added in the cells for 48 hrs and cytoplasmic and nuclear protein was extracted from the cells with the help of kit (Millipore). SDS gel was run followed with western blotting to check the expression of proteins.
Figure 3
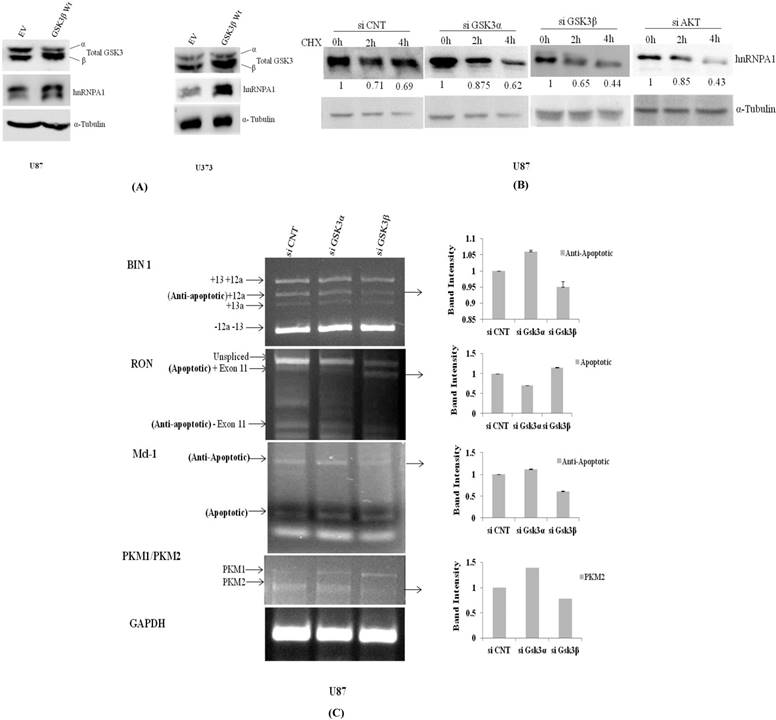
Transient over expression of GSK3β or knock down of GSK3α and GSK3β regulate expression of hnRNPA1: A) GSK3β (500ng) over expression in U87MG cells and U373 cells. B) Measured stability of hnRNPA1 in GSK3α (10nM) or GSK3β(10nM) knock down U87MG cells at different time point (0hr, 2hr, 4hr) of cycloheximide (100μg/ml) treatment. Image j available (NIH) software after normalizing each protein band with it respective loading control. C) Comparative mRNA alternative splicing analysis of various genes like Mcl1, BIN1, RON & PKM in GSK3α (10nM) and GSK3β (10nM) knock down U87 cells and GAPDH housekeeping gene for loading control. Densitometry was done using Image j available (NIH) software. (n=3, Mean±SE).
Many previous reports showed activated ERK1/2, is quite likely correlated with activated state of growth factor receptors [33], with inactivation of GSK3 kinase [34, 35]. Many evidences of ERK1/2 activation been reported in glioma, breast cancer, melanoma, head and neck cancer, lung cancer [36]. Phosphorylation of GSK3 beta kinase (Ser9) inactivates its regulatory apoptotic effect [37]. GSK3 was also proposed as a negative regulator of ERK1/2 in colon cancer [38] where both ERK1/2 and PI3kinase antagonizes GSK3β activity [39]. Phosphorylated ERK1/2 depending upon localization have diversified cytoplasmic and nuclear substrate, apparently involve in cell apoptosis or proliferation [40]. From our previous published study, we did observe inhibition of ERK1/2 activation in presence of GSK3 kinase specific inhibitors, increases apoptosis [32]. However, from our present study prominent activation of ERK1/2 and hnRNPA1 thereby increases cell proliferation and viability in GSK3α knock down cells. Similarly, in another set of experiment transient over expression of hnRNPA1 activates ERK1/2 and suppresses GSK3α expression promotes cell survival, comprehend our overall finding. Alteration in hnRNPA1 expression in glioma development context is interesting approach to look down if it is GSK3 dependent, where we observed that GSK3β cooperate with hnRNPA1 but not with GSK3α. Therefore, affecting mRNA splicing is interesting to understand to target the key pathways during glioma development.
Figure 4
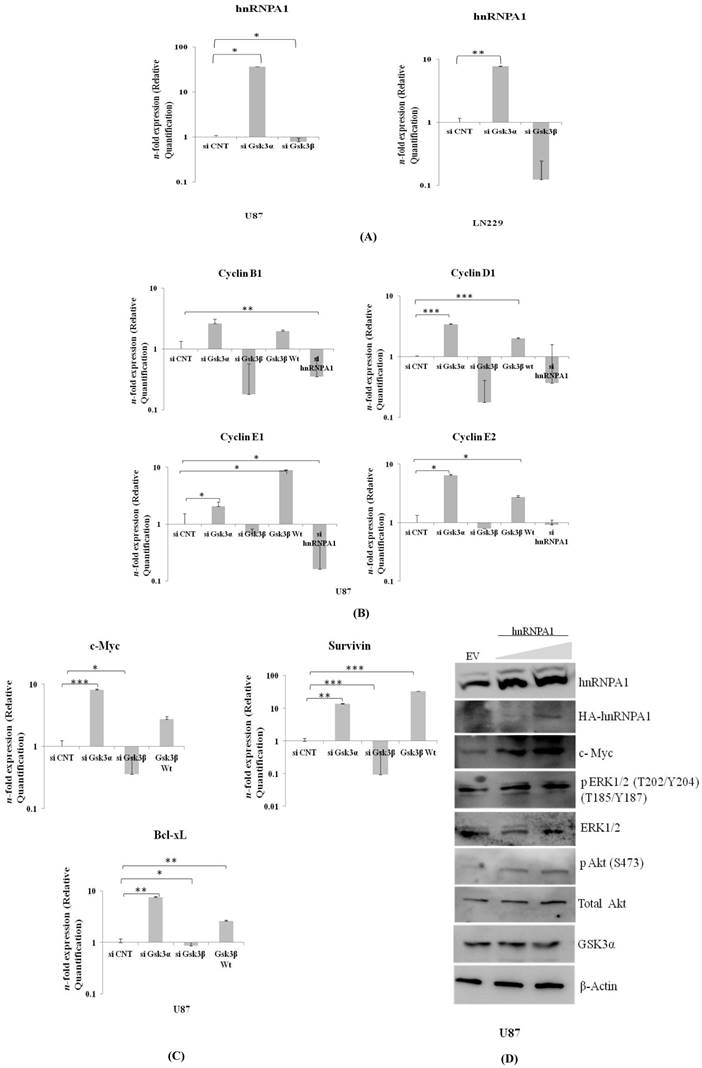
Expression of hnRNPA1 & Cell survival genes in GSK3α (10nM) & GSK3β (10nM) knock down: A) Expression of hnRNPA1 transcript in U87 & LN229, B) Cell survival genes c-Myc, Bcl-xL, Survivin were evaluated and compared with transfected U87-GSK3β (500ng) (n=3, Mean±SE). C) Expression of cyclin B1, cyclin D1, cyclin E1, cyclin E2. D) Transient over expression of hnRNPA1 (100ng & 200ng) activates survival pathway in U87 cells. Statistical analysis was performed. No significant p value >0.05, if p value <0.05 (*), <0.01(**), and <0.001(***) are considered significant.
Figure 5
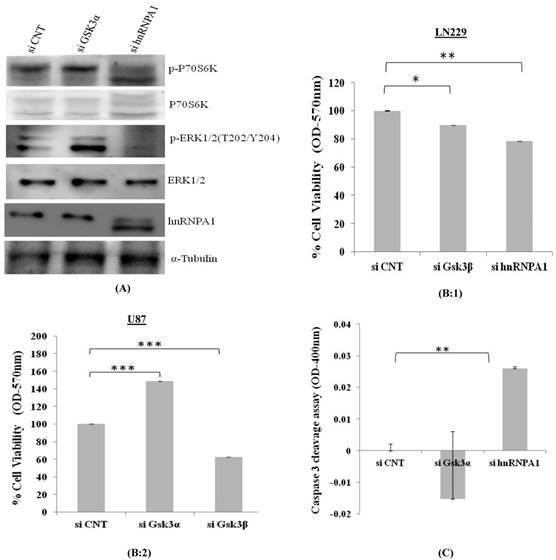
Cell viability and Caspase3 colorimetric assay in GSK3 knock down cells: A) Survival pathway were studied in LN229 cells, transfected with gene specific siRNA (GSK3α (10nM), hnRNPA1 (10nM)) compared with control siRNA (10nM). B) MTT based cell viability assay in siRNA transfected 1) LN229 and 2) U87 cells. Statistical analysis was performed. No significant p value >0.05, if p value <0.05 (*), <0.01(**), and <0.001(***) were considered significant. C) Caspase-3 colorimetric assay in siRNA transfected LN229 cells (OD at 400nm). Statistical analysis was performed. No significant p value >0.05, if p value <0.05 (*), <0.01(**), and <0.001(***) were considered significant.
Alternative splicing (AS) regulates gene expression and thereby introduces proteomic diversity [41]. Misregulation of AS is involved in cancer development and tumor maintenance [42]. Till date, AS event has been reported to be a challenging task in predicting the therapeutic gene targeted biology in gliomas, majority of gliomas reoccur with very low patient survival. Here, we have observed significant variation in the expression of hnRNP A1 after specific knockdown of GSK3α and β, which helped us to put forth our investigation to explore the alternative splicing pattern of few of the dominant genes known to undergo alternative splicing and contribute to tumorigenesis. We identified alternative splicing events modified by hnRNP A1, among them Mcl-1 an anti-apoptotic member of Bcl-2 family, undergoes AS event which produces two functionally distinct proteins, Mcl-1 (S) (pro-apoptotic) and Mcl-1 (L) (anti-apoptotic) [43, 44]. From our results, we have detected a presence of dominantly expressing anti-apoptotic transcript variant after GSK3 knockdown. In addition, several studies done in glioma's demonstrated higher expression of M2 isoform of pyruvate kinase (PK) than M1 isoform of PK expressed by normal tissues [45], thus limiting pyruvate production and activating aerobic glycolysis (Warburg effect) [46]. Mostly all glioma exhibits a high ratio of PKM2 to PKM1 RNA expression. The results of these studies are in agreement with our data in showing that after GSK3α down regulation, PKM2/PKM1 ratio is increased, indicating a possible role of GSK3α in regulation of glycolytic tumor. The present data suggests the antagonistic ability of both GSK3 isoform (α/β) in promoting apoptosis and cell viability, depending on the alternative splicing patterns of these genes modulated by hnRNPA1.
This suggests somewhere inhibiting GSK3β along with GSK3α restoration effect, could be best strategy to inhibit glioma progression because inhibiting GSK3 alpha turn out to promote splice factor expression, is promoting alternative apoptotic escape pathway.
Conclusion
Isoform specific role of GSK3α/β in gliomas has not yet been elucidated. Dissecting the role of GSK3α in glioma helped us to determine the contradictory function of both the isoforms. Silencing GSK3α led to activation of ERK1/2 as well as hnRNPA1, increases anti-apoptotic splice variants. Thus, proliferation of glioma cell increases. Our study has shed light to target GSK3 beta isoform along with restoration of GSK3 alpha in gliomas.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The authors are thankful to the Director of Dr. B.R Ambedkar Center for Biomedical Research, University of Delhi, India for research maintenance though intramural funding. This project was funded and supported by Prof. Ramalingaswami Fellowship of A.K.Y, from Department of Biotechnology, New Delhi, Research & Development and DST-Purse funding facility from University of Delhi (INDIA). This project is also supported by Science and Engineering Research Board, New Delhi (India).
Author Contribution
AKY conceived, coordinated the study, interpreted data and wrote the manuscript. V.V has done all experiments, N.J for technical assistance. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhou ZJ, Dai Z, Zhou SL. et al. Overexpression of hnrnp A1 promotes tumor invasion through regulating cd44v6 and indicates poor prognosis for hepatocellular carcinoma. International journal of cancer. 2013;132:1080-1089
2. He Y, Smith R. Nuclear functions of heterogeneous nuclear ribonucleoproteins a/b. Cellular and molecular life sciences. 2009;66:1239-1256
3. Chabot B, LeBel C, Hutchison S. et al. Heterogeneous nuclear ribonucleoprotein particle a/b proteins and the control of alternative splicing of the mammalian heterogeneous nuclear ribonucleoprotein particle a1 Pre-mRNA. Regulation of alternative splicing, Springer. 2003 pp 59-88
4. Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nature reviews Molecular cell biology. 2002;3:195-205
5. Cammas A, Pileur Fdr, Bonnal S. et al. Cytoplasmic relocalization of heterogeneous nuclear ribonucleoprotein a1 controls translation initiation of specific mrnas. Molecular biology of the cell. 2007;18:5048-5059
6. Guil S, CA¡ceres JF. The multifunctional rna-binding protein hnrnp a1 is required for processing of mir-18a. Nature structural & molecular biology. 2007;14:591-596
7. Zhang Q-S, Manche L, Xu R-M. et al. Hnrnp a1 associates with telomere ends and stimulates telomerase activity. Rna. 2006;12:1116-1128
8. David CJ, Manley JL. Alternative pre-mrna splicing regulation in cancer: Pathways and programs unhinged. Genes & development. 2010;24:2343-2364
9. Kostler WJ, Zeisel A, Korner C. et al. Epidermal growth-factor-induced transcript isoform variation drives mammary cell migration. PLoS One. 2013;8:e80566
10. Babic I, Anderson ES, Tanaka K. et al. EGFR mutation-induced alternative splicing of max contributes to growth of glycolytic tumors in brain cancer. Cell metabolism. 2013;17:1000-1008
11. Roy R, Durie D, Li H. et al. Hnrnpa1 couples nuclear export and translation of specific mrnas downstream of fgf-2/s6k2 signalling. Nucleic acids research. 2014 gku953
12. Rayasam GV, Tulasi VK, Sodhi R. et al. Glycogen synthase kinase 3: More than a namesake. British journal of pharmacology. 2009;156:885-898
13. Doble BW, Patel S, Wood GA. et al. Functional redundancy of gskα and gskβ in wnt-catenin signaling shown by using an allelic series of embryonic stem cell lines. Developmental cell. 2007;12:957-971
14. Yadav AK, Sahasrabuddhe AA, Dimri M. et al. Deletion analysis of bmi1 oncoprotein identifies its negative regulatory domain. Molecular cancer. 2010;9:158
15. Golan-Gerstl R, Cohen M, Shilo A. et al. Splicing factor hnrnp a2/b1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer research. 2011;71:4464-4472
16. Dhar R, Basu A. Constitutive activation of p70 s6 kinase is associated with intrinsic resistance to cisplatin. International journal of oncology. 2008;32:1133
17. McCubrey JA, Steelman LS, Bertrand FE. et al. Gsk-3 as potential target for therapeutic intervention in cancer. Oncotarget. 2014:2881-911
18. Rossig L, Badorff C, Holzmann Y. et al. Glycogen synthase kinase-3 couples akt-dependent signaling to the regulation of p21cip1 degradation. Journal of Biological Chemistry. 2002;277:9684-9689
19. Majewska E, Szeliga M. Akt/gsk3 signaling in glioblastoma. Neurochemical Research.:1-7.
20. Remsing Rix LL, Kuenzi BM, Luo Y. et al. Gsk3 alpha and beta are new functionally relevant targets of tivantinib in lung cancer cells. ACS chemical biology. 2014;9:353-358
21. Zeng J, Liu D, Qiu Z. et al. Gsk3β overexpression indicates poor prognosis and its inhibition reduces cell proliferation and survival of non-small cell lung cancer cells. PLoS One. 2014;9:e91231
22. Vincent EE, Elder DJE, Linda O. et al. Glycogen synthase kinase 3 protein kinase activity is frequently elevated in human non-small cell lung carcinoma and supports tumour cell proliferation. PloS One. 2014;9:e114725
23. Gao F, Al-Azayzih A, Somanath PR. Discrete functions of gsk3α and gsk3β isoforms in prostate tumor growth and micrometastasis. Oncotarget. 2015;6:5947
24. Li B, Thrasher JB, Terranova P. Glycogen synthase kinase-3: A potential preventive target for prostate cancer management. Urol Oncology. 2015 pp 456-463
25. Miyashita K, Kawakami K, Nakada M. et al. Potential therapeutic effect of glycogen synthase kinase 3β inhibition against human glioblastoma. Clinical Cancer Research. 2009;15:887-897
26. Zhao P, Li Q, Shi Z. et al. Gsk-3β regulates tumor growth and angiogenesis in human glioma cells. Oncotarget. 2015;6:31901
27. Li Y, Lu H, Huang Y. et al. Glycogen synthase kinases 3β controls differentiation of malignant glioma cells. International journal of cancer. 2010;127:1271-1282
28. Chikano Y, Domoto T, Furuta T. et al. Glycogen synthase kinase 3β sustains invasion of glioblastoma via the focal adhesion kinase, rac1, and c-jun n-terminal kinase-mediated pathway. Molecular cancer therapeutics. 2015;14:564-574
29. Kotliarova S, Pastorino S, Kovell LC. et al. Glycogen synthase kinase-3 inhibition induces glioma cell death through c-myc, nuclear factor-kappab, and glucose regulation. Cancer research. 2008;68:6643-6651
30. Cao Q, Lu X, Feng YJ. Glycogen synthase kinase-3β positively regulates the proliferation of human ovarian cancer cells. Cell research. 2006;16:671-677
31. Ougolkov AV, Fernandez-Zapico ME, Bilim VN. et al. Aberrant nuclear accumulation of glycogen synthase kinase-3β in human pancreatic cancer: Association with kinase activity and tumor dedifferentiation. Clinical Cancer Research. 2006;12:5074-5081
32. Yadav AK, Vashishta V, Joshi N. et al. AR-A014418 used against gsk3beta downregulates expression of hnrnpa1 and sf2/asf splicing factors. Journal of oncology. 2014;2014:695325
33. Katz M, Amit I, Yarden Y. Regulation of mapks by growth factors and receptor tyrosine kinases. Molecular Cell Research. 2007;1773:1161-1176
34. Cross DAE, Alessi DR, Cohen P. et al. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase b. Nature. 1995;378:785
35. Fang X, Yu SX, Lu Y. et al. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase a. Proceedings of the National Academy of Sciences. 2000;97:11960-11965
36. Caunt CJ, Sale MJ, Smith PD. et al. Mek1 and mek2 inhibitors and cancer therapy: The long and winding road. Nature reviews Cancer. 2015;15:577-592
37. Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3β by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochemical Journal. 1993;296:15-19
38. Wang Q, Zhou. Y, Wang X, et al. Glycogen synthase kinase-3 is a negative regulator of extracellular signal-regulated kinase. Oncogene. 2006;25:43-50
39. Hetman M, Hsuan SL, Habas A. et al. Erk1/2 antagonizes glycogen synthase kinase-3β induced apoptosis in cortical neurons. Journal of Biological Chemistry. 2002;277:49577-49584
40. Mebratu Y, Tesfaigzi Y. How erk1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell cycle. 2009;8:1168-1175
41. Oltean S, Bates DO. Hallmarks of alternative splicing in cancer. Oncogene. 2014;33:5311-5318
42. Venables JP. Aberrant and alternative splicing in cancer. Cancer research. 2004;64:7647-7654
43. Bae J, Leo CP, Hsu SY. et al. Mcl-1S, a splicing variant of the antiapoptotic bcl-2 family member mcl-1, encodes a proapoptotic protein possessing only the bh3 domain. Journal of Biological Chemistry. 2000;275:25255-25261
44. Quinn BA, Dash R, Azab B. et al. Targeting mcl-1 for the therapy of cancer. Expert opinion on investigational drugs. 2011;20:1397-1411
45. Mukherjee J, Phillips JJ, Zheng S. et al. Pyruvate kinase M2 expression, but not pyruvate kinase activity, is up-regulated in a grade-specific manner in human glioma. PloS one. 2013;8:e57610
46. Dong G, Mao Q, Xia W. et al. Pkm2 and cancer: The function of pkm2 beyond glycolysis (review). Oncology letters. 2016;11:1980-1986
47. Zong Z, Pang H, Yu R. et al. PCDH8 inhibits glioma cell proliferation by negatively regulating the AKT/GSK3β/β-catenin signalling pathway. Oncol Lett. 2017Sep;14(3):3357-3362
48. Kwon SJ, kwon OS, Kim KT. et al. Role of MEK partner-1 in cancer stemness through MEK/ERK pathway in cancerous neural stem cells, expressing EGFRviii. Mol Cancer. 2017Aug22;16(1):140
Author contact
![]() Corresponding author: Dr. Ajay K. Yadav, e-mail ID: ajay9774com, ayadavdu.ac.in.
Corresponding author: Dr. Ajay K. Yadav, e-mail ID: ajay9774com, ayadavdu.ac.in.
Citation styles
APA
Vashishtha, V., Jinghan, N., K.Yadav, A. (2018). Antagonistic role of GSK3 isoforms in glioma survival. Journal of Cancer, 9(10), 1846-1855. https://doi.org/10.7150/jca.21248.
ACS
Vashishtha, V.; Jinghan, N.; K.Yadav, A. Antagonistic role of GSK3 isoforms in glioma survival. J. Cancer 2018, 9 (10), 1846-1855. DOI: 10.7150/jca.21248.
NLM
Vashishtha V, Jinghan N, K.Yadav A. Antagonistic role of GSK3 isoforms in glioma survival. J Cancer 2018; 9(10):1846-1855. doi:10.7150/jca.21248. https://www.jcancer.org/v09p1846.htm
CSE
Vashishtha V, Jinghan N, K.Yadav A. 2018. Antagonistic role of GSK3 isoforms in glioma survival. J Cancer. 9(10):1846-1855.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.