Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(11):1885-1895. doi:10.7150/jca.23239 This issue Cite
Research Paper
Genomic and epigenetic signatures associated with survival rate in oral squamous cell carcinoma patients
Ilda Patrícia Ribeiro1,2, Francisco Caramelo3, Luísa Esteves1, Camila Oliveira1, Francisco Marques2,4,5, Leonor Barroso6, Joana Barbosa Melo1,2, Isabel Marques Carreira1,2 ![]()
1. Cytogenetics and Genomics Laboratory, Faculty of Medicine, University of Coimbra, 3000-354 Coimbra, Portugal
2. CIMAGO - Center of Investigation on Environment Genetics and Oncobiology - Faculty of Medicine, University of Coimbra, 3000-354 Coimbra, Portugal
3. Laboratory of Biostatistics and Medical Informatics, IBILI - Faculty of Medicine, University of Coimbra, 3000-354 Coimbra, Portugal
4. Department of Dentistry, Faculty of Medicine, University of Coimbra, 3000-075 Coimbra, Portugal
5. Stomatology Unit, Coimbra Hospital and University Centre, CHUC, EPE, 3000-075 Coimbra, Portugal
6. Maxillofacial Surgery Department, Coimbra Hospital and University Centre, CHUC, EPE, 3000-075 Coimbra, Portugal
Received 2017-10-9; Accepted 2018-2-17; Published 2018-4-27
Citation:
Ribeiro IP, Caramelo F, Esteves L, Oliveira C, Marques F, Barroso L, Melo JB, Carreira IM. Genomic and epigenetic signatures associated with survival rate in oral squamous cell carcinoma patients. J Cancer 2018; 9(11):1885-1895. doi:10.7150/jca.23239. https://www.jcancer.org/v09p1885.htm
Other stylesAbstract
Purpose: Although oral squamous cell carcinoma (OSCC) presents great mortality and morbidity worldwide, the mechanisms behind its clinical behavior remain unclear. Biomarkers are needed to forecast patients' survival and, among those patients undergoing curative therapy, which are more likely to develop tumor recurrence/metastasis. Demonstrating clinical relevance of these biomarkers could be crucial both for surveillance and in helping to establish adjuvant therapy strategies. We aimed to identify genomic and epigenetic biomarkers of OSCC prognosis as well as to explore a noninvasive strategy to perform its detection.
Methods: OSCC tumor and non-tumor tissue samples and cells scrapped from the tumor surface were genomic and epigenetically evaluated by Methylation-Specific Multiplex Ligation-dependent Probe Amplification technique.
Results: Copy number alterations in ATM, CASR, TP73, CADM1, RARB, CDH13, PAX5, RB1 genes and GATA5, PAX6, CADM1 and CHFR promoter methylation were shown to be associated with worse OSCC patients' survival. Copy number alterations in BRCA1, CDKN2A, CHFR, GATA5, PYCARD, STK11, TP53, VHL genes and GATA5, CADM1, KLLN, MSH6, PAX5, WT1 promoter methylation were shown to be associated with development of metastasis/relapses during or after OSCC patients' treatment. We also found a good agreement in the status of CDKN2A promoter methylation evaluated noninvasively or in the tumor tissue.
Conclusions: Genomic and epigenetic signatures were validated in a larger and geographically separate cohort, from TCGA database, which reinforce their clinical applicability. Noninvasive methodologies for detection of these signatures require further studies before translation in to clinical practice.
Keywords: Copy number alterations, methylation, biomarkers, OSCC survival, recurrence, TCGA data
Introduction
Oral cancer is the most common neoplasm of head and neck region, being squamous cell carcinoma (OSCC) the most frequent histological type. Although the oral cavity region is accessible to visual examination, oral cancer is still diagnosed at advanced stage when signs and symptoms are already present [1]. In contrast, in early stages oral malignant lesions are usually benign in appearance and, as they are often asymptomatic, patients tend to depreciate the small signs [2]. When oral cancer is detected at early stages, 5-year survival rate is around 90%, whereas in later stages the survival rate decreases to about 30% [3]. As so, clinical staging at the time of diagnosis is of utmost importance and can be used as a predictor of recurrence and mortality in oral cancer patients. On the other hand, the treatment of advanced tumors is frequently very mutilating, leading to visible deformations which, in turn, results in social stigmatization, speech disabilities and nutrition problems [4-6]. Additionally, the behavior of OSCC is difficult to predict using solely conventional clinical and histopathological parameters [7]. Accurate prognostic biomarkers should have immediate applicability on the clinical set, allowing to select patients for more effective tailored treatment strategies and conceivably for shorter monitoring programs. Tissue biopsy with histological assessment remains the gold standard in oral diagnosis, which needs a trained health-care provider, and is considered invasive, painful, expensive and time consuming [8]. Nowadays, several advancements have been made in the development of potentially useful diagnostic tools at the clinical and molecular level for early detection of oral cancer and its relapses, such as exfoliative cytology (consisting in cell collection from mucosal surfaces by scrapping or brushing) that presents as advantages the fact that it is painless, non-invasive, easy to perform and can be repeated several times to diagnose and follow-up the patients [9, 10]. Even so, non-invasive tools have not yet proved their value in clinical routine since they were unable to reduce the problem of late diagnosis [11].
In this study we identified a specific genomic and epigenetic profile associated with OSCC survival and risk of relapse/metastasis development and, consequently, with patients' prognosis. This genomic signature was validated using TCGA data, and may contribute for accurately stratifying patients for personalized clinical management. Additionally, we developed a noninvasive approach of identifying this genomic and epigenetic signature by evaluating in tumor samples and in cells scrapped from the tumor surface of the same OSCC patients through Methylation-Specific Multiplex Ligation-dependent Probe Amplification (MS-MLPA) technique. However, this noninvasive approach only showed good agreement for CDKN2A promoter methylation.
Materials and methods
Study population
The study protocol was approved by the Committee on Ethics in Research of the Faculty of Medicine of the University of Coimbra. All patients provided their written consent to participate in the study after being informed about the research purposes, following the regulations in the Declaration of Helsinki.
The study cohort includes tumor tissue, Tissue from surgery resection margin (macroscopically tumor-free tissue) and exfoliated cells, scrapped from the tumor surface, of 49 OSCC patients who underwent tumor resection. The surface of tumor, before surgery, was gentle scrapped with a blunt instrument to obtain exfoliated neoplastic cells. The material obtained was then taken in a falcon tube with phosphate buffered saline buffer and stored at 4ᵒC until DNA extraction, which was performed within one week after collection. The tissue samples were snap-frozen in liquid nitrogen within 30 min after resection and stored at -80ºC until use. The patients were recruited between October 2010 and January 2016 from the Maxillofacial Surgery Unit of the Coimbra Hospital and University Centre, CHUC, EPE, Portugal. Diagnosis and staging were performed in accordance with the American Joint Committee on Cancer TNM staging system. The participants in this study answered a survey regarding lifestyle and risk factors for upper aerodigestive tract malignancies, including alcohol and tobacco consumption. Patients were followed-up through hospital revisits during routine clinical appointments. The follow-up periods ranged from 1 to 63 months. Details of our study cohort are listed in Table 1. For the control group of tissue samples, gingival tissues from 16 healthy donors (6 males and 10 females, with ages ranging from 18 to 81 years) subjected to wisdom teeth removal were used. For the control group of tumor exfoliated cell samples, cells acquired by scrapping the oral surface of 14 healthy donors (4 males and 10 females, with ages ranging from 23 to 60 years) were used.
DNA extraction, HPV typing and MS-MLPA
DNA from fresh frozen tissues of patients and controls were extracted using a High Pure PCR Template Preparation Kit (Roche GmbH, Mannheim, Germany), according to the manufacturer's instructions. The DNAs were quantified by UV spectrophotometric analysis using a Nanodrop 1000 Spectrophotometer (Thermo Scientific, USA). All tumor tissue samples were analyzed for HPV infection as previous described [12, 13].
MS-MLPA analyses were performed using MS-MLPA probe set ME002 (MRC-Holland, Amsterdam, The Netherlands), which can simultaneously detect copy number alterations (CNAs) in 38 tumor suppressor genes and aberrant methylation patterns in a subset of 25 of these genes (Figure 1). All MS-MLPA reactions were performed according our previous work [14]. Three controls selected from the previously analyzed control group of 16 from tissue samples and 11 from exfoliated cells, without CNAs and methylation values below 20%, as well as a negative control (without DNA), were always included in each MS-MLPA assay. Binning of the raw data and comparative analyses were performed using Coffalyser.NET software. For each probe we determined the specific cutoff values for gain and loss, using 95% confidence intervals as determined on non-cancer subjects. A copy number gain was scored when a value exceeded 1.2 and a copy number loss was scored when a value was lower than 0.8. We considered a gene promoter as methylated when the methylation dosage ratio was ≥ 0.20, which means that at least 20% of the DNA was methylated. These cut-off values were based in our previous works [12-14].
Validation cohort from TCGA data portal
Copy number data obtained by SNP array and patients' clinical data of 314 OSCC were downloaded from the TCGA Data Portal, available at https://tcga-data.nci.nih.gov/tcga/, (accessed on the 23rd October 2015). The available copy number data was Level 3 data. The clinical-pathologic features of the validation cohort are listed in Table 2.
Statistical analysis
Data analysis was essentially divided in three parts: one focusing the relationship between genetic/epigenetic data and survival information, another attempting to obtain a genetic and epigenetic biomarker for relapses or metastases and, finally, a part regarding agreement between tissue samples and scrapped cells of the tumor surface.
Copy number and methylation data was obtained for tumor and non-tumor cells thus comparing the same genes between the two tissues, thus aiming to enlighten which genes are really contributing to tumoral features. This comparison was performed resorting to the difference between the entropy [15] computed for each gene, and genes that showed larger differences were considered to be significant to explain dissimilarities between tumor and non-tumor cells. Afterwards a factorial analysis, with two factors, was performed aiming at achieving a smaller group of genes and the relationship of them to each factor and to each observation. Additionally, a cluster analysis (two step method) was carried out and two different groups of observations were found. Finally, survival data was compared between these two groups using Kaplan-Meyer analysis and the Log Rank test.
Table 1
Clinical-pathologic characteristics of study population
| Patients (n=49) | |||
|---|---|---|---|
| n (%) | n (%) | ||
| Gender | Age at diagnosis (Years) | ||
| Male | 39 (80) | <60 | 24 (49) |
| Female | 10 (20) | ≥60 | 25 (51) |
| Anatomic Subsite | Invasion peri(neural) | ||
| Tongue | 26 (53) | Yes | 26 (53) |
| Floor of the mouth | 12 (25) | No | 20 (41) |
| Retromolar Trigone | 4 (8) | NA | 3 (6) |
| Jugal Mucosa | 2 (4) | Differentiation | |
| Palate | 2 (4) | Well | 38 (78) |
| Alveolar ridge | 3 (6) | Moderate | 9 (18) |
| Tobacco | Poor | 1 (2) | |
| Yes | 31 (63) | NA | 1 (2) |
| No | 15 (31) | Margins | |
| NA | 3 (6) | R0 | 29 (59) |
| Alcohol | R1 | 16 (33) | |
| Yes | 29 (59) | NA | 4 (8) |
| No | 15 (31) | HPV | |
| NA | 5 (10) | Positive | 1 (2) |
| TNM stage | Negative | 48 (98) | |
| I | 9 (18) | Vital status | |
| II | 14 (29) | Relapses/Metastasis in follow-up | 16 (33) |
| III | 7 (14) | Dead - OSCC | 14 (29) |
| IV | 19 (39) | Dead-non-OSCC | 1 (2) |
| Treatment | |||
| surgery alone | 13 (27) | ||
| Surgery + RT | 26 (53) | ||
| Surgery + RT + QT | 6 (12) | ||
| NA | 4 (8) | ||
NA- Not Available; RT - Radiotherapy; QT - Chemotherapy
Table 2
Clinical-pathologic characteristics of validation cohort from TCGA
| Patients (n=314) | |||
|---|---|---|---|
| n (%) | n (%) | ||
| Gender | Age at diagnosis (Years) | ||
| Male | 209 (67) | <60 | 133(42.4) |
| Female | 105 (33) | ≥60 | 180(57.3) |
| Anatomic Subsite | Invasion peri(neural) | ||
| Tongue | 131 (42) | Yes | 134(42.7) |
| Oral cavity | 73 (23) | No | 113(36) |
| Floor of the mouth | 63(20) | NA | 63(20.1) |
| Buccal Mucosa | 22(7) | Margins | |
| Alveolar Ridge | 18 (6) | R0 | 229(72.9) |
| Hard Palate | 7 (2) | R1 | 37(11.8) |
| Tobacco | Close | 34(10.8) | |
| Yes | 215 (68.5) | NA | 14(4.5) |
| No | 90 (28.6) | HPV | |
| NA | 9 (2.9) | Positive | 32 (10.2) |
| Alcohol | Negative | 281 (89.5) | |
| Yes | 203 (64,6) | NA | 1 (0.3) |
| No | 104 (33,1) | Country | |
| NA | 7 (2,3) | United States | 212 (67) |
| TNM stage | Canada | 37(11.8) | |
| I | 12 (3.8) | Brazil | 10(3.2) |
| II | 76 (24.2) | Poland | 9(2.9) |
| III | 63 (20) | Other | 9(2.9) |
| IV | 155 (49.4) | NA | 37(11.8) |
| NA | 8 (2.5) | Vital status | |
| Treatment | Relapses/Metastasis in follow-up | 189(60.2) | |
| QT | 82(26.1) | ||
| Immunotherapy | 2(0.6) | ||
| Targeted Molecular Therapy | 1(0.3) | ||
| NA | 229(72.9) | ||
NA- Not Available; RT - Radiotherapy; QT - Chemotherapy
Figure 1
Electropherograms from copy number alterations and methylation profile of (A) tumor, (B) non-tumor tissue and (C) control samples, obtained with the software GeneMapper v4.
In order to achieve a form of estimating the risk of relapses or metastases a classification algorithm was implemented. Variable importance plots based on Gini index [16] were employed to select the most informative genes and a Random Forest method was implemented with a k-fold cross validation technique.
Agreement between genetic data coming from tissues samples and scrapped cells was computed using Kappa statistics for each gene.
Statistical analysis was performed using R statistics platform (R version 3.3.2) and Matlab (R2016b, Matworks™). The statistical level of significance adopted was 0.05.
Results
Genomic signature associated with OSCC survival
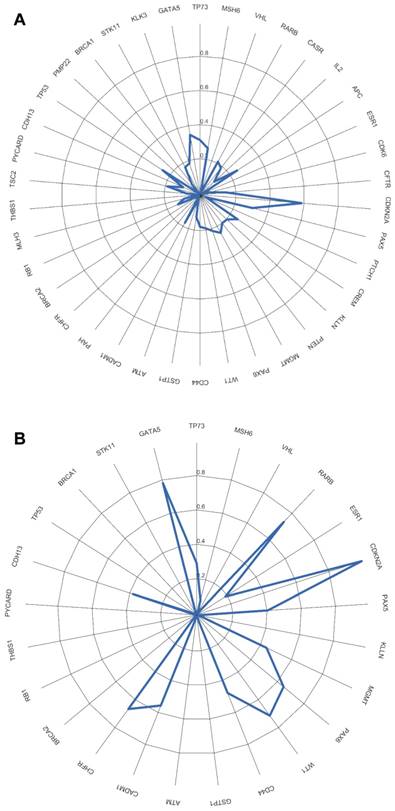
We evaluated whether the genetic differences between the tumor and non-tumor tissue samples could be used as survival predictors using entropy differences between the results of CNAs for the genes analyzed in these two kinds of samples. Firstly, we reduced data by selecting only the genes presenting a difference higher than 50%. The selected genes were: ATM, BRCA2, CADM1, CASR, CD44, CDH13, CDK6, CREM, ESR1, KLLN, MGMT, MLH3, PAX5, PAX6, PTEN, RARB, RB1, THBS, TP73 and WT1 (figure 2A).
Applying factorial and clusters analysis, we reached a division of our cohort into two clusters with a Cronbach alpha of 0.781 (internal consistency) and a silhouette Coefficient of 0.7 (cluster consistency). The genes that most contribute for this division were ATM, CASR, TP73, CADM1, RARB, CDH13, PAX5 and RB1.
Cluster 1 presented a proportion of 71% (35 patients) and cluster 2 of 29% (14 patients).
The genomic signature of these two clusters is different, cluster 1 did not presented CNA in the selected genes and cluster 2 presented gains and losses in these genes (figure 2B).
Kaplan-Meier curves for the two clusters, considering cluster 1 with all patients without CNAs for the selected genes and cluster 2 with patients that have at least one gene with CNA were performed. We observed that the average survival for cluster 1 was 48.2 months (CI95% [39.5; 56.9]) whereas for cluster 2 was 40.9 months (CI95% [25.2; 56.6]) (figure 3A). We did not reach statistical significance (Log-Rank: p=0.243).
A validation of these results using TCGA data was performed, including 58 patients in cluster 1 and 254 in cluster 2. In this validation cohort, the average survival for cluster 1 was 98.6 months and for cluster 2 was 79.9 months (figure 3B). In this validation cohort it was possible to observe 18.7 months of survival difference between the two clusters, which is higher than those observed in our cohort (7.3 months). Despite, this clinical relevant difference, we did not find statistical significance (Log-Rank: p=0.377).
Considering the observed clinical relevant difference in survival of both clusters and the absence of statistical significance, we decided to joint both databases (our database from Portuguese patients and database from TCGA) in order to try maximizing the observed differences and enlarge the number of patients. In this joint database the number of patients in cluster 1 was 93 and in cluster 2 was 268. The average survival for cluster 1 was 100 months and for cluster 2 was 77.8 months (data not shown). This joint database showed a difference of 22.2 months in the survival of both clusters. Despite this meaningful survival difference statistical significance was not reached (Log-Rank: p=0.167).
We also researched the effect of tumor stage in the survival, so we considered in this joint database, tumor stage I+II and tumor stage III + IV. Considering tumor stage I+II, Kaplan-Meier curves for the two clusters, cluster 1 with 37 patients and cluster 2 with 74 patients, showed that the average survival for cluster 1 was 135.4 months and for cluster 2 was 80.0 months (figure 4A). Patients with tumor stage I+II with and without CNAs (cluster 1 and 2) in the selected genes, exhibited a marginal statistical significance difference (Log-Rank: p=0.049) of 55.4 months in the survival of both clusters.
Considering tumor stage III+IV, Kaplan-Meier curves for the two clusters, cluster 1 with 55 patients and cluster 2 with 187 patients, showed that the average survival for cluster 1 was 84.9 months and for cluster 2 was 71.0 months (figure 4B). We verified 13.9 months of difference in survival between these clusters; however, we did not reach statistical significance (Log-Rank: p=0.809).
The distribution of patients' age is very similar in both clusters, which do not constitute a bias in the observed statistical significance survival differences.
Figure 2
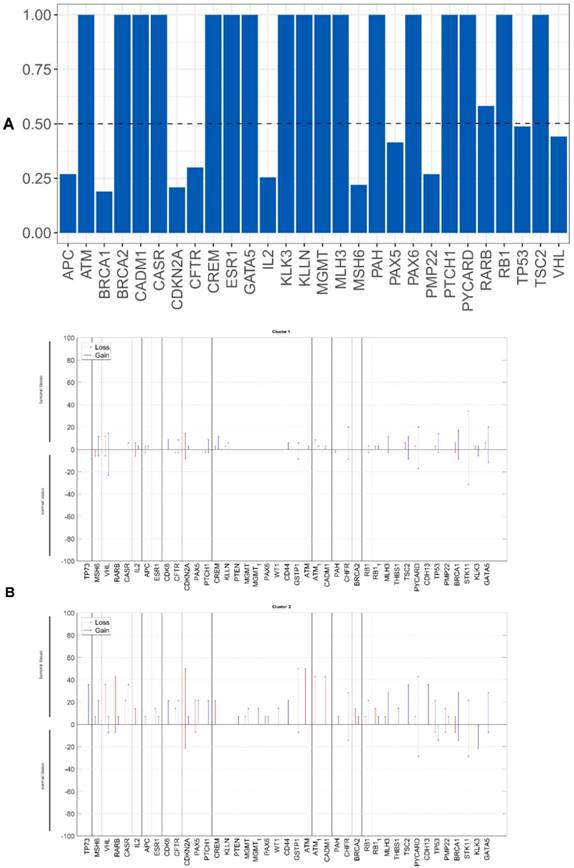
A) Differences of entropy observed among CNA results for the genes analyzed in the OSCC tumor and non-tumor tissue samples. B) Copy number gains and losses detected in tumor and non-tumor tissue samples of cluster 1 and cluster 2. Loss is represented by red and gain by blue.
Figure 3
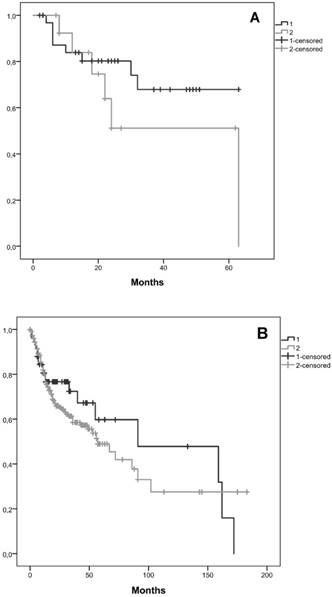
Kaplan-Meier curves for the two clusters identified, A) in our cohort, B) in the validation cohort from the TCGA database. Cluster 1 is represented by 1 and cluster 2 by 2.
Epigenetic signature associated with OSCC survival
We evaluated whether the epigenetic differences between the tumor and non-tumor tissue samples could be used as survival predictors using entropy differences between the results of gene promoter methylation in these two kinds of samples. We reduced the data, selecting only the genes with a difference higher than 50%. The selected genes were: ATM, BRCA1, BRCA2, CADM1, CDKN2A, CHFR, ESR1, GATA5, MGMT, PAX5, PAX6, TP53 and TP73 (figure 5A).
Applying factorial and clusters analysis with these selected genes, we reached a division of our cohort into two clusters with a Cronbach alpha of 0.718 and a silhouette Coefficient of 0.7. The cluster 1 presented a proportion of 57% with 28 patients and the cluster 2 of 43% with 21 patients.
The epigenetic profile of these two clusters is different. We observed that cluster 1 did not present any gene promoter methylation and cluster 2 presented methylation in these genes (figure 5B).
Kaplan-Meier curves for the two clusters were performed, being 47.8 months the average survival of cluster 1 (CI95% [38.5; 57.1]) and 44.8 months of cluster 2 (CI95% [31.8; 57.7]) (figure 6). We did not reach statistical significance (Log-Rank: p=0.330).
Validation using TCGA data was not done because methylation data is not comparable with our own data.
Genomic and epigenetic signatures associated with OSCC relapse/metastasis risk
Random Forest model using CNA and gene promoter methylation results to search biomarkers with capability of predicting the relapse/metastases development risk in our OSCC cohort was performed. For CNA, we verified that a set of eight genes has the power to discriminate between patients that developed relapse/metastases during clinical follow up from those without recurrence. The genomic signature with potential to perform this discrimination comprises the following genes: BRCA1, CDKN2A, CHFR, GATA5, PYCARD, STK11, TP53 and VHL (figure 7A). We did not reach statistical significance.
A validation of genomic results using TCGA data was performed, but we did not find statistical significance (data not shown).
For gene promoter methylation, we verified that a set of six genes has the power to discriminate between patients that developed relapse/metastases during clinical follow up from those without recurrence.
The epigenetic signature with potential to perform this discrimination comprises the following genes: CADM1, GATA5, KLNN, MSH6, PAX5 and WT1 (figure 7B).
We did not reach a statistical significance.
Agreement between results obtained from tumor tissue samples and scrapped cells of the tumor surface
Evaluation of CDKN2A gene promoter methylation status in both tumor tissue and scrapped cells exhibited a total agreement, Kappa = 1 (figure 8A). From all analyzed genes, in our cohort only the promoter methylation status of CDKN2A gene can be assessed using a non-invasive approach with the same result as in the tumor tissue.
For the overall copy number alterations (0.1568929 IC95% [0.0764; 0.2368]) and methylation status (0.005028447 IC95% [-0.0872; 0.1150]) of the analyzed genes in the tumor tissue and in the scrapped cells (figure 8 A, B) we observed a reduced agreement.
Figure 4
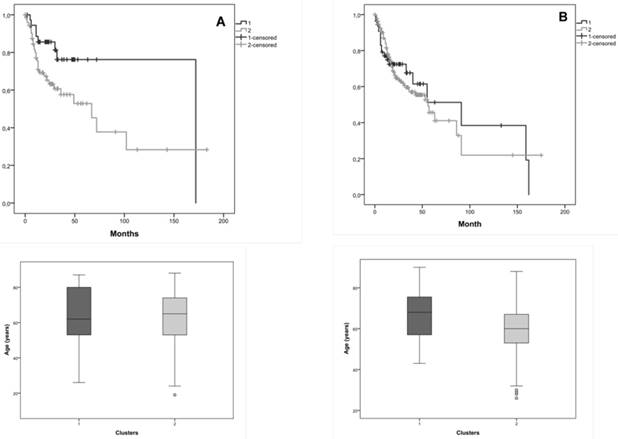
Kaplan-Meier curves for the two clusters identified in the joint database (our and validation cohorts) and the distribution of patients' age in both clusters, A) for tumor stage I+II, B) for tumor stage III+IV. Cluster 1 is represented by 1 and cluster 2 by 2.
Discussion
Using a clinically well characterized OSCC cohort, followed during 1 to 63 months, we identified a specific genomic and epigenetic signature associated with survival and risk of metastasis/relapse development during/after treatment. Nowadays we are witnessing an explosion in the knowledge of biological markers related to pathogenesis and progression of OSCC, which might emerge as a possibility to complement the selection of patients to more aggressive treatment modalities. Several chromosomal regions and genes have been pointed out as potential biomarkers for predicting clinical outcome and as therapeutic targets [17-19]. However, single markers have proved insufficient predictive power, so, no biomarker was yet translated to clinical practice [20]. Our study gathers novelty since we identified and validate in an independent OSCC cohort a specific set of genes that can predict the patients' prognosis, with great impact in the survival of the patients. Copy number alterations in ATM, CASR, TP73, CADM1, RARB, CDH13, PAX5, RB1 genes and GATA5, PAX6, CADM1 and CHFR promoter methylation were shown to be associated with worse patient survival in our OSCC patients. Copy number alterations in BRCA1, CDKN2A, CHFR, GATA5, PYCARD, STK11, TP53, VHL genes and GATA5, CADM1, KLNN, MSH6, PAX5 and WT1 promoter methylation were shown to be associated with development of metastasis/relapses during or after treatment of our OSCC patients. These genomic signatures were validated in a geographically separated cohort (from TCGA database), reinforcing their potential for clinical application, since they were associated with differences in patient survival around 20 months and more than 50 months if we considered only tumor stage I+II. The presence of these set of genes with copy number alterations could guide the clinical management of OSCC patients, indicating which patients have a better prognosis and also which patients must be monitored in closer follow up or should have an intensification of therapy due to the major risk of metastasis/relapse.
Focusing on the individual gene members of the genomic and epigenetic signatures related to survival and metastasis/relapse risk, we see that these genes are highly relevant to OSCC disease. Chief among them is TP53 gene, in which aberrations are the most frequent molecular events in human cancers. This gene encodes the p53 protein, which is involved in many key events in the cell like regulation of cell cycle and glucose metabolism in cancer cells, DNA-repair, apoptosis, and senescence, which are induced by various stress signals, including DNA-damage and inflammation [21, 22]. Another gene is CDKN2A, in which inactivation was found in 57% of HPV-negative head and neck cancer [23]. This gene regulates cell cycle progression by blocking the activity of CCND1 and its associated kinases, CDK6 and CDK4, which phosphorylate and inactivate the tumor suppressor RB1 [24, 25]. Patients that harbor simultaneous amplified CCND1 and deleted CDKN2A presented worse prognosis [24]. BRCA1 overexpression in leukoplakia followed by subsequent underexpression in tongue squamous cell carcinoma was also already described [26]. CADM1 downregulation through epigenetic silencing or loss of heterozygosity is related to tumor cell invasion and metastatic potential [27, 28]. ATM plays a role in maintaining genome integrity, being its aberrant promoter methylation linked to lost or greatly diminished expression of several tumor suppressors, namely BRCA1 [29], CDKN2A [30] and VHL [31]. ATM promoter hypermethylation showed a significant correlation with decreased overall OSCC patient survival [32]. We previously reported the association of MSH6 and GATA5 promoter methylation with OSCC worse prognosis, being the later gene also significantly associated with shorter survival rate [14]. PAX5 promoter methylation was considered useful to identify patients at high risk of locoregional recurrence, after surgical treatment [33].
Figure 5
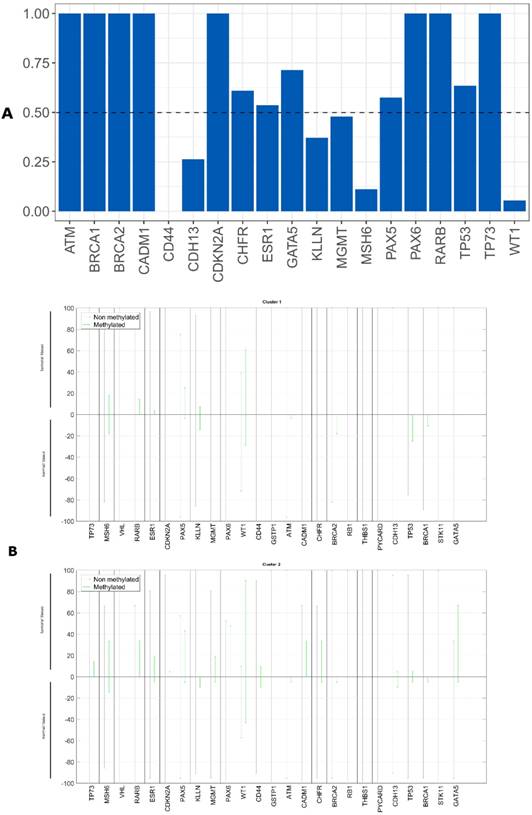
A) Differences of entropy observed among methylation status for the analyzed genes in the OSCC tumor and non-tumor tissue samples, B) Methylation profile detected in tumor and non-tumor tissue samples of our cohort for the cluster 1 and the cluster 2.
Figure 6
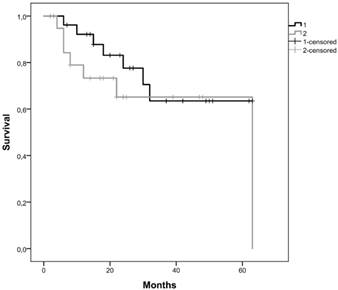
Kaplan-Meier curves for the two clusters identified using gene promoter methylation results of our cohort. Cluster 1 is represented by 1 and cluster 2 by 2.
Figure 7
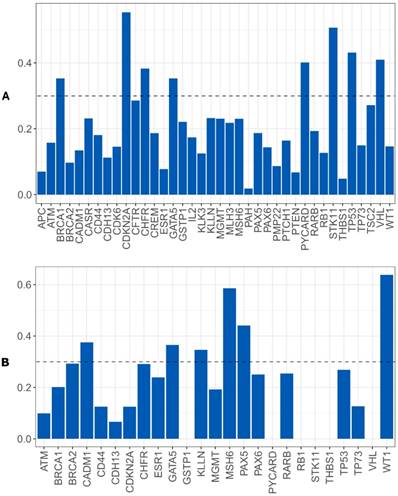
Importance level of the genes analyzed to discriminate between the patients that develop or not metastases/relapses, A) using CNA data, B) using gene promoter methylation genes.
Even with some limitations, such as, the different platform used to obtain the genomic results (MS-MLPA in our study cohort and SNP-microarray in TCGA cohort) and also the different clinical follow-up frame time of the two cohorts, our results were in general validated in this larger and geographical distinct cohort (from TCGA database). This validation proves the clinical relevance of the identified signatures for OSCC patients. The reduced follow-up time of our patients (1-63 months) and also the junction of two databases with different patients' follow-up time could explain the absence of survival statistical significance results; nevertheless, the survival difference between the two identified clusters is clear, representing a huge difference in the life time of these patients. Considering the genomic signature to predict the risk of relapses/ metastases, the absence of statistical significance in both cohorts could be due to the reduced number of patients that developed metastases and relapses during the time of follow-up.
Figure 8
Agreement measured by Kappa value between tumor tissue and scrapped cells. A) using CNAs, B) using methylation status.
Screening oral cancer implies searching oral potentially malignant and cancerous/recurrence lesions, typically before symptoms occur in people belonging to groups of risk. In 2005 Bremmer et al [9] showed that the Multiplex Ligation-dependent Probe Amplification (MLPA) technique is suitable for detection of genetic alterations in noninvasive samples, opening the door to improve the early diagnostic of oral potential malignant lesions. Four years later, the same group [34] compared the results from exfoliated cells and oral lesions biopsies of the same patients showing a high sensitive rate of this noninvasive technique. However, up to now, this technique was not validated, being still a promise for clinical practice without evidence of a truly practical application. We showed in our cohort that using MS-MLPA technique we could only evaluate the CDKN2A gene promoter methylation status with same result either in scrapped cells from surface of tumor or in tumor tissue. The major limitation of these non-invasive samples could be the admixture with normal cells which hampers the detection of genetic and epigenetic alterations of tumor cells. Further studies regarding the collection of these non-invasive samples and also the technologies used to perform the molecular evaluations are needed, since in this moment this non-invasive methodology is not reliable for cancer detection.
Conclusions
Our results suggest the involvement of a specific set of genes that together has the ability to predict the patients' prognosis. The proposed genomic and epigenetic signatures include genes that individually were already related to OSCC and together proved to be associated to survival and risk of metastasis/relapse development. These findings are of clinical relevance since accurate prognostic biomarkers are still urgently needed in the clinical management of OSCC patients. Our genomic signatures were validated in a larger and geographically separate cohort, from the TCGA database, which reinforce their strength to future biomarker-driven clinical trials. We obtained the same result from CDKN2A gene promoter methylation status either in scrapped cells from the surface of tumor or in tumor tissue. Non-invasive methodologies to perform molecular screening in the high-risk populations are promising; however, further studies are needed before a translation to clinical practice, namely regarding sample collection in this hard to reach population.
Acknowledgements
Ribeiro I.P. is a recipient of a PhD fellowship (SFRH/BD/52290/2013) from the Portuguese Foundation for Science and Technology. This work was in part supported by CIMAGO (Center of Investigation on Environment Genetics and Oncobiology - Faculty of Medicine, University of Coimbra).
Competing Interests
The authors have declared that no competing interest exists.
References
1. van der Waal I, de Bree R, Brakenhoff R, Coebergh JW. Early diagnosis in primary oral cancer: is it possible? Medicina oral, patologia oral y cirugia bucal. 2011;16:e300-5
2. Cowpe JG, Longmore RB, Green MW. Quantitative exfoliative cytology of abnormal oral mucosal smears. Journal of the Royal Society of Medicine. 1988;81:509-13
3. Omar E. Current concepts and future of noninvasive procedures for diagnosing oral squamous cell carcinoma-a systematic review. Head & face medicine. 2015;11:6
4. Campbell BH, Spinelli K, Marbella AM, Myers KB, Kuhn JC, Layde PM. Aspiration, weight loss, and quality of life in head and neck cancer survivors. Archives of otolaryngology-head & neck surgery. 2004;130:1100-3
5. Holloway RL, Hellewell JL, Marbella AM, Layde PM, Myers KB, Campbell BH. Psychosocial effects in long-term head and neck cancer survivors. Head & neck. 2005;27:281-8
6. Meyer TK, Kuhn JC, Campbell BH, Marbella AM, Myers KB, Layde PM. Speech intelligibility and quality of life in head and neck cancer survivors. The Laryngoscope. 2004;114:1977-81
7. Oliveira LR, Ribeiro-Silva A. Prognostic significance of immunohistochemical biomarkers in oral squamous cell carcinoma. International journal of oral and maxillofacial surgery. 2011;40:298-307
8. Nair DR, Pruthy R, Pawar U, Chaturvedi P. Oral cancer: Premalignant conditions and screening-an update. Journal of cancer research and therapeutics. 2012;8(Suppl 1):S57-66
9. Bremmer JF, Braakhuis BJ, Ruijter-Schippers HJ, Brink A, Duarte HM, Kuik DJ. et al. A noninvasive genetic screening test to detect oral preneoplastic lesions. Laboratory investigation; a journal of technical methods and pathology. 2005;85:1481-8
10. Verma R, Singh A, Badni M, Chandra A, Gupta S, Verma R. Evaluation of exfoliative cytology in the diagnosis of oral premalignant and malignant lesions: A cytomorphometric analysis. Dental research journal. 2015;12:83-8
11. Messadi DV. Diagnostic aids for detection of oral precancerous conditions. International journal of oral science. 2013;5:59-65
12. Ribeiro IP, Marques F, Caramelo F, Ferrao J, Prazeres H, Juliao MJ. et al. Genetic imbalances detected by multiplex ligation-dependent probe amplification in a cohort of patients with oral squamous cell carcinoma-the first step towards clinical personalized medicine. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35:4687-95
13. Ribeiro IP, Marques F, Caramelo F, Pereira J, Patricio M, Prazeres H. et al. Genetic gains and losses in oral squamous cell carcinoma: impact on clinical management. Cellular oncology. 2014;37:29-39
14. Ribeiro IP, Caramelo F, Marques F, Domingues A, Mesquita M, Barroso L. et al. WT1, MSH6, GATA5 and PAX5 as epigenetic oral squamous cell carcinoma biomarkers - a short report. Cellular oncology. 2016;39:573-82
15. Hausser J, Strimmer K. Entropy inference and the James-Stein estimator, with application to nonlinear gene association networks. J Mach Learn Res. 2009;10:1469-84
16. Menze BH, Kelm BM, Masuch R, Himmelreich U, Bachert P, Petrich W. et al. A comparison of random forest and its Gini importance with standard chemometric methods for the feature selection and classification of spectral data. BMC bioinformatics. 2009;10:213
17. Chen C, Zhang Y, Loomis MM, Upton MP, Lohavanichbutr P, Houck JR. et al. Genome-Wide Loss of Heterozygosity and DNA Copy Number Aberration in HPV-Negative Oral Squamous Cell Carcinoma and Their Associations with Disease-Specific Survival. PloS one. 2015;10:e0135074
18. Melchers LJ, Clausen MJ, Mastik MF, Slagter-Menkema L, van der Wal JE, Wisman GB. et al. Identification of methylation markers for the prediction of nodal metastasis in oral and oropharyngeal squamous cell carcinoma. Epigenetics. 2015;10:850-60
19. Naganuma K, Hatta M, Ikebe T, Yamazaki J. Epigenetic alterations of the keratin 13 gene in oral squamous cell carcinoma. BMC cancer. 2014;14:988
20. Thomas GR, Nadiminti H, Regalado J. Molecular predictors of clinical outcome in patients with head and neck squamous cell carcinoma. International journal of experimental pathology. 2005;86:347-63
21. Bensaad K, Vousden KH. p53: new roles in metabolism. Trends in cell biology. 2007;17:286-91
22. Hussain SP, Harris CC. p53 biological network: at the crossroads of the cellular-stress response pathway and molecular carcinogenesis. Journal of Nippon Medical School = Nippon Ika Daigaku zasshi. 2006;73:54-64
23. Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576-82
24. Beck TN, Kaczmar J, Handorf E, Nikonova A, Dubyk C, Peri S. et al. Phospho-T356RB1 predicts survival in HPV-negative squamous cell carcinoma of the head and neck. Oncotarget. 2015;6:18863-74
25. Burke JR, Hura GL, Rubin SM. Structures of inactive retinoblastoma protein reveal multiple mechanisms for cell cycle control. Genes & development. 2012;26:1156-66
26. Vora HH, Shah NG, Patel DD, Trivedi TI, Choksi TJ. BRCA1 expression in leukoplakia and carcinoma of the tongue. Journal of surgical oncology. 2003;83:232-40
27. van den Berg RM, Snijders PJ, Grunberg K, Kooi C, Spreeuwenberg MD, Meijer CJ. et al. Comprehensive CADM1 promoter methylation analysis in NSCLC and normal lung specimens. Lung cancer. 2011;72:316-21
28. van Kempen PM, van Bockel L, Braunius WW, Moelans CB, van Olst M, de Jong R. et al. HPV-positive oropharyngeal squamous cell carcinoma is associated with TIMP3 and CADM1 promoter hypermethylation. Cancer medicine. 2014;3:1185-96
29. Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E. et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. Journal of the National Cancer Institute. 2000;92:564-9
30. Costello JF, Berger MS, Huang HS, Cavenee WK. Silencing of p16/CDKN2 expression in human gliomas by methylation and chromatin condensation. Cancer research. 1996;56:2405-10
31. Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S. et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:9700-4
32. Ai L, Vo QN, Zuo C, Li L, Ling W, Suen JY. et al. Ataxia-telangiectasia-mutated (ATM) gene in head and neck squamous cell carcinoma: promoter hypermethylation with clinical correlation in 100 cases. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2004;13:150-6
33. Hayashi M, Guerrero-Preston R, Sidransky D, Koch WM. Paired box 5 methylation detection by droplet digital PCR for ultra-sensitive deep surgical margins analysis of head and neck squamous cell carcinoma. Cancer prevention research. 2015;8:1017-26
34. Bremmer JF, Graveland AP, Brink A, Braakhuis BJ, Kuik DJ, Leemans CR. et al. Screening for oral precancer with noninvasive genetic cytology. Cancer prevention research. 2009;2:128-33
Author contact
![]() Corresponding author: Isabel Marques Carreira, Cytogenetics and Genomics Laboratory, Faculty of Medicine, University of Coimbra, Polo Ciências da Saúde, 3000-354 Coimbra, Portugal. Tel. +351 239 480 027; Fax +351 239 480 035; E-Mail: citogeneticauc.pt; icarreirauc.pt
Corresponding author: Isabel Marques Carreira, Cytogenetics and Genomics Laboratory, Faculty of Medicine, University of Coimbra, Polo Ciências da Saúde, 3000-354 Coimbra, Portugal. Tel. +351 239 480 027; Fax +351 239 480 035; E-Mail: citogeneticauc.pt; icarreirauc.pt
Citation styles
APA
Ribeiro, I.P., Caramelo, F., Esteves, L., Oliveira, C., Marques, F., Barroso, L., Melo, J.B., Carreira, I.M. (2018). Genomic and epigenetic signatures associated with survival rate in oral squamous cell carcinoma patients. Journal of Cancer, 9(11), 1885-1895. https://doi.org/10.7150/jca.23239.
ACS
Ribeiro, I.P.; Caramelo, F.; Esteves, L.; Oliveira, C.; Marques, F.; Barroso, L.; Melo, J.B.; Carreira, I.M. Genomic and epigenetic signatures associated with survival rate in oral squamous cell carcinoma patients. J. Cancer 2018, 9 (11), 1885-1895. DOI: 10.7150/jca.23239.
NLM
Ribeiro IP, Caramelo F, Esteves L, Oliveira C, Marques F, Barroso L, Melo JB, Carreira IM. Genomic and epigenetic signatures associated with survival rate in oral squamous cell carcinoma patients. J Cancer 2018; 9(11):1885-1895. doi:10.7150/jca.23239. https://www.jcancer.org/v09p1885.htm
CSE
Ribeiro IP, Caramelo F, Esteves L, Oliveira C, Marques F, Barroso L, Melo JB, Carreira IM. 2018. Genomic and epigenetic signatures associated with survival rate in oral squamous cell carcinoma patients. J Cancer. 9(11):1885-1895.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.