Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(21):3939-3949. doi:10.7150/jca.26041 This issue Cite
Research Paper
Overexpression of Peroxiredoxin 6 (PRDX6) Promotes the Aggressive Phenotypes of Esophageal Squamous Cell Carcinoma
Yan He1,2,3,#, Wanglei Xu1,2,#, Yuji Xiao1, Lu Pan1, Guangxia Chen4, Yiting Tang5, Jundong Zhou3, Jinchang Wu3, Wei Zhu1,2, Shuyu Zhang6,1, ![]() , Jianping Cao1,2,
, Jianping Cao1,2, ![]()
1. School of Radiation Medicine and Protection, Medical College of Soochow University, Suzhou 215123, China
2. State Key Laboratory of Radiation Medicine and Protection, Collaborative Innovation Center of Radiation Medicine of Jiangsu Higher Education Institutions, Soochow University, Suzhou 215123, China
3. Suzhou Cancer Center Core Laboratory, Nanjing Medical University Affiliated Suzhou Hospital, Suzhou 215001, China
4. Department of Gastroenterology, First People's Hospital of Xuzhou, Xuzhou 221002, China
5. Department of Radiotherapy, Changzhou Tumor Hospital, Soochow University, Changzhou 213032, China
6. Department of Oncology, The Affiliated Changzhou No.2 People's Hospital of Nanjing Medical University, Changzhou 213100, China
# The first two authors contributed equally to this work.
Received 2018-3-13; Accepted 2018-9-4; Published 2018-10-10
Citation:
He Y, Xu W, Xiao Y, Pan L, Chen G, Tang Y, Zhou J, Wu J, Zhu W, Zhang S, Cao J. Overexpression of Peroxiredoxin 6 (PRDX6) Promotes the Aggressive Phenotypes of Esophageal Squamous Cell Carcinoma. J Cancer 2018; 9(21):3939-3949. doi:10.7150/jca.26041. https://www.jcancer.org/v09p3939.htm
Other stylesAbstract
Esophageal squamous cell carcinoma (ESCC) is one of the most common malignancies. Peroxiredoxin 6 (PRDX6), a member of peroxidase superfamily, has a function of eliminating the reactive oxygen species (ROS), and participates in development of multiple diseases, including tumors. The purpose of this study was to investigate the expression of PRDX6 in normal and cancerous esophageal tissues and to characterize its role in ESCC progression. We found significantly higher expression of PRDX6 in ESCC tissues than in normal esophageal tissues or tumor-adjacent tissues and that the PRDX6 expression level was positively correlated with the proliferation-related markers. In ESCC cells, PRDX6 distribution was more pronounced in the nucleus region. PRDX6 overexpression by an adenovirus significantly promoted cell proliferation, migration and invasion in TE-1 and Eca-109 cells. Conversely, lentivirus-mediated knock-down of PRDX6 expression significantly reduced cell growth, colony formation and metastasis in ESCC cells. PRDX6 modulated the phosphorylation of Akt and Erk1/2, and the expression of MMP2. We also found that PRDX6 and Erk1/2 pathway were mutually regulated in ESCC cells. In addition, PRDX6 overexpression eliminated radiation-induced ROS and decreased consequent cell apoptosis, indicative of a role in radioresistance. Finally, the role of PRDX6 in promoting tumor growth was further confirmed in nude mice with ESCC xenografts. Taken together, we demonstrated that overexpression of PRDX6 promotes the progression of ESCC through Erk1/2, which provides a potential therapeutic target for human ESCC.
Keywords: cell proliferation, esophageal squamous cell carcinoma (ESCC), MAP kinase signaling system, reactive oxygen species (ROS), Peroxiredoxin 6 (PRDX6)
Introduction
Esophageal cell carcinoma is one of the most common and deadliest malignancies worldwide, with elevated incidence and mortality rates in developing countries [1,2]. China is the world's highest incidence of esophageal cancer, and esophageal squamous cell carcinoma (ESCC) constitutes more than 95% of esophageal cancer diagnoses [2,3]. The prognosis of this cancer remains poor because of late diagnosis and the rapid metastasis, despite the currently multiple methods of treatment for ESCC [4]. In addition to surgery, simultaneous radiotherapy and chemotherapy is the current standard treatment of non-surgical treatment of esophageal cancer [5]. Although a variety of dysregulated molecules and potential targets of ESCC have gradually been characterized, the molecular mechanism driving the progression of ESCC remains complex and largely unknown [6,7].
A variety of reactive oxygen species (ROS), such as H2O2 or O2-, are found to be the by-products of cellular aerobic metabolism, ongoing stress and exposure to X-rays [8,9]. Cancer cells are likely to have increased ROS levels compared with normal counterparts, which contributes to the biochemical and molecular changes necessary for the tumor initiation, promotion and progression, as well as tumor resistance to radio- and chemotherapy [10,11]. Peroxiredoxins (PRDXs), a family of non-seleno peroxidases, have gained increasing attention since their recognition in recent years [12,13]. Six members of the PRDX family have been described in mammalian, named PRDX 1-6. PRDX6 is the sole mammalian 1-Cys PRDX [12-14].
PRDX6 was first demonstrated for protein isolated from the ciliary body of the bovine eye [15]. Although PRDX6 shares structural and functional properties with other PRDX members, it has a single conserved Cys residue causing a different catalytic cycle, and it uses glutathione (GSH) instead of thioredoxin as the physiological reductant. Furthermore, PRDX6 exhibits a bifunctional enzyme with both phospholipase A2 (PLA2) activity and peroxidase function [14,16]. PRDX6 overexpression is associated with a number of degenerative diseases such as Alzheimer's, Parkinson's and Crosti syndrome [17,18]. Accumulating studies have shown that aberrant expression of PRDX6 is associated with cancer progression. Jo M et al reported that PRDX6-overexpressing transgenic (Tg) mice displayed a greater increase in the growth of lung tumor compared with normal mice [19]. PRDX6 is highly expressed in melanoma [20], lung [21] and bladder cancers [22]. In these cancers, overexpression of PRDX6 promotes cancer cell proliferation and the invasive phonotype [23,24].
However, the expression and functional significance of PRDX6 in ESCC have not been reported yet. In this study, we investigated the expression of PRDX6 and its functional consequences in ESCC. We found that the PRDX6 expression was significantly increased in ESCC tumor tissues. Furthermore, PRDX6 overexpression increased the proliferation and metastasis capacity in vitro and in vivo. Thus, PRDX6 may serve as a therapeutic target for the treatment of ESCC.
Materials and Methods
Reagents, Vectors and Viruses
The anti-PRDX6 antibody was purchased from Abcam (Cambridge, MA). The antibodies against phospho-Akt, Akt, phospho-Erk1/2, Erk1/2, matrix metalloproteinase 2 (MMP-2), MMP-9 and GAPDH were obtained from Cell Signaling Technology (Beverly, MA). The MEK inhibitor PD0325901 was purchased from Cell Signaling Technology (Beverly, MA).
The human PRDX6 (GenBank accession no. ENSG00000117592) coding region (ENST00000340385.5) was amplified by PCR using a primer pair specific to PRDX6. The amplified fragment was inserted into the pEGFP-N1 vector. The plasmid was then sequenced for confirmation.
Adenovirus overexpressing PRDX6 (Ad-PRDX6) and control adenovirus overexpressing RFP (Ad-RFP) were constructed by Hanbio (Shanghai, China). Both adenoviruses were propagated in 293A cells. Cells were infected with a multiplicity of infection (MOI) of 20 virus particles ⁄ cell 24 h before being used in experimentation. For knock-down of PRDX6, shRNA-expressing lentivirus (sh-PRDX6-1, sh-PRDX6-2 and sh-PRDX6-3) or non-silencing control lentivirus (shRNA-NC) were designed and constructed by Hanbio (Shanghai, China).
Tissue Samples
For immunohistochemistry analysis, 95 ESCC samples and 26 matched, adjacent normal esophagus tissues were collected from 95 patients, as previously reported [25,26]. All patients gave signed, informed consent for their tissues to be used for scientific research. Ethical approval for the study was obtained from the Jiangyin People's Hospital. All diagnoses were based on pathological and/or cytological evidence; the histological features of the specimens were evaluated by a senior pathologist according to the classification criteria from the WHO [27].
Immunohistochemistry (IHC)
IHC staining and the stained section scoring was performed as described previously [25,26].
Cell Culture and Irradiation
The human esophageal cancer cells (TE-1 and Eca-109) and esophageal epithelial HET-1A cells were maintained in DMEM supplemented with 10% FBS and antibiotics (Gibco, Grand Island, NY). Cells were grown in a 37°C incubator with 5% CO2.
For irradiation, cells were exposed to 4 or 8 Gy of ionizing radiation using X-ray linear accelerator (Rad Source, Suwanee, GA) at a fixed dose rate of 1.15 Gy/min.
Western Blotting
Eca-109 and TE-1 cells were lysed in Lysis buffer (Promega, Madison, WI) and centrifuged at 4 °C for 10 min. The supernatant was collected and subjected for Western blotting as described previously [25,26].
Cell Viability Assay
Eca-109 and TE-1 cells were infected with adenoviruses or lentiviruses according to the experimental design. Cell viability was evaluated using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H- tetrazolium bromide (MTT) assay. The cells were incubated with 20 μL MTT (5 mg/ml) for 4 h. After the medium was removed, 100 μL DMSO was added and the optical density (OD) at 490 nm was measured using a microplate reader (Bio-Rad, Hercules, CA). Three independent experiments were performed in quadruplicate.
EdU Assay
The proliferative cells were determined by uptake of 5-Ethynyl-2'-deoxyuridine (EdU) into DNA. Cells were labeled with 50 μM EdU (RiboBio Co., Ltd, Guangzhou, China) for 4 h. Then, the cells were fixed with formaldehyde for 15 min and treated with 0.5% Triton X-100 for 20 min at room temperature. After 3 washes with PBS, the cells were treated with 100 μL of 1x ApolloR reaction cocktail for 30 min. Subsequently, the DNA of each well of cells were stained with Hoechst33342 for 30 min and observed under a fluorescence microscope (Olympus, Tokyo, Japan).
Colony Formation
Cells infected with the indicated viruses were plated at low density (1,000 cells per 6-cm plate), incubated for 10 days and fixed and stained with crystal violet. Foci and colonies containing more than 50 cells were counted using a microscope.
Wound Healing Migration and Matrigel Invasion Assays
After virus infection, cells were scratched with the tip of a 200 μL pipette and then washed twice with PBS to remove the floating and detached cells. Then, fresh serum-free medium was added, and photos were taken at 0, 12 24 and 36 h to assess cell migration using a microscope (Olympus, Tokyo, Japan).
The invasive potential of ESCC cells was assessed using 24-well Matrigel invasion chambers (pore size 8 μm, Costar, New York, NY). Inserts were pre-coated with 40 μL Matrigel (1:4 dilution; BD Biosciences, San Jose, CA). Then, 5 × 104 cells/mL ESCC cells in serum-free medium were added to the upper chambers. The lower chambers were filled with medium that contained 10% fetal bovine serum. After incubation for 12 h, the cells remaining in the upper chambers were scraped off, and the invading cells in the lower chambers were fixed with 3.7% paraformaldehyde. Then, the cells were stained with crystal violet at room temperature and photographed under a microscope.
Measurement of Apoptosis
Cells were infected with viruses 24 h prior to treatment with sham or 8 Gy X-ray irradiation. Apoptosis was measured using propidium iodide (PI)/Annexin-V double staining following manufacturer's instructions (BD Biosciences, San Jose, CA). Cells were harvested 48 h after irradiation and apoptotic fractions were measured using flow cytometry (BD Biosciences, San Jose, CA). The Annexin-V1/PI- cells are early in the apoptotic process, the Annexin-V1/PI1cells indicating late apoptosis. The percentage of both Annexin-V+/PI- and Annexin-V+/PI+ cells was counted.
Reactive Oxygen Species (ROS) Generation Assay
ROS levels were determined using the ROS-sensitive dye 2',7'- dichlorofluorescein diacetate (DCF-DA), which is converted by ROS into the highly fluorescent 2',7'-dichlorofluorescein (DCF). Eca-109 cells were incubated with DCF-DA (10 μM) for 30 min. The level of DCF fluorescence was measured by a fluorescence microscope. For quantification, the level of DCF fluorescence was measured at 488 nm using a 96-well plate reader.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde for 10 min, treated with 1% Triton X-100 for 10 min, and then incubated with blocking serum for 1 h at room temperature. After washing with PBS, samples were incubated with PRDX6 antibody over night at 4°C, followed by FITC-conjugated goat-anti-rabbit secondary antibody (Beyotime, Nantong, China) for 2 h. Nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, St. Louis, MO). The cells were observed using an Olympus fluorescence microscope (Olympus, Tokyo, Japan).
Animals and Treatments
4-week-old BALB/c nude mice were purchased from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China). The mice were maintained under standard laboratory conditions on a 12-h light-dark cycle and given access to sterilized food and water under specific pathogen free environment. For the subcutaneous injection, Eca-109 cells (1x107) were suspended in 200 μL PBS and then inoculated subcutaneously into the right posterior flank region of BALB/c nude mice. The mice were divided into 6 groups and injected Eca-109 cells, respectively. The six groups are as follows: 1) Control group without treatment; 2) Ad-RFP group; 3) Ad-PRDX6 group; 4) Lenti-sh-NC group; 5) Lenti-sh-PRDX6-2 group; 6) Lenti-sh-PRDX6-3 group. Each group consisted of five nude mice. Two-dimensional measurements were taken with an electronic caliper every 3 days, and the tumor volume in mm3 was calculated using the formula: volume=axb2/0.52, where a is the longest diameter and b is the shortest diameter. When a tumor reached 150-200 mm3 in volume or 27 days after injected, the mice were sacrificed and tumors were frozen in -80°C or fixed in 10% formalin overnight and subjected to routine histological examination. This study was approved by the Institutional Animal Care and Use Committee of Soochow University.
Statistical Analysis
Data are expressed as the mean ± standard error of the mean (SEM) of at least three independent experiments. Standard error bars were included for all data points. The data were then analyzed using Student's t-test when only two groups were present or assessed by one-way analysis of variance (ANOVA) when more than two groups were compared. SPSS software (Release 17.0, SPSS Inc.) was utilized for statistical analysis. Data were considered significant if P < 0.05.
Results
Expression of PRDX6 is upregulated in esophageal cancer tissues
To explore the expression of PRDX6 in normal and cancerous esophageal tissues, we measured PRDX6 expression by immunohistochemistry. Immunohistochemical staining was performed in 15 paraffin-embedded normal esophageal tissues and 95 ESCC tissues, 26 of which had corresponding adjacent tumor tissues. An overall stronger staining for PRDX6 was frequently observed in the ESCC tissues, whereas very weak staining of PRDX6 was observed in the normal and tumor adjacent tissues (Fig.1A). PRDX6 protein is expressed in both the nucleus and cytoplasm of esophageal cancer tissues (Fig.1A).
The samples were further scored by a pathologist, as described in the Materials and Methods section. As shown in Fig. 1B, PRDX6 expression in adjacent tumor tissues was higher than that in normal esophageal tissues (P = 0.0048, Fig. 1B). In contrast, there were significant differences in the expression of PRDX6 between tumor tissues and adjacent tumor tissues (P < 0.001, Fig. 1B). We also found that proliferation-related markers Ki67, PCNA and CyclinD1 expression were positively correlated with that of PRDX6 in esophageal cancer tissues (Fig.1C). These results indicated an increase in the expression of PRDX6 in ESCC, suggesting a characteristic of this malignancy.
PRDX6 promoted the growth of esophageal cancer cells
To explore the functional significance of PRDX6 in ESCC progression, we first measured the expression of PRDX6 in ESCC cancer cells and a noncancerous esophageal cell line.
Result shows that ESCC cell lines (TE-1 and Eca-109) expressed higher level of PRDX6 compared with esophageal epithelial HET-1A cells (Fig. 2A), which confirmed the increased expression of PRDX6 in ESCC cells. To investigate the distribution of PRDX6 in cultured cells, immunofluorescence assay was performed. Result showed that PRDX6 was present more in the nucleus than in the cytoplasm of ESCC cells (Fig. 2B). To confirm this result, Eca-109 cells were transfected with a PRDX6-EGFP overexpression vector (pcDNA.3.1-PRDX6-EGFP) and the fluorescence were observed under a confocal microscope. Images showed that the fluorescence was more pronounced in the nucleus (Fig. 2C). The above results indicated a preferred nuclear distribution of PRDX6 in ESCC cells.
To explore the role of PRDX6 in esophageal cancer cell growth, Eca-109 and TE-1 cells were first infected with a PRDX6-overexpressing adenovirus (Ad-PRDX6) or a control adenovirus (Ad-RFP). Western blotting assay confirmed that Ad-PRDX6-infected cells showed significantly increase of PRDX6 in the two esophageal cancer cell lines (Fig. 2D). We further knocked down PRDX6 expression using lentiviruses. Three different shRNAs lentiviruses were designed to silence PRDX6 and sh-PRDX6-2 and sh-PRDX6-3 showed strongest inhibitory effect by Western blotting (Fig. 2E). Cells were then seeded in 96-well plates and incubated for additional 48 or 72 h. The results showed that PRDX6 overexpression significantly increased cell growth in both Eca-109 and TE-1cells (Fig. 3A). Moreover, forced expression of PRDX6 resulted in a significant increase of EdU-positive proportion by 12.9 % in Eca-109 cells (Fig. 3B). In contrast, the downregulation of PRDX6 by sh-PRDX6-2 or sh-PRDX6-3 significantly suppressed cell proliferation as measured by an MTT assay (Fig. 3C) and EdU assay (Fig 3D). Consistently, silencing of PRDX6 reduced the percentage of colony formation and the size of colonies in the two ESCC cells (Fig. 3E and 3F). Taken together, these results indicate that PRDX6 promotes the growth of ESCC cells.
PRDX6 promotes the migration and invasion of esophageal cancer cells
Next, we investigated the effect of PRDX6 on ESCC cell migration and invasion using wound-healing and Matrigel invasion assays. In wound-healing assay, confluent Eca-109 cell cultures were scraped to create a wound, and cell migration was assessed 12, 24 and 36 h later. As shown in Fig. 4A, the wound area of Ad-PRDX6-infected Eca-109 cells was significantly narrower than that of the control group at 24 and 36 h after the scratch, indicating that PRDX6 overexpression promoted cell migration. In contrast, silencing of PRDX6 by sh-PRDX6-3 significantly reduced the wound area by 13.06% at 24 h and by 14.24% at 36 h in Eca-109 cells (Fig. 4B).
Fig 1
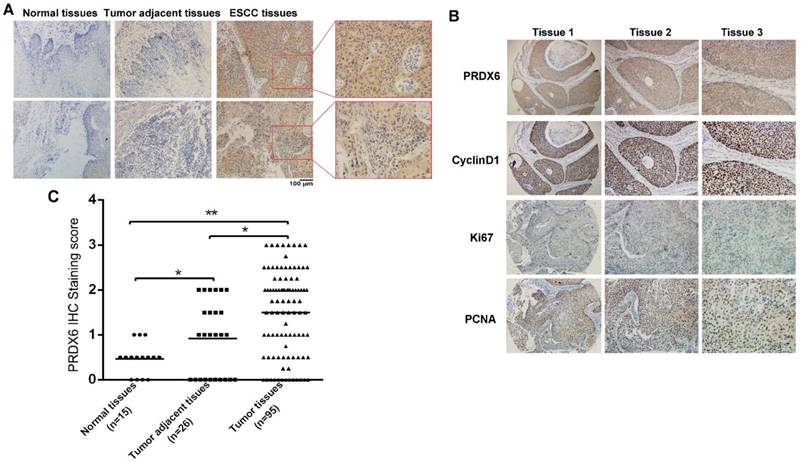
Upregulation of PRDX6 in ESCC samples. (A) Representative IHC staining of PRDX6 expression in normal esophageal tissue specimen and ESCC tissues and tumor adjacent tissues. (B) PRDX6 immunohistochemical (IHC) staining score in normal esophageal tissues (n = 15), ESCC tissues (n = 95) and tumor adjacent tissues (n = 26). * P < 0.05; ** P < 0.01. (C) Co-expression of PRDX6 with proliferation-related proteins Ki67, Cyclin D and PCNA in ESCC tissues.
Fig 2
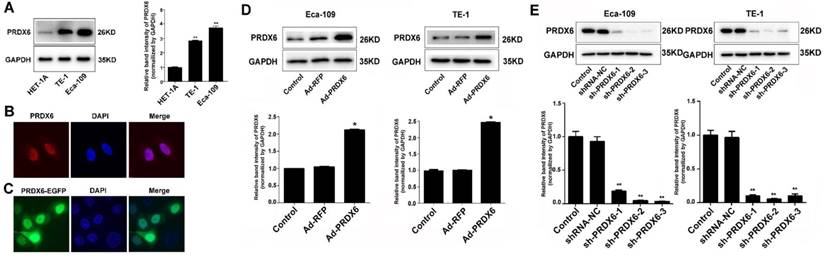
The overexpression and nuclear distribution of PRDX6 in ESCC cells. (A) Western blotting of PRDX6 expression in ESCC cell lines (TE-1 and Eca-109) and an esophageal epithelial cell line (HET-1A). (B) Immunofluorescence assay of PRDX6 expression in Eca-109 cells. (C) Eca-109 cells were transfected with pEGFP-PRDX6. Green fluorescence reflecting the distribution of PRDX6 was observed by a confocal microcope. (D) Eca-109 and TE-1 cells were infected with Ad-RFP or Ad-PRDX6. The expression of PRDX6 was detected by Western blotting. (E) Eca-109 and TE-1 cells were mock infected or infected with indicated lentiviruses targeting PRDX6. The expression of PRDX6 was detected by Western blotting. Relative expression of PRDX6 in each group was quantified by Image J software. Data are presented as mean ± SEM and were normalized to the control cells, * P < 0.05; ** P < 0.01.
Fig 3
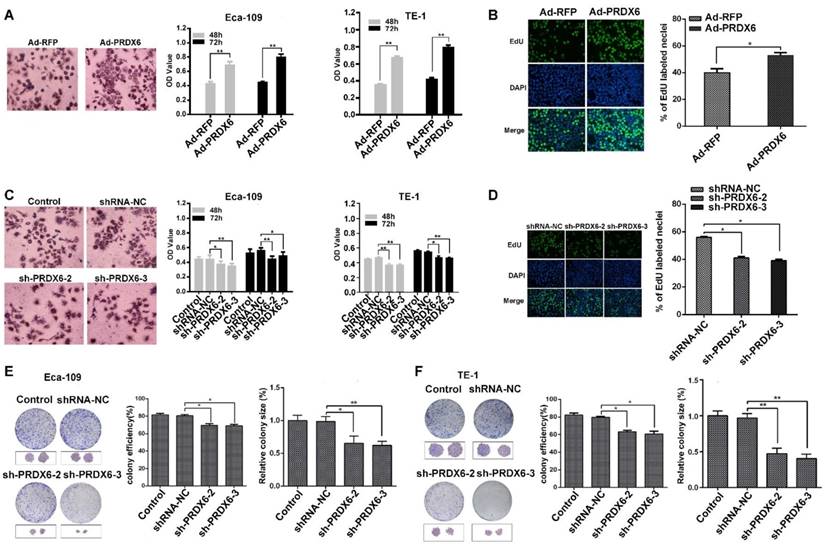
PRDX6 promotes the proliferation and colony formation of ESCC cells. (A) Eca-109 and TE-1 cells were infected with Ad-RFP or Ad-PRDX6. Cells were seeded in a 96- well plate and incubated for 48 h. Cell proliferation was measured by an MTT assay. The left panel shows the images of cells after MTT staining. (B) EdU assay of Eca-109 cell proliferation. Representative images of EdU positive cells from an EdU incorporation assay. The images were acquired using a fluorescence microscope under the same conditions. (C) Eca-109 and TE-1 cells were mock infected or infected with indicated lentiviruses. Cell proliferation was measured by an MTT assay. (D) Eca-109 cells were infected with lentiviruses targeting PRDX6. Cell proliferation was measured by an EdU assay. The effect of PRDX6 silencing on colony formation efficiency and colony size in (E) Eca-109 and (F) TE-1 cells. The left panel shows the representative colonies, and the right panel shows the data analysis. Relative colony size in each group was quantified by Image J software. Data are presented as mean ± SEM and were normalized to the control cells, * P < 0.05; ** P < 0.01.
Esophageal cancer cells were then subjected to Matrigel invasion assay. After the treatment for 12 h, cells that migrated to the bottom surface of the membrane were stained with crystal violet and the number of invading cells was calculated manually. Forced expression of PRDX6 by Ad-PRDX6 induced a ~1.80-fold increase of cells that invade through the Matrigel and membrane compared with the control Ad-RFP-infected cells (Fig. 4C). Knock-down of PRDX6 by shRNA-3 significantly resulted in a 1.34-fold decrease of the number of invaded cells, compared with the control group (Fig. 4D). Western blot analysis was performed to measure the signaling pathways involved the PRDX6-induced migration and invasiveness of ESCC cells. Results showed that overexpression of PRDX6 increased the phosphorylation of Akt and Erk1/2, whereas knock-down of PRDX6 reversed their phosphorylation in Eca-109 cells (Fig. 4E). Moreover, PRDX6 modulated the expression of downstream MMP2 but not MMP9 in ESCC cells (Fig. 4E). These results indicated that PRDX6 enhanced the migration and the invasiveness of ESCC through Akt and Erk1/2 pathway.
PRDX6 modulates the radiosensitivity of ESCC cells
Radiotherapy is a major modality for ESCC and ionizing radiation elicit ROS to induce cancer cell death [28, 29]. We therefore investigated whether PRDX6 affected the radiosensitivity of ESCC cells. Cellular ROS levels in Eca-109 cells were measured using the DCFH-DA probe. Results revealed that ROS level was significantly increased 4 h after 4Gy X-ray irradiation, whereas PRDX6-overexpressing cells (Ad-PRDX6) showed significantly reduced ROS level, compared with the control group (Fig. 5A). Conversely, downregulation of PRDX6 significantly increased ROS level in Eca-109 cell (Fig. 5B). These results indicate that PRDX6 modulates radiation-induced ROS.
Fig 4
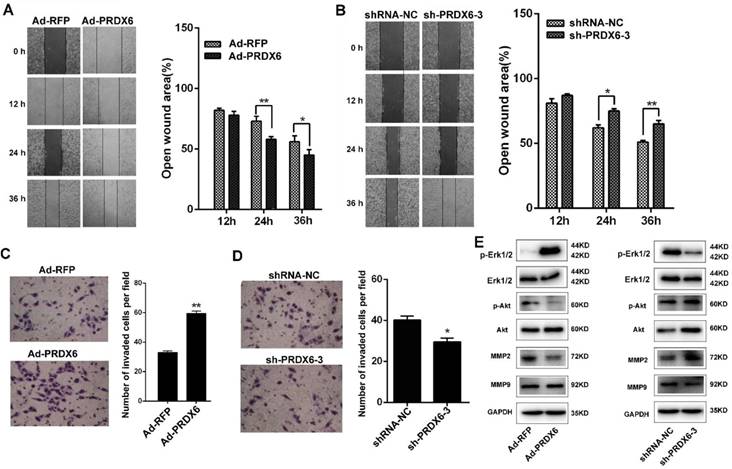
PRDX6 regulates cell migration and invasion in ESCC cells. (A) Wound healing assay of Eca-109 cells infected with Ad-RFP or Ad-PRDX6. Wound healing was observed 12, 24 and 36 h after the treatment, and the open wound area was normalized to the area at the initial time that the wound was made. (B) The effect of PRDX6 silencing on wound healing in Eca-109 cells. Eca-109 cells were infected with indicated lentivirus. (C) Transwell assay of cells infected with Ad-RFP or Ad-PRDX6. Cells migrated to the bottom chamber were stained with crystal violet. (D) Eca-109 cells were infected with indicated lentivirus. Cells migrated to the bottom chamber were stained with crystal violet. Data are presented as mean ± SEM and were normalized to the control cells, * P < 0.05; ** P < 0.01. (E) Eca-109 cells were transfected with the indicated vectors or shRNAs. Cell extracts were subjected to Western blotting analysis for the expression of PRDX6, Erk1/2, phosphor-Erk1/2, Akt, phosphor-Akt, MMP2 and MMP9.
We then assessed the effect of PRDX6 on the apoptosis induced by irradiation. Eca-109 cells were infected with PRDX6 overexpression or knock-down virus followed by 8 Gy X-ray irradiation. The proportion of apoptotic cells was determined by flow cytometry. Compared with cells treated without irradiation, the group of cells after 8 Gy X ray irradiation had a clear increase of apoptosis percentage. However, PRDX6-overexpression group (Ad-PRDX6) significantly reduced radiation-induced apoptosis by 15.7 ± 1.22%, compared with the control group (Fig. 5C). For PRDX6 downregulation group (sh-PRDX6-3), apoptosis percentage was significantly higher than that of control group (shRNA-NC; Fig. 5D). Altogether, PRDX6 modulated the radiosensitivity of ESCC cells.
PRDX6 and Erk1/2 pathway is mutually regulated
Activation of Erk1/2 is implicated in the progression and resistance numerous cancers, making it an attractive therapeutic target [30]. We therefore investigated the relationship between PRDX6 and Erk1/2 pathway in esophageal cancer cells. Cells were infected with lentiviruses targeting PRDX6 and Western blotting assay showed that silencing of PRDX6 either by shRNA-2 or shRNA-3 decreased the phosphorylation of Erk1/2, indicating that PRDX6 regulates Erk1/2 pathway in ESCC cells (Fig.6A). Conversely, forced expression of PRDX6 increased Erk1/2 phosphorylation (Fig. 6B). Furthermore, we also found that addition of MEK (upstream regulator of Erk1/2) inhibitor PD0325901 reduced the expression of PRDX6 expression in a dose-dependent manner, suggesting that Erk1/2 also regulates the PRDX6 expression in ESCC cells (Fig. 6C and 6D). Taken together, the above results indicate that PRDX6 and Erk1/2 pathway was mutually regulated.
PRDX6 promotes esophageal cancer growth in vivo
To determine the effect of PRDX6 on ESCC growth in vivo, nude mice were first inoculated with Eca-109 cells infected with Ad-PRDX6 or Ad-RFP. Western blot confirmed a marked increase of PRDX6 in Ad-PRDX6-infected group (Fig. 7A). Mice in each group showed no difference in body weight and no gross pathologic abnormality, indicating no observable toxicity (Fig. 7B). Nude mice inoculated with Eca-109 cells infected with PRDX6 overexpression adenovirus developed tumors faster than that of the control Ad-RFP-infected group (Fig. 7C). On day 27, the PRDX6 group (Ad-PRDX6) showed an increased tumor volume by 25.28% (Fig. 7D). Then, we detect proliferation marker Ki67 in tumor to verify the increase effect of PRDX6 in vivo. In Ad-PRDX6-infected xenografts, the Ki67-positive percentage was significantly increased, compared with Ad-RFP-infected group (Fig. 7E).
Fig 5
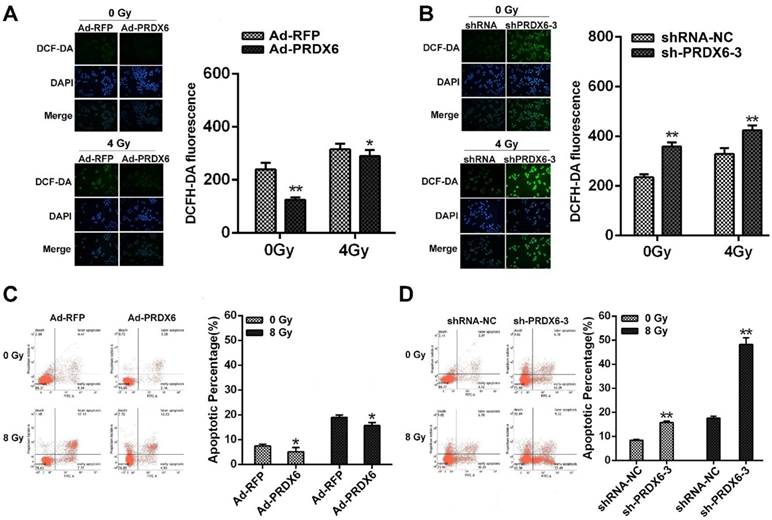
PRDX6 modulates the radiosensitivity of ESCC cells. (A) The effect of PRDX6 overexpression on ROS levels of Eca-109 cells. Cells infected with Ad-RFP or Ad-PRDX6. Fluorescent signals, reflecting the concentration of ROS, were measured by a fluorescence microscope under the same conditions. (B) Eca-109 cells were infected with indicated lentivirus. ROS levels were measured as mentioned above. (C) The effect of PRDX6 on cell apoptosis in Eca-109 cells. Cells were infected with Ad-PRDX6 or Ad-RFP then exposed to 8 Gy of irradiation. (D) Cells were infected with shRNA-NC or sh-PRDX6-3 lentivirus followed by 8 Gy irradiation. Data are presented as mean ± SEM and were normalized to the control cells, * P < 0.05; ** P < 0.01.
Fig 6
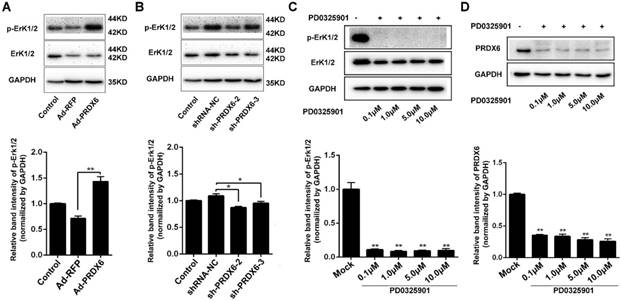
PRDX6 and Erk1/2 pathway are mutually regulated. (A) Eca-109 cells were mock infected or infected with Ad-RFP or Ad-PRDX6. The expression of p-Erk1/2 and Erk1/2 were subjected to Western blotting. Relative expression of p-Erk1/2 in each group was quantified by Image J software. (B) Eca-109 cells were mock infected or infected with lentivirus expressing shRNA-NC, sh-PRDX6-2, sh-PRDX6-3. The expression of p-Erk1/2 and Erk1/2 were subjected to Western blotting. (C) Eca-109 cells were incubated with at concentrations of 0.1, 1.0, 5.0 or 10 μM PD0325901 (Erk1/2 pathway inhibitor). The expression of p-Erk1/2 and Erk1/2 were detected by Western blotting. (D) After treatment with various concentration of PD0325901, the expression of PRDX6 was measured by Western blotting in Eca-109 cells. Data are presented as mean ± SEM and were normalized to the control cells, * P < 0.05; ** P < 0.01.
Fig 7
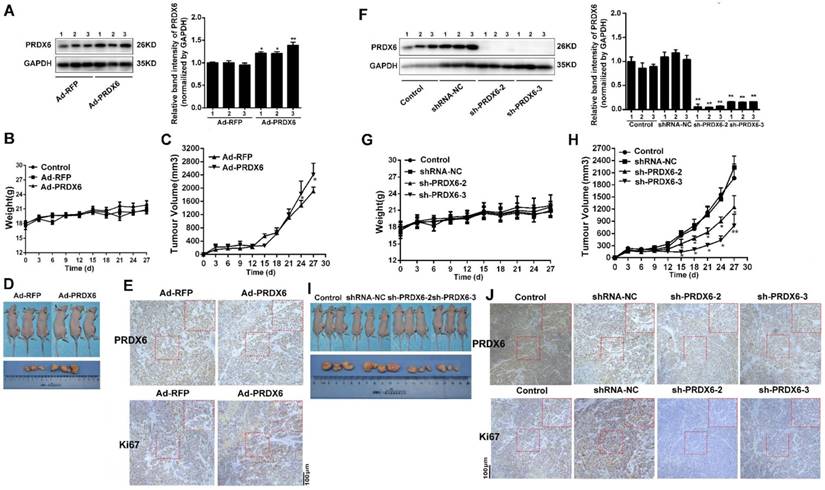
PRDX6 promoted esophageal cancer growth in vivo. Each group of mice was composed of five nude mice. Cells were infected with indicated virus. Bal b/c nude mice were subcutaneously grafted with 1 × 106 Eca-109 cells. Tumor volumes were measured every 3 days for 27 days, as described in the Materials and Methods. (A) Verification the overexpression of PRDX6 by Ad-PRDX6 in vivo. Western blotting showing the expression of PRDX6 in each group (Ad-RFP and Ad-PRDX6). Relative expression of PRDX6 of each group was quantified by Image J. (B) Body weight of nude mice of each group. (C) Tumor volume growth curve of each group. (D) Representative tumors from the mice of each group at the end time-point. (E) Immunohistochemical staining for PRDX6 and Ki67 in tumors from each group. (F) Verification the silencing of PRDX6 by lentiviruses in vivo. Western blotting showing the expression of PRDX6 in each group. (G) Body weight of nude mice of each group. (H) Tumor volume growth curve of each group. (I) Representative tumors from the mice of each group at the end time-point. (J) Immunohistochemical staining for PRDX6 and Ki67 in xenografts from each group.
Then, nude mice were inoculated subcutaneously with Eca-109 cells pre-infected with lentivirus targeting PRDX6 or control lentivirus (Fig. 7F). No differential body weight between groups was observed (Fig. 7G). As shown in Fig. 7H, downregulation of PRDX6 significantly decreased growth of Eca-109 xenografts, compared with the control group. On day 27, compared with shRNA-NC group, downregulation of PRDX6 (sh-PRDX6-2/3) inhibited 42.07% and 76.08% of tumor volume, respectively (Fig. 7I). Ki67-positive cells were significantly decreased in Eca-109 xenografts in the PRDX6 knock-down group (Fig. 7J). Taken together, PRDX6 promoted esophageal cancer growth in vivo.
Discussion
Aberrant expression of PRDX family enzymes has been reported in various kinds of cancers, and thought to be biomarkers and targets of cancer cells [31]. ROS are toxic to cancer cells and PRDX6 plays an important role in peroxide detoxification. PRDX6 has been shown to be dysregulated in breast, prostate and lung cancers and modulate their progression [20,23,24]. Fujita et al reported that PRDX6 autoantibody is a serum marker in esophageal squamous cell carcinoma [32]. In the present study, we found that the expression of PRDX6 was significantly increased compared with normal esophageal tissues or adjacent tumor tissues. ESCC cell lines (TE-1 and Eca-109) also showed higher PRDX6 level than non-cancerous esophageal cells (HET-1A). Moreover, the expression of PRDX6 and proliferation-related markers was correlated. In ESCC cells and tumors, preferred nuclear distribution of PRDX6 was observed as evidenced immunofluorescence and EGFP fused PRDX6. In a recent study, Liu G et al found that PRDX6 interacts with phosphoprotein nucleophosmin, which is mainly located in the nucleus [33]. In renal α-intercalated cells, PRDX6 distribution is more widespread in both cytoplasm and at the cell periphery, which binds to the C-terminal tail of anion exchanger 1 (AE1) [34]. These results indicate that PRDX6 distribution may be cell-type specific and may interact with different partner protein(s).
Besides eliminating ROS, PRDX6 has exhibited multiple functions in the progression and treatment of different cancers. In lung cancer, PRDX6 positively regulates promotion of tumor development via the activation of the JAK2/STAT3 pathway [35]. Consistent evidence is found in breast cancer cells [24]. However, reduce levels of PRDX6 has been found in papillary thyroid carcinomas, especially with BRAF V600E cases [36]. In addition, PRDX6 expression is decreased in hepatocellular carcinoma, which is associated with poor prognosis [37]. In human ovarian cancer cells, PRDX6 overexpression attenuates cisplatin-induced apoptosis [38]. However, PRDX6 promoted apoptosis when treated with tumor necrosis factor alpha (TNF-α) in hepatocellular carcinoma cells, indicating a tumor suppressor [37]. In this study, using human ESCC derived TE-1 and Eca-109 cell lines, we found that PRDX6 promoted cell proliferation and invasion. Without affecting the body weight of nude mice, the proliferation of PRDX6 was confirmed in ESCC xenograft model in vivo. We also demonstrated that PRDX6 is involved in the radioresistance of ESCC cells. Moreover, PRDX6 regulated the phosphorylation of Erk1/2. Yun HM et al has reported that mutant PRDX6 (C47S) attenuated Erk1/2 activity in lung cancer cells and nude mice [23]. However, we also demonstrated that Erk1/2 activation positively affects PRDX6 expression, which constitutes a positive feedback loop.
In summary, we found that the PRDX6 expression was significantly increased in ESCC tumor tissues. Furthermore, PRDX6 overexpression increased the proliferation, metastasis and radioresistance of ESCC cells. Thus, PRDX6 is involved in the progression and radiosensitivity, which represents a novel therapeutic target for ESCC treatment.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (81673100, 81272920 and 81602101), Key Scientific Development Program of China (2016YFC0904702), the Suzhou Key Medical Center (SZZX201506), Changzhou Municipal Commission of Health and Family Planning (QN201604) and Social Development Program of Jiangsu Province (BE2017634 and BE2017652).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7-30
2. Chen W, Zheng R, Baade PD. et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115-32
3. Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349(23):2241-52
4. D'Amico TA. Outcomes after surgery for esophageal cancer. Gastrointest Cancer Res. 2007;1(5):188-96
5. Ma Z, Cai H, Cui Y. Progress in the treatment of esophageal neuroendocrine carcinoma. Tumour Biol. 2017;39(6):1010428317711313
6. Hao JJ, Lin DC, Dinh HQ. et al. Spatial intratumoral heterogeneity and temporal clonal evolution in esophageal squamous cell carcinoma. 2016;48(12):1500-1507.
7. Zhang L, Zhou Y, Cheng C. et al. Genomic analyses reveal mutational signatures and frequently altered genes in esophageal squamous cell carcinoma. Am J Hum Genet. 2015;96(4):597-611
8. Manda G, Isvoranu G, Comanescu MV, Manea A, Debelec Butuner B, Korkmaz KS. The redox biology network in cancer pathophysiology and therapeutics. Redox Biol. 2015;5:347-57
9. Pérez S, Taléns-Visconti R, Rius-Pérez S, Finamor I, Sastre J. Redox signaling in the gastrointestinal tract. Free Radic Biol Med. 2017;104:75-103
10. Galadari S, Rahman A, Pallichankandy S, Thayyullathil F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic Biol Med. 2017Mar;104:144-164
11. Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Semin Cell Dev Biol. 2017 pii: S1084-9521(16)30383-4. doi: 10.1016/j.semcdb.2017.05.023. [Epub ahead of print]
12. Nicolussi A, D'Inzeo S, Capalbo C, Giannini G, Coppa A. The role of peroxiredoxins in cancer. Mol Clin Oncol. 2017;6(2):139-153
13. Hofmann B, Hecht HJ, Flohé L. Peroxiredoxins. Biol Chem. 2002;383(3-4):347-64
14. Manevich Y, Fisher AB. Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radic Biol Med. 2005;38(11):1422-32
15. Shichi H, Demar JC. Non-selenium glutathione peroxidase without glutathione S-transferase activity from bovine ciliary body. Exp Eye Res. 1990;50:513-520
16. Fisher AB. Peroxiredoxin 6: a bifunctional enzyme with glutathione peroxidase and phospholipase A₂ activities. Antioxid Redox Signal. 2011;15(3):831-44
17. Power JH, Asad S, Chataway TK. et al. Peroxiredoxin 6 in human brain: molecular forms, cellular distribution and association with Alzheimer's disease pathology. Acta Neuropathol. 2008;115(6):611-22
18. Power JH, Shannon JM, Blumbergs PC, Gai WP. Nonselenium glutathione peroxidase in human brain: elevated levels in Parkinson's disease and dementia with lewy bodies. Am J Pathol. 2002;161(3):885-94
19. Jo M, Yun HM, Park KR. et al. Lung tumor growth-promoting function of peroxiredoxin 6. Free Radic Biol Med. 2013;61:453-63
20. Schmitt A, Schmitz W, Hufnagel A, Schartl M, Meierjohann S. Peroxiredoxin 6 triggers melanoma cell growth by increasing arachidonic acid-dependent lipid signalling. Biochem J. 2015;471(2):267-79
21. Schremmer B, Manevich Y, Feinstein SI, Fisher AB. Peroxiredoxins in the lung with emphasis on peroxiredoxin VI. Subcell Biochem. 2007;44:317-44
22. Quan C, Cha EJ, Lee HL, Han KH, Lee KM, Kim WJ. Enhanced expression of peroxiredoxin I and VI correlates with development, recurrence and progression of human bladder cancer. J Urol. 2006;175:1512-1516
23. Yun HM, Park KR, Lee HP. et al. PRDX6 promotes lung tumor progression via its GPx and iPLA2 activities. Free Radic Biol Med. 2014;69:367-76
24. Chang XZ, Li DQ, Hou YF. et al. Identification of the functional role of peroxiredoxin 6 in the progression of breast cancer. Breast Cancer Res. 2007;9(6):R76
25. Luo J, Zhou X, Ge X. et al. Upregulation of Ying Yang 1 (YY1) suppresses esophageal squamous cell carcinoma development through heme oxygenase-1. Cancer Sci. 2013;104(11):1544-51
26. Liu P, Gao Y, Huan J. et al. Upregulation of PAX2 promotes the metastasis of esophageal cancer through interleukin-5. Cell Physiol Biochem. 2015;35(2):740-54
27. Yamasaki M, Miyata H, Miyazaki Y. et al. Evaluation of the nodal status in the 7th edition of the UICC-TNM classification for esophageal squamous cell carcinoma: proposed modifications for improved survival stratification: impact of lymph node metastases on overall survival after esophagectomy. Ann Surg Oncol. 2014;21(9):2850-6
28. Holley AK, Miao L, St Clair DK, St Clair WH. Redox-modulated phenomena and radiation therapy: the central role of superoxide dismutases. Antioxid Redox Signal. 2014;20(10):1567-89
29. Koritzinsky M, Wouters BG. The roles of reactive oxygen species and autophagy in mediating the tolerance of tumor cells to cycling hypoxia. Semin Radiat Oncol. 2013;23(4):252-61
30. Germann UA, Furey BF, Markland W. et al. Targeting the MAPK Signaling Pathway in Cancer: Promising Preclinical Activity with the Novel Selective Erk1/2 Inhibitor BVD-523 (Ulixertinib). Mol Cancer Ther. 2017;16(11):2351-2363
31. Zhang B, Wang Y, Su Y. Peroxiredoxins, a novel target in cancer radiotherapy. Cancer Lett. 2009;286(2):154-60
32. Fujita Y, Nakanishi T, Hiramatsu M. et al. Proteomics-based approach identifying autoantibody against peroxiredoxin VI as a novel serum marker in esophageal squamous cell carcinoma. Clin Cancer Res. 2006;12(21):6415-20
33. Liu GY, Shi JX, Shi SL. et al. Nucleophosmin Regulates Intracellular Oxidative Stress Homeostasis via Antioxidant PRDX6. J Cell Biochem. 2017;118(12):4697-4707
34. Sorrell SL, Golder ZJ, Johnstone DB, Frankl FEK. Renal peroxiredoxin 6 interacts with anion exchanger 1 and plays a novel role in pH homeostasis. Kidney Int. 2016;89(1):105-112
35. Yun HM, Park KR, Park MH. et al. PRDX6 promotes tumor development via the JAK2/STAT3 pathway in a urethane-induced lung tumor model. Free Radic Biol Med. 2015;80:136-44
36. Nicolussi A, D'Inzeo S, Mincione G. et al. PRDX1 and PRDX6 are repressed in papillary thyroid carcinomas via BRAF V600E-dependent and -independent mechanisms. Int J Oncol. 2014;44(2):548-56
37. Xu X, Lu D, Zhuang R. et al. The phospholipase A2 activity of peroxiredoxin 6 promotes cancer cell death induced by tumor necrosis factor alpha in hepatocellular carcinoma. Mol Carcinog. 2016;55(9):1299-308
38. Pak JH, Choi WH, Lee HM. et al. Peroxiredoxin 6 overexpression attenuates cisplatin-induced apoptosis in human ovarian cancer cells. Cancer Invest. 2011;29(1):21-8
Author contact
![]() Corresponding authors: Zhang S, No. 68 Gehuzhong Rd, The Affiliated Changzhou No.2 People's Hospital of Nanjing Medical University, Changzhou 213100, China. Tel./Fax: +86-512-65883941; E-mail: zhang.shuyucom. Cao J, No. 199 Ren'ai Rd, Medical College of Soochow University, Suzhou 215123, Tel./Fax:+86-512-65880037; E-mail: jpcaoedu.cn.
Corresponding authors: Zhang S, No. 68 Gehuzhong Rd, The Affiliated Changzhou No.2 People's Hospital of Nanjing Medical University, Changzhou 213100, China. Tel./Fax: +86-512-65883941; E-mail: zhang.shuyucom. Cao J, No. 199 Ren'ai Rd, Medical College of Soochow University, Suzhou 215123, Tel./Fax:+86-512-65880037; E-mail: jpcaoedu.cn.
Citation styles
APA
He, Y., Xu, W., Xiao, Y., Pan, L., Chen, G., Tang, Y., Zhou, J., Wu, J., Zhu, W., Zhang, S., Cao, J. (2018). Overexpression of Peroxiredoxin 6 (PRDX6) Promotes the Aggressive Phenotypes of Esophageal Squamous Cell Carcinoma. Journal of Cancer, 9(21), 3939-3949. https://doi.org/10.7150/jca.26041.
ACS
He, Y.; Xu, W.; Xiao, Y.; Pan, L.; Chen, G.; Tang, Y.; Zhou, J.; Wu, J.; Zhu, W.; Zhang, S.; Cao, J. Overexpression of Peroxiredoxin 6 (PRDX6) Promotes the Aggressive Phenotypes of Esophageal Squamous Cell Carcinoma. J. Cancer 2018, 9 (21), 3939-3949. DOI: 10.7150/jca.26041.
NLM
He Y, Xu W, Xiao Y, Pan L, Chen G, Tang Y, Zhou J, Wu J, Zhu W, Zhang S, Cao J. Overexpression of Peroxiredoxin 6 (PRDX6) Promotes the Aggressive Phenotypes of Esophageal Squamous Cell Carcinoma. J Cancer 2018; 9(21):3939-3949. doi:10.7150/jca.26041. https://www.jcancer.org/v09p3939.htm
CSE
He Y, Xu W, Xiao Y, Pan L, Chen G, Tang Y, Zhou J, Wu J, Zhu W, Zhang S, Cao J. 2018. Overexpression of Peroxiredoxin 6 (PRDX6) Promotes the Aggressive Phenotypes of Esophageal Squamous Cell Carcinoma. J Cancer. 9(21):3939-3949.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.