Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(11):1925-1931. doi:10.7150/jca.23968 This issue Cite
Research Paper
The innovative regularity and role of p16 methylation in blood during HCC development
Cheng-Gang Jiang1, Qun Chen2, Lina Wu3, Guang Wang4, Jianzhong Ma5 ![]()
1. Department of Surgical Oncology, The First Affiliated Hospital of China Medical University, Shenyang 110001, Liaoning Province, China;
2. School of Sciences, Ningxia Medical University, Yinchuan 750004 China;
3. Department of medical laboratory, Shengjing Hospital of China Medical University, Shenyang 110004 China;
4. Department of General Surgery, The First Affiliated Hospital, China Medical University, Shenyang 110001, China;
5. Department of public foundation, China Medical University, Shenyang 110122, China
Received 2017-11-21; Accepted 2018-3-6; Published 2018-4-27
Citation:
Jiang CG, Chen Q, Wu L, Wang G, Ma J. The innovative regularity and role of p16 methylation in blood during HCC development. J Cancer 2018; 9(11):1925-1931. doi:10.7150/jca.23968. https://www.jcancer.org/v09p1925.htm
Other stylesAbstract
Purpose: This study was performed to examine the regularity and role of p16 methylation in hepatocellular carcinoma (HCC) blood.
Methods: Big data of the case-control studies due to blood p16 methylation detection were collected in English and Chinese Journals. The risk of HCC's histologic process was investigated using both Meta-analysis and the quantitative correlation analysis.
Results: p16 methylation frequencies in blood were gradually increased from 0 % in normal to 10 % in benign disease, and to 60 % in HCC development. Based on p16 methylation of normal control blood, p16 methylation between normal and benign disease had no risk, and the methylation risk in HCC was significantly increased from normal to HCC through benign disease OR, 95% CI =16.23 ( 11.66, 22.58 ). Compared with the benign disease matched by HCC patient, the methylation risk of p16 in HCC was found, with the pooled OR value of 10.06 (95% IC = 7.64, 13.21) in blood. In addition, the regulatory mechanism affecting p16 methylation risk in normal blood had no role, and the strength of p16 methylation risk was rapidly increased between benign diseases and HCC blood. p16 methylation risk started from the patients with benign disease in blood. These results in blood and tissue detection were basically consistent.
Conclusions: HCC pathogenesis affecting p16 methylation don't work during normal blood, when from benign diseases to HCC bloods, can produce powerful role. The transcriptional inactivation associated with p16 methylation might start from benign liver disease, and might be increased from benign liver disease to HCC process. p16 methylation in blood can be used as a promising non-invasive biomarker to HCC's prediction and diagnosis.
Keywords: hepatocellular carcinoma, DNA promoter methylation, p16 gene, pathogenesis, prevention
Introduction
Hepatocellular carcinoma (HCC) is one of the most common solid tumors, and is one of the major causes of cancer death worldwide [1]. The aberrant DNA methylation is the primary mediator of changes in HCC, and may become a promising biomarker for the early diagnosis of cancer diseases [2]. In clinical practice, due to the onset of symptoms, about the 60% of patients with HCC had advanced and surgical treatment of poor prognosis, easy to relapse, the mortality and morbidity of HCC were rapid extension, alpha fetoprotein (AFP) was still recognized diagnosis and screening index, some patients with hepatitis and liver cirrhosis AFP also increased, some advanced HCC patients serum AFP was normal, and the poor sensitivity and specific degree [3]. In order to reduce the mortality and morbidity of HCC, to improve the cure incidence, and to well solve the clinical diagnosis problem of HCC, the early diagnosis played a very pivotal role in clinical practice, due to the cancer cells can release a lot of free DNA in the serum or plasma, through the blood methylation was researched, the early diagnosis target of HCC without damage detection may be helped in clinical practice. Little effort has been spent on the system study of p16 methylation in the blood of HCC's formation and development process. As far as we know, HCC detection still lacked a useful routine biomarker making its early detection easier than it is currently.
HCC is the main pathological type of primary liver cancer, the molecular mechanism of its pathogenesis is still unclear. In recent years, p16 gene 5'abnormal promoter CpG island methylation in many patients with tumor tissue and serum or plasma circulating nucleic acid had a higher detection incidence [1-3]. The studies of molecular biology had found that a lot of susceptibility genes methylations associated with the occurrence and development of HCC, in which the expression of p16 gene inactivation was closely related to the occurrence of HCC, p16 methylation, a cyclin-dependent kinase inhibitor gene that regulated cell cycling, had frequently been detected in human cancers including HCC [4]. Epigenetic changes in gene expression affecting methylation is accepted as the main cause of inactivation of the p16 gene, 5'CpG islands methylation might be the major mechanism of p16 and p15 genes inactivation in primary HCC in the studied population [5]. The detecting methylation of tumor suppressor genes in the serum or plasma is very important for understanding the mechanisms of oncogenesis, with corresponding, HCC pathogenesis may directly response to the methylation changes of p16 gene in blood, for the study of mechanism, yet little is known about the epigenetic relationship between the abnormal methylation in blood and HCC pathogenesis. The present study was discussed in blood that there was a change characteristic through the performance of methylation process, and that HCC pathogenesis played a role in where was the strongest in the process of HCC histology evolution.
We hoped that in near future, the methylation biomarker could be applied to clinical application, how HCC patients at risk size could be diagnosed early, and that the progression of HCC disease could be more correctly assessed. Several studies had shown that p16 methelation of the tumor suppressor gene frequently displays genetic alteration in HCC tissues [6]. The results of some meta-analysis indicated that aberrant DNA methylation was associated with the risk of HCC in tissue. Aberrant DNA methylation might become the useful biomarkers for the prediction and prognostication of HCC in tissue [7], but not in blood samples. No study was performed to examine the gradual changing regularity of p16 methylation in HCC blood. The present study was performed to examine the incidence frequency of p16 methylation in the sera of normal, benign disease and HCC bloods, and to investigate the risk regularity of the gradual changing of p16 methylation in blood, and to evaluate both p16 methylation role and the relationship between p16 methylation epigenetics and HCC pathogenesis. These studies are essential to early identification of HCC and precursor lesions, and to the HCC pathogenesis.
Materials and Methods
Data sources
The published studies were included with an English or Chinese language. The databases screened were used with Pubmed, EMBASE, Cochrane Library, Science Citation Index, Wanfang Literature Batabase, China National Knowledge Infrastructure, and the Chinese BioMedical Literature Database. The keywords combinations queried were used by p16, methylation, hypermethylation, liver cancer, HCC, blood, serum, plasma, tissue, hepatocirrhosis and benign diseases. The benign diseases group was included in chronic hepatitis, cirrhosis, hepatitis syndrome, hepatitis C disease (HCV) and hepatitis B virus (HBV). Human blood included serum, plasma and peripheral blood. Disagreement was adjudicated by author discussion. Quality assessment was performed by the author discussion. These different methods of meyhelation detection were recorded in this paper.
Data extraction
Data extraction was independently conducted using a standardized approach. Data for year of publication and name of first author, geographical populations, the numbers of cases and controls, and incidences of p16 methylation were collected using standard data extraction form. The literature with repeating and poor quality as well as low coverage information could not be used in the systematic literature search. Each published article must be extracted from the data of case-control study in blood, and must be extracted from the normal group, the precancerous lesion group and the liver cancer group data that came from the original article. A total of 14 papers were collected in normal, benign disease and HCC blood (Table 1). The data of three groups in Table 1 were completely determined by the previous articles, three groups were comparatively objective and true.
Table 1
Both characteristics and data of references in peripheral blood
| References | Countries | Normal | BL | HCC | Peripheral blood | DET | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| M | N | M | N | M | N | |||||
| 1 | Wong1999[8] | Hong Kong | 10 | 0 | 38 | 0 | 22 | 13 | plasma/serum | MSP |
| 2 | Wong2000[9] | Hong Kong | 20 | 0 | 15 | 0 | 15 | 8 | plasma/serum | MSP |
| 3 | Kejun2004[10] | China | 9 | 0 | 17 | 0 | 39 | 20 | serum | MSP |
| 4 | Zhang2004[11] | China | 30 | 0 | 30 | 0 | 30 | 20 | plasma | MSP |
| 5 | Chu2004[12] | Korea | 23 | 4 | 46 | 22 | serum | MSP | ||
| 6 | Chen2006[13] | China | 20 | 0 | 15 | 0 | 64 | 49 | serum | MSP |
| 7 | Zhang2006[14] | China | 30 | 0 | 40 | 2 | 44 | 29 | plasma | MSP |
| 8 | Ahmed2010[15] | Egyptian | 62 | 16 | 25 | 23 | serum | MSP | ||
| 9 | He2010[16] | China | 30 | 0 | 60 | 0 | 100 | 65 | plasma | MSP |
| 10 | Ye2013[17] | China | 54 | 20 | 55 | 26 | plasma | MSP | ||
| 11 | Yang2013[18] | China | 40 | 0 | 80 | 0 | 56 | 39 | serum | MSP |
| 12 | El-M2014[19] | Egyptian | 8 | 0 | 20 | 1 | 30 | 2 | blood | MSP |
| 13 | Wen2015[20] | China | 10 | 3 | 31 | 13 | plasma | MSP | ||
| 14 | Dong2016[21] | China | 60 | 0 | 60 | 41 | plasma | MSP | ||
BL: Benign lesions; MSP: methylation-specific PCR; HCC: hepatocellular carcinoma; DET: Dedetection;
M: total number; N: p16 methylation occurrence number.
Statistical analysis
Odds ratios (ORs) and corresponding 95% CIs were used as the risk measures of the relationship. Both OR > 1 and the interval 95% CIs > 1 indicated a higher risk. When there was no statistically significant heterogeneity (P > 0.05), a pooled effect was calculated with a fixed-effects model; otherwise, a random-effects model was employed. We also assessed the publication bias with funnel plots. In bias drawer file the publication of forest plot was conducted by the formula below:
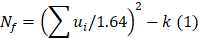
where 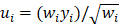 , yi = InORi, wi stood for weight, Nf stood for fail-safe number and k stood for the number of included studies. The larger the Nf was, especially when Nf was significantly greater than the 5k+10, the better reliability the result in Meta-analysis was, and the less likely the conclusion which was impacted by publication bias was [22]. Meta-analysis was performed by software Rev Man 4.2. If there was a small probability of the event in case-control groups, the Peto OR model of Meta-analysis was taken. In addition, all statistical analyses were conducted with Significant Hypothesis Test (SPSS 15.0 software). Differences at P < 0.05 were considered significant.
, yi = InORi, wi stood for weight, Nf stood for fail-safe number and k stood for the number of included studies. The larger the Nf was, especially when Nf was significantly greater than the 5k+10, the better reliability the result in Meta-analysis was, and the less likely the conclusion which was impacted by publication bias was [22]. Meta-analysis was performed by software Rev Man 4.2. If there was a small probability of the event in case-control groups, the Peto OR model of Meta-analysis was taken. In addition, all statistical analyses were conducted with Significant Hypothesis Test (SPSS 15.0 software). Differences at P < 0.05 were considered significant.
Results
A total of 14 eligible studies were included in this meta-analysis. The case-control studies of p16 methylation published from 1999 to 2016 included 617 HCC bloods, 464 benign disease bloods and 257 normal bloods in Table 1. The frequencies (approximately equaled probability) of p16 methylation in blood were gradually increased from 0 % (0/257) in normal to 10 % (42/464) in benign disease, and to 60 % (370 /617) in HCC. Our results revealed that there was a significant difference (P<0.05) in the frequency of p16 methylation between normal and benign disease bloods, and that there was a significant difference (P<0.05) in the frequency of p16 methylation between benign disease and HCC bloods. Our results showed that the changing frequency of p16 methylation from normal to HCC development was gradually increased in body blood.
As shown in Table 1, based on the methylation of normal blood, a total of 9 eligible studies were included, this meta-analysis of p16 methylation was involved in 315 benign disease and 197 normal bloods. Because the incidences probabilities of normal and benign disease methylations were (0/257 and 42/464) all the small probability events in Table 1, Prto OR model was taken in this mete-analysis. These results were shown with the forest plot Fig.1 (a) and the funnel plots Fig.1(b). Due to the methylation numbers of the 7 studies which came from the 9 eligible studies of benign diseases in Fig.1 (a) were all zero, they can't be assessed in Fig 1(a), but because the benign diseases methylation of 7 studies didn't happen, they showed clearly that there was no risk in the 7 of the 9 eligible studies between normal and benign disease bloods. And in the other 2 of the 9 eligible studies in Fig.1(a), their interval of 95% IC=(0.56, 56.21) was not bigger than 1, this result can not prove that there was a risk in the other 2 of 9 eligible studies. Ultimately, the results in 9 eligible studies showed that there was no risk (P = 0.17) in p16 methylation affecting HCC between normal and benign disease bloods.
As shown in Table 1, based on the methylation of normal blood, a total of 10 eligible studies were included with 460 HCC and 267 normal bloods. Because the incidence probability (approximately equaled frequency) of p16 methylation in normal blood was zero (0/257) in Table 1, and was a small probability event, Prto OR model was took in this meta-analysis. These results were shown with the forest plot Fig.2 (a) and the funnel plots Fig.2 (b). Our results revealed that p16 methylation risk in HCC was significantly increased from normal to HCC though benign disease bloods OR, 95% CI =16.23 (11.66, 22.58) in Fig.2 (a). In Fig.2 (b), because all points in Fig.2 (b) were concentrated above 95% IC area, and were symmetrical, there was clearly no publication bias in Fig.2 (b).
As shown in Table 1, based on the methylation of benign disease blood, a total of 13 eligible studies were included in 557 HCC and 464 benign disease bloods. Because the incidence of p16 methelation of benign disease was obviously the small probability event (42/464), Prto OR model was taken in this meta-analysis. The result of the meta-analysis was shown with the forest plot Fig.3 (a) and the funnel plots Fig.3 (b). Compared the benign diseases with HCC patients matched in blood, the methylation risk of p16 in HCC blood was found with OR, 95% CI =10.06 (7.64, 13.21) in Fig.3 (a). In Fig.3 (b), because the three points in Fig.3 (b) were beyond 95% IC area, the publication bias of Fig.3 (b) in bias drawer file was conducted by the formula (1).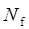 (fail-safe number ) was calculated by us, and it was 2247.67, it was significantly greater than the 5k+10 (75), the conclusion which was impacted by publication bias was reliability in Fig.(a) and Fig.3 (b).
(fail-safe number ) was calculated by us, and it was 2247.67, it was significantly greater than the 5k+10 (75), the conclusion which was impacted by publication bias was reliability in Fig.(a) and Fig.3 (b).
Discussions
Many investigators had turned to the association and diagnostic value of p16 methylation in HCC tissue. However, little effort had been spent on the study of both changing regularity and role of p16 methylation incidence in the process of HCC's formation and development, especially in the blood. In this study, the association between the incidence frequency of blood p16 methylation and the clinicopathological characteristics of HCC was assessed, the statistical results showed that there were the significant differences (P<0.05) in methylation frequency between 0% in normal and 9.9% in benign disease bloods, between 9.9% in benign disease and 60% in HCC bloods, and between HCC patients 60% and no HCC patients of both normal and benign disease 5.8% (42/721 in Table 1). This result showed that the regularity of p16 methylation frequencies in blood were gradually increased from 0 % in normal to 9.9% in benign disease, to 60 % in HCC development. All results above revealed that in the histological process of HCC's formation and development, the normal blood didn't appear on p16 methylation, p16 methylation frequencies from benign disease to HCC emerged dramatic rise, and because the incidence of P 16 methylation was affected to HCC pathogenesis [4], HCC pathogenesis was not working in normal blood, when from benign diseases to HCC blood, produced a powerful role. Therefore, we got the right conclusion that HCC pathogenesis affecting p16 methylation in blood did not play a role from normal person, and began to work from benign liver disease. At the same time, these results revealed in frequency that the prevention of HCC should start from the patients of benign liver disease.
Fig 1
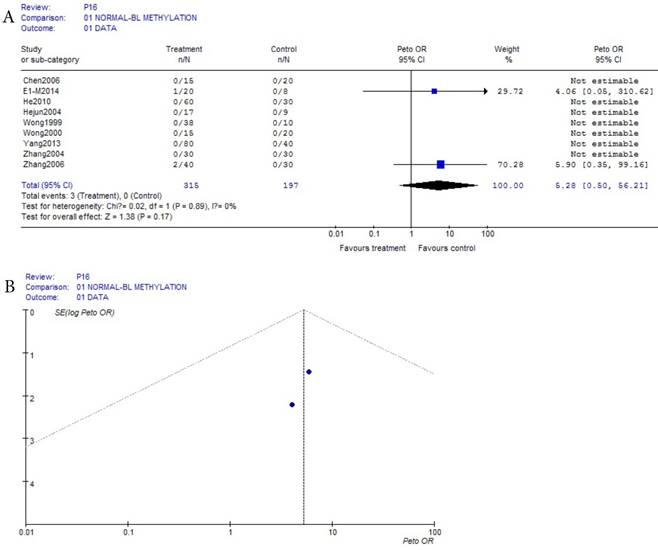
(a) Forest plot between normal control and benign disease risk in blood Fig.1 (b) Funnel plot of corresponding figure 1 (a)
Fig 2
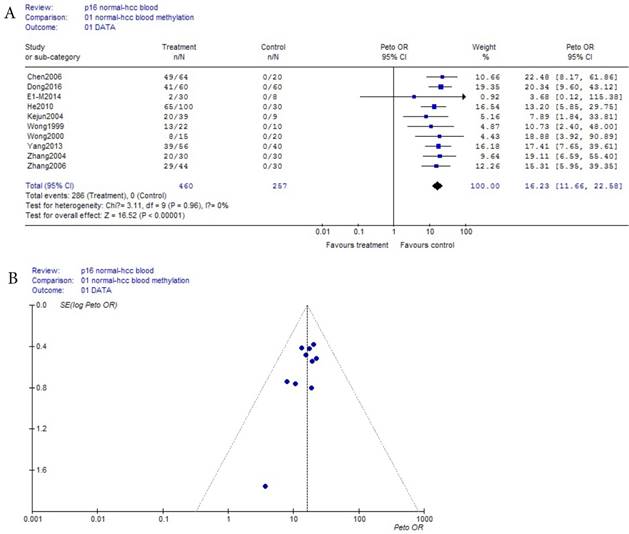
(a) Forest plot between normal control and HCC risk in blood Fig.2 (b) Funnel plot of corresponding figure 2 (a)
Fig 3
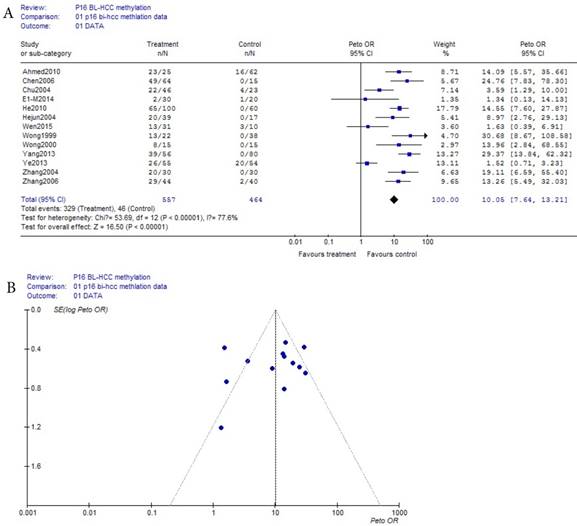
(a) Forest plot between benign disease and HCC risk in blood Fig.3 (b) Funnel plot of corresponding figure 3 (a)
In recent year, some researchers had interested in that the risk size of HCC were related to blood p16 methylation between normal and HCC bloods. However, no was observed that the changing regularity and role of p16 methylation risk in HCC histological process. We had studied this issue from the following two aspects. First, based on p16 methylation of normal blood, p16 methylation between normal and benign disease was no risk in Fig.1(a), and the methylation risk in HCC was significantly increased from normal to HCC though benign diseases OR, 95% CI =16.23 ( 11.66, 22.58 ) in Fig.2(a). Secondly, based on benign disease matched by HCC in blood, the methylation risk in HCC OR, 95% CI =10.06 (7.64, 13.21) in Fig.3(a) had increased dramatically. Because 5'CpG islands methylation of p16 and p15 genes might be an early event in HCC [4-5], and because the gene expression due to extensive CpG island methylation had accepted as the main cause of inactivation of the p16 gene [6], the risk of p16 methylation directly response to the pathogenesis of HCC. According to this analysis above, HCC pathogenesis associated with p16 methylation risk did not work during normal person, its role started from benign liver disease, while developing HCC, patients in HCC increased risk quickly, HCC pathogenesis had dramatically a powerful role.
The study of aberrant DNA methylation of p16 gene was very important to understand the mechanisms of oncogenesis [23]. Due to p16 gene transcriptional inactivation might play an important role in HCC tissue [4-5]. According to that 5'CpG islands methylation might be the major mechanism of p16 gene inactivation in primary HCC [5-6], and that the significant correlation between 5'CpG islands methylation and p16/p15 mRNA expression suppression was found [6], and according to the changing regulation of both frequency and risk size of p16 methylation in blood studied by us above, we might suggested that the transcriptional inactivation of p16 gene was increased dramatically between benign liver disease and HCC process, and began to produce from benign liver disease, and eventually impacted HCC occurrence. In addition, based on our find that the transcriptional inactivation of p16 gene began to produce from benign liver disease, we may suggested that the prevention of HCC should start from patients with benign liver disease, and that the benign liver disease in blood was an important time window for the prevention of HCC.
The previous studies were performed to investigate the effect of p16 methylation on HCC patients using the meta-analysis of available case-control studies in tissue [7,24]. Meta-analysis was relatively rare in blood study, so far, the study between normal and benign liver diseases in blood have not been reported, our findings suggested that the combination of p16 methylation and AFP level may be a promising non-invasive discrimination in HCC patients. Previous study identified the correlations between p16 aberrant methylation and HCC [OR, 95% CI =13.41 (9.18, 19.5) in carcinoma tissues/normal tissues, and OR, 95% CI =123.15 (49.12, 308.74) in carcinoma serums/normal serums] [24]. Both tumor tissues of HCC patients and healthy liver tissues of patients with other diseases (OR 12.17, 95% CI: 6.64%-22.31%) and tumor tissues of HCC patients and liver tissues of patients with non-tumorous liver diseases (OR 6.82, 95% CI: 4.31%-10.79%) were got with the study of the meta-analysis [7]. Our results shown that there was no risk in between normal and benign disease bloods, and that there were the risks OR, 95% CI =16.23 (11.66, 22.58) in between normal control and HCC bloods, and OR, 95% CI =10.06 (7.64,13.21) in between benign disease and HCC bloods. In the meta-analysis of one paper, the correlation risk of p16 promoter methylation between HCC and chronic hepatitis blood was OR , 95% CI =12.94 ( 2.29,73.02), and in three papers, the correlation risk of p16 promoter methylation between HCC and liver cirrhosis blood was OR ,95% CI = 6.44 ( 1.16,35.66) [25]. In addition to the risk of this research [OR, 95% CI =123.15 (49.12, 308.74) in carcinoma serums/normal serums] was too large [24], in comparison with other results, our results in blood were basically consistent with tissue results which were previously published. Our results suggested that p16 methylation positive in tissue and blood might predict the clinical progression of HCC patients, and that aberrant p16 methylaition in blood detection was an important way for screening and diagnosis of HCC.
The more data Meta-analysis is used, the more reliable its results is. The HCC test data of p16 methylation in the case-control blood was very scarce. So far, the data used in this paper were much more than in other literature. To make full use of data to verify substantial heterogeneity, the sensitivity analysis in this paper was performed to analyze the influence of the recalculated OR value. In Figure 3(a), there were 13 references, three references with the minimum lower bounds of OR, 95%CI in Figure 3(a) were removed, that was, the data of [10],[19] and [20] references were removed, and the remaining 10 references of 13 were subjected to the meta-analysis. The result of sensitivity analysis of lower bound was showed as OR , 95% CI =15.23 ( 11.25,20.61). Similarly, [8],[9] and [13] references with the maximum three upper bounds of OR, 95%CI in Figure 3(a) were removed. The meta-analysis showed that the result of sensitivity analysis of upper bound was OR , 95% CI =8.82 ( 6.57, 11.83). There was OR, 95% CI =10.06 (7.64, 13.21) in Figure 3(a) above. The interval of sensitivity analysis of the recalculated OR value was got as 8.82<OR<15.23. All the upper and lower bounds values were far larger than 1, the result in sensitivity analysis fully proved that there was a significant big risk between benign liver disease and HCC.
In the most recent Meta-analysis, p16 methylation in HCC tissue was associated with the development, age, hepatic viruses infection of HCC [25]. However, the amount of case-control samples which we had got in the serum was not large. Some results were based on the sample size of the minority population. We will increase the sample size in future research to enhance the reliability of our results. Everyone don't know that P16 protein expression reduced by methylation was related to clinicopathological features of HCC patients with regard to AFP level, tumor metastasis and presence of multiple nodules. We shall hope that with the aid of p16 epigenetics, these problems will be discussed in the future, and that p16 methylation non-invasive in application value of HCC needs to be further confirmed.
Conclusions
HCC pathogenesis affecting p16 methylation was no work during normal person blood, when from benign diseases to HCC, produced a powerful role. The transcriptional inactivation of p16 gene associated with p16 methylation in blood might start from the benign liver disease, and was increased dramatically between benign liver diseases and HCC process, and eventually they synchronously impacted HCC occurrence. p16 methylation in blood may be a promising non-invasive biomarker for HCC's prediction and diagnosis.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bosch FX, Ribes J, Borràs J. Epidemiology of primary liver cancer. Semin Liver Dis. 1999;19:271-285
2. Lambert MP, Paliwal A, Vaissiere T, Chemin I. et al. Aberrant DNA methylation distinguishes hepatocellular carcinoma associated with HBV and HCV infection and alcohol intake. J Hepatol. 2011;54:705-715
3. Dengler M, Staufer K, Huber H. et al. Soluble Axl is an accuincidence biomarker of cirrhosis and hepatocellular carcinoma development: results from a large scale multicenter analysis. Oncotarget. 2017;8(28):46234-46248
4. Ezhevsky SA, Ho A, Becker-Hapak M. et al. Differential regulation of retinoblastoma tumor suppressor protein by G(1) cyclin-dependent kinase complexes in vivo. Mol Cell Biol. 2001;21(14):4773-4784
5. Qin Y, Liu JY, Li B, Sun Zl. et al. Association of low p16INK4a and p15INK4b mRNAs expression with their CpG islands methylation with human hepatocellular carcinogenesis. World J Gastroenterol. 2004;10(9):1276-1280
6. Zhang YJ, Ahsan H, Chen Y. et al. High frequency of promoter hypermethylation of RASSF1A and p16 and its relationship to aflatoxin B1-DNA adduct levels in human hepatocellular carcinoma. Mol Carcinog. 2002;35(2):85-92
7. Zhang C, Li J, Huang T. et al. Meta-analysis of DNA methylation biomarkers in hepatocellular carcinoma. Oncotarget. 2016;7(49):81255-81267
8. Wong IH, Lo YM, Zhang J. et al. Detection of Aberrant p16 Methylation in the Plasma and Serum of Liver Cancer Patients. Cancer Res. 1999;59(1):71-73
9. Wong IH, Lo YM, Yeo W. et al. Frequent p15 Promoter Methylation in Tumor and Peripheral Blood from Hepatocellular Carcinoma Patients. Clin Cancer Res. 2000;6(9):3516-3521
10. Na K. et al. p16 methylation detection in the serum of liver caner patients. Chin J Oncol. 2004;26(4):569-571
11. Zang FC. et al. Methylation of p16 gene in plasma in diagnosis of primary hepatocellular carcinoma. LUO Su1, JOU RN AL OF JILIN UNIVERSITY ( SCIENCE EDITION). 2004;42(2):298-300
12. Chu HJ, Heo J, Seo SB. et al. Detection of aberrant p16INK4A methylation in sera of patients with liver cirrhosis and hepatocellular carcinoma. J Korean Med Sci. 2004;19(1):83-86
13. Chen LB. et al. Analysis of Promoter Hypermethylation of p16 Gene and DAPK Gene in Sera from Primary Liver Cancer Patients. Tumor Prevention Research. 2006;33(3):175-177
14. Zhang J. et al. p16 promoter hypermethylation in the plasma DNA and its possible application in molecular diagnosis of hepatocellular carcinoma. Chin J Lab Med. 2006;29(10):895-898
15. Ahmed R, Salama H, Fouad A. et al. Detection of aberrant p16INK4A methylation in sera of patients with HCV-related liver diseases: An Egyptian study. Med Sci Monit. 2010;16(9):CR410-CR415
16. He QF. et al. Promoter hypermethylation of RASSF1A and p16 gene in plasma of patients with hepatocellular carcinoma. Chin J Public Health. 2010;26(7):819-821
17. Ye Z. et al. Methylation profile of tumor suppressor genes in the cell-free DNA of plasma in hepatocellular carcinoma. Chin J Clin Oncol. 2013;40(23):14361440
18. Yang J. et al. The gene methylation of p16, Runx3 in patients with hepatocellular carcinoma. MODERN ONCOLOGY. 2013;21(5):1081-1084
19. El-Mougy Fatma A, Mohammed M et al. Aberrant p16INK4A methylation: Relation to viral related chronic liver disease and hepatocellular carcinoma. South Asian J Cancer. 2014;3(1):1-4
20. Huang W. et al. Analysis of DNA Methylation in Plasma for Monitoring Hepatocarcinogenesis. Genet Test Mol Biomarkers. 2015;19(6):295-302
21. Dong XG. et al. Detection of methylation of p16 and RASSF1A gene in plasma and tumor tissues from patients with hepatocellular carcinoma and its clinical significance. Chin Med Biotechnol. 2016;11(3):252-258
22. Sui C, Wang G, Chen Q. et al. Variation risks of SFRP2 hypermethylation between precancerous disease and colorectal cancer. Tumour Biol. 2014;35(10):10457-65
23. Toiyama Y, Okugawa Y, Goel A. DNA methylation and microRNA biomarkers for noninvasive detection of gastric and colorectal cancer. Biochem Biophys Res Commun. 2014;455(1-2):43-57
24. Zang JJ, Xie F, Xu JF. et al. p16 gene hypermethylation and hepatocellular carcinoma: a systematic review and meta-analysis. World J Gastroenterol. 2011;17(25):3043-3048
25. Lv X, Ye G, Zhang X. et al. p16 Methylation was associated with the development, age, hepatic viruses infection of hepatocellular carcinoma, and p16 expression had a poor survival: A systematic meta-analysis. Medicine (Baltimore). 2017;96(38):e8106
Author contact
![]() Corresponding author: Jianzhong Ma, Department of public foundation, China Medical University, Shenyang 110122, China; Phone: 086-13386885893. Fax: 86-024-23865539. E-mail: jzmaedu.cn
Corresponding author: Jianzhong Ma, Department of public foundation, China Medical University, Shenyang 110122, China; Phone: 086-13386885893. Fax: 86-024-23865539. E-mail: jzmaedu.cn
Citation styles
APA
Jiang, C.G., Chen, Q., Wu, L., Wang, G., Ma, J. (2018). The innovative regularity and role of p16 methylation in blood during HCC development. Journal of Cancer, 9(11), 1925-1931. https://doi.org/10.7150/jca.23968.
ACS
Jiang, C.G.; Chen, Q.; Wu, L.; Wang, G.; Ma, J. The innovative regularity and role of p16 methylation in blood during HCC development. J. Cancer 2018, 9 (11), 1925-1931. DOI: 10.7150/jca.23968.
NLM
Jiang CG, Chen Q, Wu L, Wang G, Ma J. The innovative regularity and role of p16 methylation in blood during HCC development. J Cancer 2018; 9(11):1925-1931. doi:10.7150/jca.23968. https://www.jcancer.org/v09p1925.htm
CSE
Jiang CG, Chen Q, Wu L, Wang G, Ma J. 2018. The innovative regularity and role of p16 methylation in blood during HCC development. J Cancer. 9(11):1925-1931.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.