Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(18):3407-3416. doi:10.7150/jca.24201 This issue Cite
Research Paper
Autophagy Inhibition Promotes Bevacizumab-induced Apoptosis and Proliferation Inhibition in Colorectal Cancer Cells
Zhi Zhao1#, Guanggai Xia2#, Ni Li3, Ruping Su1, Xiao Chen1, Li Zhong1 ![]()
1. Department of Gastrointestinal and Hernia Surgery, People's Hospital of Guilin, Guilin, China, 541002
2. Department of General Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai, China, 200233
3. Health Management Center, People's Hospital of Guilin, Guilin, China, 541002
# Zhi Zhao and Guanggai Xia contributed equally to this work.
Received 2017-12-5; Accepted 2018-7-2; Published 2018-9-8
Citation:
Zhao Z, Xia G, Li N, Su R, Chen X, Zhong L. Autophagy Inhibition Promotes Bevacizumab-induced Apoptosis and Proliferation Inhibition in Colorectal Cancer Cells. J Cancer 2018; 9(18):3407-3416. doi:10.7150/jca.24201. https://www.jcancer.org/v09p3407.htm
Other stylesAbstract
Aim: Anti-VEGF therapy plays an important role in the treatment of malignant tumors, especially metastatic malignant tumors. However, resistance and an inefficient response to anti-VEGF therapy exist. The current study aimed to investigate whether autophagy plays a part in the anti-tumor effect of bevacizumab in colorectal cancer cells.
Methods: VEGF-A expression was measured by immunohistochemical methods. Cell viability and cell apoptosis were detected using 3-(4,5-dimethylthiazol-2-yl)-3,5-diphenylformazan (MTT) and flow cytometry. Autophagy was assessed by a western blot, fluorescence microscopy and transmission electron microscopy. HIF-1α was measured using a western blot. A xenograft tumor model of colorectal cancer was constructed to determine the efficacy of the treatment of bevacizumab and chloroquine.
Results: VEGF-A protein was upregulated in colorectal cancer tissue. Anti-VEGF (bevacizumab) inhibited cell viability and induced apoptosis. Moreover, bevacizumab induced autophagy. The inhibition of autophagy by chloroquine or by small interfering RNA promoted bevacizumab-induced apoptosis and proliferation inhibition. Further study showed that bevacizumab treatment significantly augmented HIF-1α. Furthermore, cells pretreated with YC-1, a HIF-1α inhibitor, displayed significantly attenuated bevacizumab-induced autophagy. Finally, a combinatory treatment of bevacizumab and chloroquine synergistically inhibited tumor growth in a xenograft tumor model of colorectal cancer cells.
Conclusions: Our results showed that the inhibition of autophagy promoted the anti-tumor effect of bevacizumab and may offer a promising therapeutic strategy for colorectal cancer.
Keywords: Autophagy, Anti-VEGF, Bevacizumab, Colorectal cancer
Introduction
It is estimated that colorectal cancer is the third most frequently diagnosed cancer in males and the second in females [1]. In 2008, an estimated 12 million new cases of colorectal cancer were diagnosed, and approximately 608,700 people died from colorectal cancer [2]. In 2010, the death rate from colorectal cancer declined by 46%. These improvements in mortality are thought to be a result of cancer prevention, earlier diagnosis and better treatment methods, especially with the progress of surgical procedures and chemotherapy. However, approximately 50% to 60% of colorectal cancer patients will develop distant metastases, and 50% will die from colorectal cancer recurrence [3].
Recently, targeted therapy is recognized as an effective therapeutic strategy to treat human cancer, especially anti-vascular endothelial growth factor (VEGF) therapy [4-6]. VEGF, which was identified by Judah Folkman, is recognized as a key regulator of normal and abnormal angiogenesis [7]. VEGF stimulates blood vessel growth and recruits a neovascular supply to promote tumor growth. Moreover, it is reported that VEGF expression is overexpressed in most solid tumors [8, 9]. Bevacizumab, a humanized monoclonal anti-VEGF antibody, dramatically suppresses tumor growth by blocking angiogenesis. At the present time, bevacizumab is widely applied in metastatic colorectal cancer, lung cancer and renal cell carcinoma [10-12]. However, it is reported that resistance and an inefficient response to bevacizumab exist [6, 13]. Therefore, it is necessary to find new therapeutic approaches and an optimized combined treatment.
Macroautophagy (herein referred to as autophagy) is a highly conserved self- degradation process whereby cellular proteins and organelles are captured and digested in double membrane-bound vesicles and delivered to lysosomes for degradation and recycling [14, 15]. Under normal conditions, autophagy plays a key role to maintain cellular homeostasis. Recently, increasing evidence demonstrates that autophagy plays a protective role in established tumor cells, especially in the conditions of nutrient, oxygen and growth factor deprivation and chemotherapeutics [16-18]. Therefore, autophagy inhibition represents a promising strategy to treat human cancer.
However, it is unknown whether the inhibition of angiogenesis using bevacizumab induces autophagy and whether autophagy inhibition increases the anticancer effects of bevacizumab in colorectal cancer cells. In the present study, we investigated whether inhibiting angiogenesis using bevacizumab induces autophagy. In addition, we investigated whether the inhibition of autophagy promotes the anticancer effects of bevacizumab.
Materials and Methods
Patient samples
Patient samples from 110 patients were obtained from the pathology department at the people's hospital of Guilin between January 2004 and November 2009. Patients were included in the present study if they fulfilled the following criteria: (a) a pathological diagnosis of colorectal cancer; (b) no previous malignancy or second primary tumor; (c) they did not receive any treatment prior to the curative excision of the primary tumor. The 7th TNM colorectal cancer classification of the American Joint Committee on Cancer was used for pathological staging.
Immunohistochemistry
Tissue section immunohistochemistry was performed according to the manufacturer's instructions as previously described. Briefly, 4 μm thick tissue sections were deparaffinized and rehydrated. After antigen retrieval was performed, the tissue sections were incubated with an anti-VEGF antibody (dilution 1:500) overnight at 4 °C. Next, the slides were incubated with a secondary antibody for 30 min at room temperature. Immunoreactivity was revealed using DAB (3,3-diaminobenzidine). Finally, Mayer's hematoxylin was used for counterstaining.
Assessment of immunostaining
Immunostaining was semiquantitatively evaluated based on both the percentage of positive cells (0, 0-10%; 1, 10-25%; 2, 26-50%; 3, 51-70%; 4, >70%) and the intensity of the stain (0, negative; 1, weak; and 2, strong). Finally, an immunoreactivity score was calculated by adding the immunostaining intensity score and the percentage of positive cells. Tumors with immunoreactivity scores less than 4 were considered low VEGF expression, whereas cases in which the scores were equal or higher than 4 were considered high VEGF expression. The immunostaining results were evaluated by two independent pathologists.
Cell culture
The human colorectal cancer cells lines HT-29 was cultured in DMEM medium (Gibco), supplemented with 10% fetal bovine serum (FBS), 100 UI/ml penicillin, and 100 mg/ml streptomycin in a 37°C humidified atmosphere containing 5% CO2.
Reagents and antibodies
The antibodies were all purchased from commercial sources and included VEGF-A (Abcam, UK), LC3 (CST, USA), Beclin-1 (CST, USA), and HIF-1α (Abcam, UK). The reagents used in this study included chloroquine (CQ) (Sigma-Aldrich, USA), 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) (Sigma-Aldrich, USA), YC-1 (Sigma-Aldrich, USA), bevacizumab (Roche, Switzerland), the Annexin V-FITC Assay Kit (Kengen, China) and the LipofectamineTM 2000 reagent (Invitrogen, USA). The Annexin V-FITC Apoptosis Detection Kit (Kengen, China) was also used. The human colon cancer cell line HT29 was purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai).
Apoptosis analysis using flow cytometry
Apoptotic cells were assessed according to the protocol of the Annexin V-FITC Apoptosis Detection Kit as described previously. Briefly, the cells were seeded in 6-well plates and were treated with bevacizumab in the absence or presence of CQ for 24 h. After harvesting, the cells were resuspended in 500 µl of binding buffer supplemented with 5 μl of AnnexinV-FITC and 5 μl of propidium iodide (PI). Finally, the cells were analyzed using flow cytometry (Becton Dickinson, USA).
MTT assay
Cellular viability was assessed using an MTT assay. The cells were seeded in 96-well plates at 5×103 cells per well and were treated with bevacizumab in the absence or presence of chloroquine for 24 h. Each dose of the compound was tested in quadruplicate. After treatment, the cell viability was evaluated by an MTT assay. The absorbance was measured at 540 nm with a microplate reader (Bio-Rad, USA).
GFP-LC3 transient transfection
The cells were transfected with the pEGFP-LC3 plasmid using LipofectamineTM 2000 reagent according to the manufacturer's instruction and were maintained on coverslips in 6-well plates. After transfection for 48 h, the cells were treated with bevacizumab in the absence or presence of CQ for 24 h. Subsequently, the cells were observed using fluorescence microscopy (BD Bioscience).
Western blot
Briefly, the cells were lysed in RIPA buffer (50 mM Tris-base, 1.0 mM EDTA, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate, and 1 mM PMSF). The lysates were cleared by centrifugation at 12000 g for 10 min at 4 °C. The protein concentrations were measured using the Bradford assay. Equal amounts of protein were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and were then transferred to PVDF membranes (Millipore, USA). The membrane was washed, blocked, and, subsequently, incubated overnight with a primary antibody at 4°C followed by incubation with a horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h at room temperature. The protein bands were detected using an enhanced chemiluminescence assay (Millipore, USA). β-actin was used as an internal control. Band intensities were determined using Image J software.
RNA interference
Small interfering RNA (siRNA) targeting human Beclin-1 was synthesized by RiBOBIO Co. (Guangzhou, China) together with control siRNA. The siRNA sequences against Beclin 1 were previously described [16]. The cells were transfected with either Beclin-1 or control siRNA at 100 nM using the LipofectamineTM 2000 reagent for 48 h according to the manufacturer's instructions.
Transmission electron microscopy
After the treatment with bevacizumab in the absence or presence of CQ for 24 h, the cells were fixed with 2.5% glutaraldehyde with 0.1 mol/l sodium cacodylate, post-fixed in 1% phosphate-buffered osmium tetroxide for 1.5 h, washed and, finally, were stained for 1 h in 3% aqueous uranyl acetate. The samples were then dehydrated with graded alcohol and embedded. After sectioning using a Reichert Ultramicrotome, the samples were stained with lead citrate and analyzed using a Philips EM420 transmission electron microscope.
Tumor xenograft model
The experiment was performed with the permission of the Animal Ethics Committee of Guilin people's Hospital. Female nude mice (BALB/c, 6-8 weeks of age, nonfertile and 18-20 g each) were purchased from the pathogen-free animal breeding facility of the Guangdong province (Guangzhou, China), maintained under specific pathogen-free conditions. The HT-29 cells (5×106 cells/mouse), in 0.2 mL of PBS, were injected subcutaneously into the left lower flank region. Once the tumors reached a measurable size (tumor diameter was approximately 5 mm), the mice were randomly assigned to receive one of the following four groups: (i) the control group received an intraperitoneal injection of PBS 5 times a week (n=4); (ii) the chloroquine group received an intraperitoneal injection of chloroquine 60 mg/kg 5 times a week (n=4); (iii) the bevacizumab group received an intraperitoneal injection of bevacizumab 5 mg/kg 5 times a week (n=4); or (iv) a combination of treatments ii+iii (n=4). The tumors were measured with calipers every 5 d, and the tumor volumes were calculated using the formula: volume (mm3) =length×width2/2, where the length represents the largest diameter, and the width is the smallest diameter. The animals were sacrificed by day 28. The tumor tissues were completely removed and photographed.
Statistical analysis
All statistical analyses were performed using SPSS 17.0 statistical software. The variance analysis between the groups was performed by an independent t-test or a one-way ANOVA. The categorical data were analyzed using a chi-square test. The Kaplan-Meier test was used to evaluate the survival rate, and the survival rate curve was compared using the log-rank test. The Cox proportional hazards model was used for the multivariate survival analysis. The differences with P<0.05 were considered statistically significant.
Table 1
Clinicopathological correlations with VEGF-A expression in colorectal cancer
| Variable | VEGF-A low expression (52) | VEGF-A high expression (58) | P |
|---|---|---|---|
| Age (y) | 0.169 | ||
| < 60 | 36 | 34 | |
| ≥ 60 | 16 | 24 | |
| Gender | 0.201 | ||
| Male | 38 | 37 | |
| Female | 14 | 21 | |
| Tumor size (cm) | 0.416 | ||
| ≤4 | 24 | 29 | |
| >4 | 28 | 29 | |
| Histological type | 0.326 | ||
| Well | 3 | 0 | |
| Moderately | 13 | 15 | |
| Poorly | 35 | 42 | |
| Undifferentiated | 1 | 1 | |
| pT stage | 0.327 | ||
| T1 | 4 | 1 | |
| T2 | 3 | 3 | |
| T3 | 4 | 2 | |
| T4 | 41 | 52 | |
| pN stage | 0.047 | ||
| N0 | 18 | 7 | |
| N1 | 9 | 14 | |
| N2 | 12 | 18 | |
| N3 | 13 | 19 | |
| pTNM stage | 0.012 | ||
| I | 3 | 1 | |
| II | 17 | 7 | |
| III | 32 | 50 |
Results
VEGF protein is upregulated in colorectal cancer
Representative images of VEGF staining are shown in Fig. 1A. VEGF-A expression was observed in the cytoplasm. Fifty-eight patients exhibited high VEGF expression, and 52 patients displayed low VEGF-A expression. The correlations between VEGF-A expression and the clinical variables are listed in Table 1. VEGF expression was closely associated with the pathological (p) TNM stage (P=0.012) and pN stage (P=0.047). Moreover, patients with a high VEGF-A expression showed a significantly shorter overall survival (OS) (P=0.000) and disease-free survival (DFS) (P=0.007) (Fig. 1B) However, no significant correlations were observed between VEGF-A expression and age, gender, tumor size, Borrmann type, histological type, and pT stage (Table 1).
Fig 1
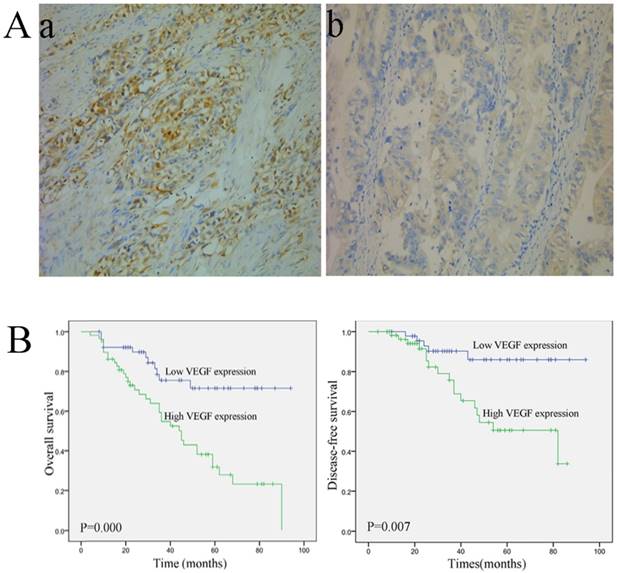
Immunohistochemical staining and survival curve for VEGF-A. (A) VEGF-A protein is expressed in colorectal cancer tissue. (a, 200×) High VEGF-A expression in colorectal cancer tissue; (b, 200×) Low VEGF-A expression in colorectal cancer tissue; (B) A Kaplan-Meier analysis shows that patients with a high VEGF-A expression have a significantly poorer overall survival (a) and disease-free survival (b) compared to those with a low VEGF-A expression (P < 0.01 for both).
Bevacizumab inhibits cell viability
Colorectal cancer cells were treated with bevacizumab for 24, 48 or 72 h. As shown in Fig. 2, bevacizumab inhibited cell growth in a dose- and time-dependent manner. The IC50 value for bevacizumab in the HT-29 cells was 22.50 mg/mL.
Fig 2
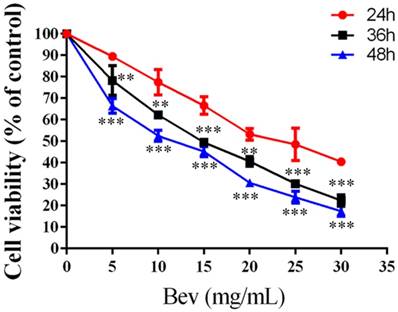
Bevacizumab inhibits cell viability. Cell proliferation was analyzed by MTT in colorectal cancer cells treated with bevacizumab at the indicated concentrations for 24, 36 or 48 h. Bevacizumab inhibited the proliferation of the colorectal cancer cells in a dose- and time-dependent manner. Bev=bevacizumab; **, P<0.01 compared with 24h; ***, P<0.001 compared with 24h.
Bevacizumab induces autophagy in colorectal cancer cells
The lipidated form of LC3 transforming from LC3-I to LC3-II is a characteristic of autophagy. As indicated in Fig. 3A, an increased level of LC3-II was observed in the colorectal cancer cells after exposure to bevacizumab. Moreover, Beclin-1, a key autophagy-related gene, was also activated after exposure to bevacizumab (Fig. 3A). To further confirm that LC3 was involved in bevacizumab-induced autophagy, the pEGFP-LC3 plasmid was transiently transfected into the HT-29 cells. As shown in Fig. 3B, the un-treated control group cells exhibited diffuse and weak LC3 punctate dots, whereas the bevacizumab-treated cells exhibited intense and granular LC3 punctate dots in the cytoplasm. In addition, we further observed the ultrastructure of the colorectal cancer cells treated with bevacizumab for 24 h by transmission electron microscopy. As shown in Fig. 3C, numerous autophagic vesicles were observed in the cytoplasm of the bevacizumab treated group, whereas autophagic vesicles were rare in the control group. Taken together, our results indicated that bevacizumab induced autophagy in colorectal cancer cells.
Autophagy inhibition promotes bevacizumab-induced apoptosis and proliferation inhibition in colorectal cancer cells
Increasing evidence suggests that autophagy acts as a double-edged sword in the control of cell death and survival under various conditions. To determine the biological role of autophagy in cell survival or death, the autophagy inhibitor CQ was utilized to block the autophagic process in the colorectal cancer cells. To this end, the cells were treated with CQ, bevacizumab alone or in combination. Our results showed that the co-treatment with bevacizumab and CQ significantly promoted bevacizumab-induced colorectal cancer cells growth suppression (Fig. 4A). In line with this observation, the co-treatment with bevacizumab and CQ also significantly increased apoptotic cell death (Fig. 4B). To further confirm that bevacizumab-induced autophagy had a protective role on cell apoptosis, an alternate approach was applied to inhibit autophagy using RNA interference to knock down the expression of Beclin 1. Figure 4C shows that the expression level of Beclin 1 was markedly reduced in the Beclin 1 siRNA-treated cells. As a result, silencing the expression of beclin 1 markedly enhanced the inhibition of cell growth (Fig. 4D) and promoted apoptotic cell death (Fig. 4E). These findings suggested that autophagy might play a protective role against apoptotic cell death in colorectal cancer cells.
HIF-1α accumulation mediates bevacizumab-induced autophagy in colorectal cancer cells
It is well known that angiogenesis promotes tumor initiation and progression by bringing large amounts of nutrient metabolism, oxygen and energy. The inhibition of angiogenesis results in a deficiency of oxygen and energy. Intratumoral hypoxia leads to HIF-1α overexpression. Thus, we determined whether HIF-1α was involved in bevacizumab-induced autophagy. As shown in Fig. 5A, a western blot analysis revealed that HIF-1α generation was upregulated in the bevacizumab-treated group. To better understand the functional role of HIF-1α in autophagy induced by bevacizumab, we utilized YC-1, a HIF-1α inhibitor, to block HIF-1α generation. As a result, in the presence of YC-1, there was autophagic protein accumulation (Fig. 5B), and the average number of GFP-LC3 dots per cell (Fig. 5C) significantly decreased. Moreover, after treated with YC-1, bevacizumab-induced apoptotic cell death was significantly increased (Fig. 5D). Consistent with this observation, cell viability assays showed that YC-1 promotes bevacizumab-induced tumor cell growth suppression (Fig. 5E). These results suggested that HIF-1α plays a critical role in the induction of autophagy.
Fig 3
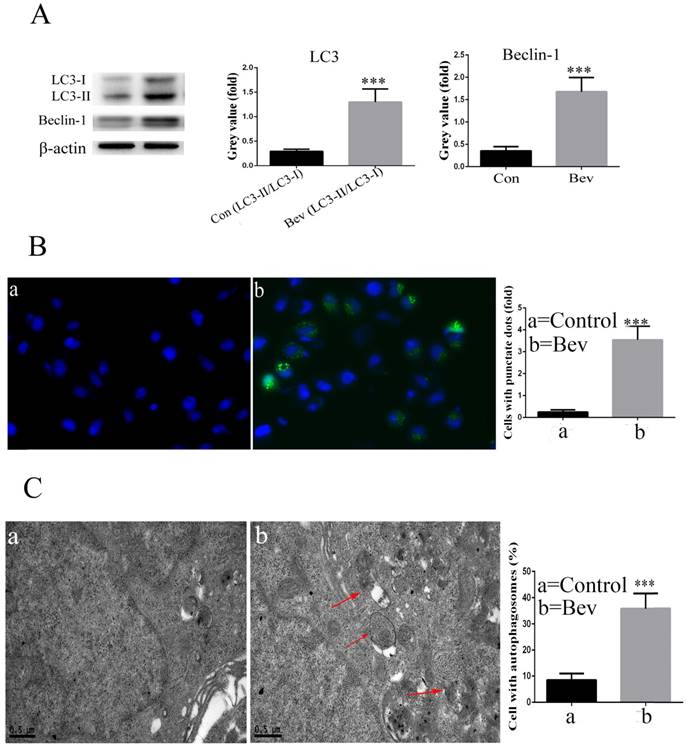
Bevacizumab induces autophagy in HT-29 cells. (A) After the cells were exposed to Bevacizumab for the indicated times, the cell lysates from the HT-29 cells were subjected to a western blot analysis for LC3 and Beclin-1. (B) The HT-29 cells were transiently transfected with the pEGFP-LC3 plasmid and treated with Bevacizumab. The number of GFP-LC3 puncta in each cell was counted. The percentage of GFP-LC3 puncta containing cells was assessed by fluorescence microscopy. (C) Representative transmission electron microscopy (TEM) images depicting ultrastructures in the HT-29 cells treated with Bevacizumab. Red arrows=autophagic vacuoles. Con=Control; Bev=bevacizumab; ***, P<0.001 compared with control.
Autophagy inhibition enhances the anticancer activity of bevacizumab in vivo
To confirm the synergistic effect of bevacizumab and CQ for colorectal cancer in vivo, HT-29 cells were injected into nude mice. As shown in Fig. 6A-B, at day 28, the mice did not show a significant difference in tumor volume after chloroquine administration compared with the vehicle controls. However, the tumor volumes in the bevacizumab group and combined group were all reduced compared to the control group (Fig. 6A-B). At 28 days, the mean tumor volume in the control group was 968 ± 96.64 mm3. In comparison to the control group, the tumor inhibition rates were 45.2% and 61.9% in the bevacizumab and combined groups, respectively. The tumor weight measurements were consistent with those of the tumor size (Fig. 6C). Moreover, bevacizumab-induced apoptotic cell death was significantly increased in the presence of CQ (Fig. 6D). In addition, immunohistochemical analysis confirms autophagy induction as evidenced by increased LC3 staining in bevacizumab-treated tumors. Co-treatment with CQ, an inhibitor of autophagy that blocks the final steps of autophagic degradation, enhanced the bevacizumab-induced accumulation of LC3 (Fig. 6E). These results demonstrated that bevacizumab and CQ acted synergistically against colorectal cancer in vivo.
Discussion
A century ago, Rudolf Virchow first observed that tumor growth is accompanied by increased vascularity, which suggests that blocking new blood vessel formation in tumors will stop or slow their growth [19]. Tumors induce the sprouting of new vessels from the surrounding vasculature and achieve this process via the expression of pro-angiogenic growth factors, such as members of the VEGF family of ligands. Bevacizumab, a humanized monoclonal antibody that binds specifically to VEGF-A alone, is approved by the FDA and is applied as the first line treatment in many solid tumors, such as metastatic colorectal cancer, metastatic non-small-cell lung cancer and metastasis breast cancer. However, resistance and an inefficient response to anti-angiogenic therapy exist in some solid tumors. Recently, it was reported that VEGF-A expression is upregulated in many solid tumors. In our study, we also found that VEGF-A expression was upregulated in colorectal cancer. Moreover, the high VEGF-A expression in colorectal cancer tissue was closely associated with the pTNM stage, pN stage, and poor prognosis. These findings are consistent with previous reports [20, 21]. In addition, our results showed that bevacizumab inhibited proliferation and induced apoptosis in colorectal cancer cells in vitro.
Fig 4
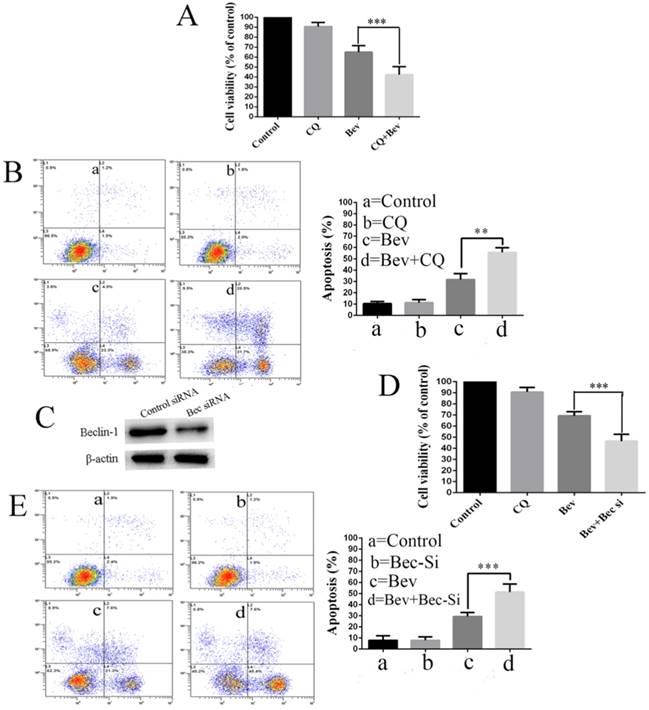
Autophagy inhibition promotes bevacizumab-induced proliferation inhibition and apoptosis in colorectal cancer cells. (A) Cell proliferation was analyzed by MTT in colorectal cancer cells treated with bevacizumab (20 mg/mL) in the absence or presence of 20 µM CQ for 24 h. (B) Cells apoptosis was analyzed by flow cytometry in colorectal cancer cells treated with bevacizumab (20 mg/mL) in the absence or presence of 20 µM CQ for 24 h. (C) The cells were transfected with Beclin-1 siRNA for 48 h, and the expression of Beclin-1 was detected by a western blot. β-actin was used as the loading control. (D) After transfection for forty-eight hours, the cells were treated with bevacizumab (20 mg/mL) for another 24 h, and cell proliferation was analyzed by MTT. (E) After transfection for forty-eight hours, cell apoptosis was analyzed by flow cytometry. Con=Control; CQ=chloroquine; Bev=Bevacizumab; **, P<0.01; ***, P<0.001.
Fig 5
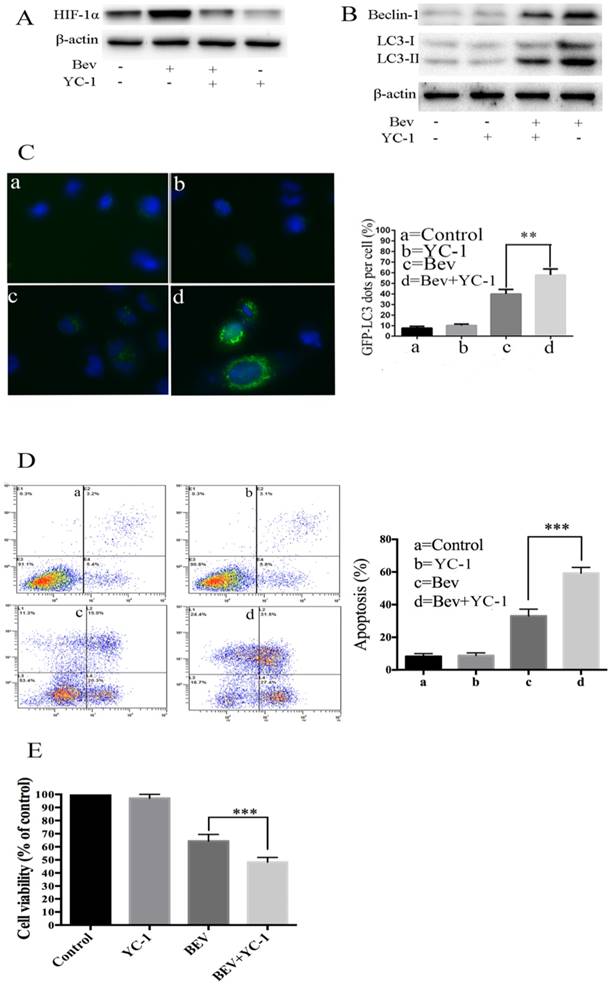
HIF-1α accumulation mediates bevacizumab-induced autophagy in colorectal cancer cells. (A) Cellular HIF-1α expression was assayed by a western blot after treatment with bevacizumab (20 mg/mL) for 24 h in colorectal cancer cells. (B) The cells were pretreated with YC-1 (5 µM) for 12 hours. Following pretreatment with YC-1, the cells were exposed to bevacizumab (20 mg/mL) for 24 h. The cell lysates were subjected to a western blot analysis for LC3 and Beclin-1. (C) The cells were transiently transfected with the pEGFP-LC3 plasmid. After pretreatment with YC-1 (5 µM), the cells were treated with bevacizumab (20 mg/mL) for 24 h. The number of GFP-LC3 puncta in each cell was counted. The percentage of GFP-LC3 puncta containing cells was assessed from 100 random fields. (D) After pretreatment with YC-1 (5 µM), the cells were treated with bevacizumab (20 mg/mL) for 24 h, and apoptosis was analyzed by flow cytometry. (E) After pretreatment with YC-1 (5 µM), the cells were treated with bevacizumab (20 mg/mL) for 24 h, and cell viability was analyzed by MTT. β-actin was used as the loading control; Con=Control; CQ=chloroquine; Bev=Bevacizumab; **, P<0.01; ***, P<0.001.
Fig 6
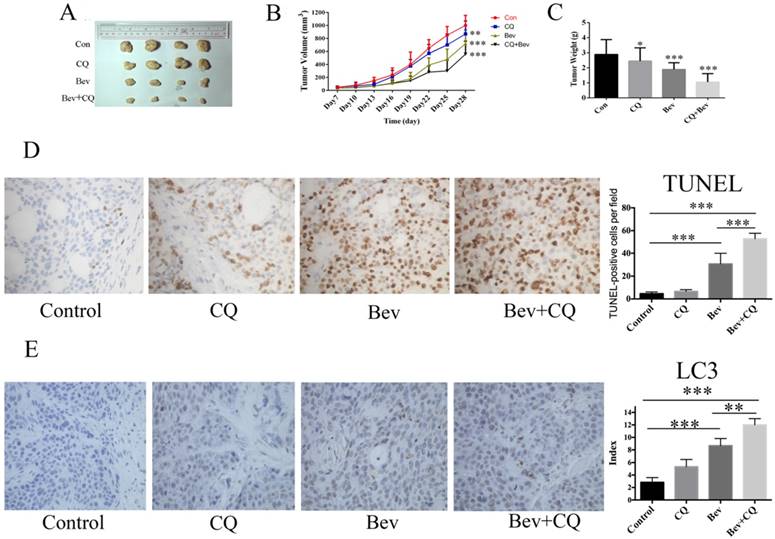
Bevacizumab and CQ act synergistically against xenograft tumor proliferation in nude mice. (A) Xenograft tumors of four groups; (B) The tumor volume of each group was measured every 3 days by a caliper; (C) The tumor weight was calculated after the nude mice were sacrificed; (D) Cell apoptosis was measured by TUNEL staining of tumor tissue sections. TUNEL-positive cells were quantified at magnification (×400) for 8 fields of each tumor sample. (E) Immunohistochemical detection of LC3 in HT29 tumor section. Con=Control; CQ=chloroquine; Bev=Bevacizumab; NS, no significance; *, P<0.05 compared with the control; **, P<0.01 compared with the control;***, P<0.001 compared with the control.
Autophagy is a highly conserved, dynamic and lysosome-mediated process that degrades and recycles cellular components within the lysosome to generate cellular energy and building blocks for biosynthesis [21]. Autophagy plays a key role in the process of tumor initiation and growth [22, 23]. Increasing evidence indicates that many tumor cells upregulate autophagy to promote tumor growth under the conditions of nutrition starvation, hypoxic, and therapeutic stress. In our study, we showed that bevacizumab induced autophagy, as evidenced by the appearance of autophagic vacuoles, punctate patterns of LC3 and the accumulation of Beclin 1. Moreover, inhibiting autophagy using CQ or RNA interference promoted bevacizumab-induced apoptosis and proliferation inhibition, suggesting that autophagy plays a protective role in tumor cells.
We not only found that the inhibition of autophagy significantly enhanced the bevacizumab-induced inhibition of proliferation in vitro, but we also found the same results in vivo. After inhibiting autophagy by CQ or using small interfering RNA, bevacizumab significantly inhibited tumor cell proliferation in vivo in contrast to the bevacizumab-treated alone group. Our results are consistent with previous reports. Muthu Selvakumaran et al reports that inhibition of autophagy by chloroquine sensitizes mouse colon cancer xenografts to bevacizumab [24]. However, they did not prove that autophagy inhibition promotes the anti-tumor effect of bevacizumab in vitro. Moreover, they did not elucidate the exact mechanism of bevacizumab-induced autophagy.
It is well known that antiangiogenic therapy inhibits tumor growth by blocking new blood vessel formation. Decreases in vessel numbers could result in increases in intra-tumor hypoxia. Given that HIF-1α plays a vital role in the hypoxia microenvironment, we focused our attention on HIF-1α generation. Our results showed that intracellular HIF-1α production was significantly increased after treatment with bevacizumab. Moreover, after treatment with the HIF-1α inhibitor YC-1, autophagy induced by bevacizumab was markedly reduced. Since autophagy is induced as a response to cellular stress, our results of hypoxia-induced autophagy in tumor cells may function as an adaptive response to the hypoxia caused by anti-angiogenic therapy. This finding is consistent with previous studies [25, 26].
In conclusion, in our study, we demonstrated that bevacizumab inhibited proliferation and induced apoptosis in colorectal cancer cells. Moreover, we also found that bevacizumab induced autophagy by activating HIF-1α. In addition, the inhibition of autophagy significantly enhanced bevacizumab-induced apoptosis and the inhibition of proliferation in vitro. Similarly, we also found that inhibited autophagy significantly enhanced the inhibition of proliferation in vivo. Taken together, our results suggest that combining autophagy inhibitors with bevacizumab may provide a promising approach for the treatment of colorectal cancer. However, further clinical studies are necessary to validate the combined effects.
Abbreviations
VEGF: anti-vascular endothelial growth factor; CQ: chloroquine; DAB: 3,3-diaminobenzidine; FBS: fetal bovine serum; MTT: 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide; EDTA: ethylenediaminetetraacetic acid; PMSF: phenylmethanesulfonyl fluoride; SDS: sodium dodecyl sulfate; PAGE: polyacrylamide gel electrophoresis; PVDF: polyvinylidene fluoride; HRP: horseradish peroxidase; siRNA: Small interfering RNA; p: pathological; OS: overall survival; DFS: disease-free survival.
Acknowledgements
This study was supported by the Wu Jieping medical fund (No. 320.6750.13390).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Torre LA, Bray F, Siegel RL. et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87-108
2. Jemal A, Center MM, DeSantis C. et al. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19:1893-1907
3. Islami F, Goding Sauer A, Miller KD. et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J Clin. 2017;68:31-54
4. Vasudev NS, Reynolds AR. Anti-angiogenic therapy for cancer: current progress, unresolved questions and future directions. Angiogenesis. 2014;17:471-494
5. Chebib R, Verlingue L, Cozic N. et al. Angiogenesis inhibition in the second-line treatment of metastatic colorectal cancer: A systematic review and pooled analysis. Semin Oncol. 2017;44:114-128
6. Van Beijnum JR, Nowak-Sliwinska P, Huijbers EJ. et al. The great escape; the hallmarks of resistance to antiangiogenic therapy. Pharmacol Rev. 2015;67:441-461
7. Kerbel RS. A Decade of Experience in Developing Preclinical Models of Advanced- or Early-Stage Spontaneous Metastasis to Study Antiangiogenic Drugs, Metronomic Chemotherapy, and the Tumor Microenvironment. Cancer J. 2015;21:274-283
8. Mohamed SY, Mohammed HL, Ibrahim HM. et al. Role of VEGF, CD105, and CD31 in the Prognosis of Colorectal Cancer Cases. J Gastrointest Cancer. 2017;6:1-10
9. Rubatt JM, Darcy KM, Hutson A. et al. Independent prognostic relevance of microvessel density in advanced epithelial ovarian cancer and associations between CD31, CD105, p53 status, and angiogenic marker expression: A Gynecologic Oncology Group study. Gynecol Oncol. 2009;112:469-474
10. Giampieri R, Caporale M, Pietrantonio F. et al. Second-line angiogenesis inhibition in metastatic colorectal cancer patients: Straightforward or overcrowded? Crit Rev Oncol Hematol. 2016;100:99-106
11. Stratigos M, Matikas A, Voutsina A. et al. Targeting angiogenesis in small cell lung cancer. Transl Lung Cancer Res. 2016;5:389-400
12. Jain RK, Gandhi S, George S. Second-line systemic therapy in metastatic renal-cell carcinoma: A review. Urol Oncol. 2017;35:640-646
13. Sathornsumetee S, Rich JN. Antiangiogenic therapy in malignant glioma: promise and challenge. Curr Pharm Des. 2007;13:3545-3558
14. Gómez-Sintes R, Villarejo-Zori B, Serrano-Puebla A. et al. Standard Assays for the Study of Autophagy in the Ex Vivo Retina. Cells. 2017;22:6 1-10
15. Ngabire D, Kim GD. Autophagy and Inflammatory Response in the Tumor Microenvironment. Int J Mol Sci. 2017;18:1-15
16. Zhao Z, Han F, Yang S. et al. Oxamate-mediated inhibition of lactate dehydrogenase induces protective autophagy in gastric cancer cells: Involvement of the Akt-mTOR signaling pathway. Cancer Lett. 2015;358:17-26
17. Poillet-Perez L, Despouy G, Delage-Mourroux R. et al. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015;4:184-192
18. Zhu J, Zheng Y, Zhang H. et al. Low concentration of chloroquine enhanced efficacy of cisplatin in the treatment of human ovarian cancer dependent on autophagy. Am J Transl Res. 2017;9:4046-4058
19. Ebos JML, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nature Reviews Clinical Oncology. 2011;8:210-221
20. Mahecha AM, Wang H. The influence of vascular endothelial growth factor-A and matrix metalloproteinase-2 and -9 in angiogenesis, metastasis, and prognosis of endometrial cancer. Onco Targets Ther. 2017;10:4617-4624
21. Zhan P, Wang J, Lv XJ. et al. Prognostic value of vascular endothelial growth factor expression in patients with lung cancer: a systematic review with meta-analysis. J Thorac Oncol. 2009;4:1094-1103
22. White E, Mehnert JM, Chan CS. Autophagy, Metabolism, and Cancer. Clin Cancer Res. 2015;21:5037-5046
23. White E. The role for autophagy in cancer. J Clin Invest. 2015;125:42-46
24. Selvakumaran M, Amaravadi RK, Vasilevskaya IA. et al. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clin Cancer Res. 2013;19:2995-3007
25. Zhou J, Yao W, Li C. et al. Administration of follicle-stimulating hormone induces autophagy via upregulation of HIF-1alpha in mouse granulosa cells. Cell Death Dis. 2017;8:3001-3012
26. Gui L, Liu B, Lv G. Hypoxia induces autophagy in cardiomyocytes via a hypoxia-inducible factor 1-dependent mechanism. Exp Ther Med. 2016;11:2233-2239
Author contact
![]() Corresponding author: Li Zhong. E-mail: Zhongli0302com; Telephone number: +86-773-2822152; Fax number: +86-773-2822152
Corresponding author: Li Zhong. E-mail: Zhongli0302com; Telephone number: +86-773-2822152; Fax number: +86-773-2822152
Citation styles
APA
Zhao, Z., Xia, G., Li, N., Su, R., Chen, X., Zhong, L. (2018). Autophagy Inhibition Promotes Bevacizumab-induced Apoptosis and Proliferation Inhibition in Colorectal Cancer Cells. Journal of Cancer, 9(18), 3407-3416. https://doi.org/10.7150/jca.24201.
ACS
Zhao, Z.; Xia, G.; Li, N.; Su, R.; Chen, X.; Zhong, L. Autophagy Inhibition Promotes Bevacizumab-induced Apoptosis and Proliferation Inhibition in Colorectal Cancer Cells. J. Cancer 2018, 9 (18), 3407-3416. DOI: 10.7150/jca.24201.
NLM
Zhao Z, Xia G, Li N, Su R, Chen X, Zhong L. Autophagy Inhibition Promotes Bevacizumab-induced Apoptosis and Proliferation Inhibition in Colorectal Cancer Cells. J Cancer 2018; 9(18):3407-3416. doi:10.7150/jca.24201. https://www.jcancer.org/v09p3407.htm
CSE
Zhao Z, Xia G, Li N, Su R, Chen X, Zhong L. 2018. Autophagy Inhibition Promotes Bevacizumab-induced Apoptosis and Proliferation Inhibition in Colorectal Cancer Cells. J Cancer. 9(18):3407-3416.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.