Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(21):4029-4038. doi:10.7150/jca.25005 This issue Cite
Research Paper
CIP2A Promotes Proliferation, Invasion and Chemoresistance to Cisplatin in Renal Cell Carcinoma
Yongzhen Zhang1,2, Liang Fang1, Yuanwei Zang1, Juchao Ren1 ![]() , Zhonghua Xu1
, Zhonghua Xu1 ![]()
1. Department of Urology, Qilu Hospital, Shandong University, 107# Wenhua Xi Road, Jinan 250012, PRC.
2. Department of Cancer Biology, University of Cincinnati, Cincinnati 45219, USA.
Received 2018-1-18; Accepted 2018-8-23; Published 2018-10-17
Citation:
Zhang Y, Fang L, Zang Y, Ren J, Xu Z. CIP2A Promotes Proliferation, Invasion and Chemoresistance to Cisplatin in Renal Cell Carcinoma. J Cancer 2018; 9(21):4029-4038. doi:10.7150/jca.25005. https://www.jcancer.org/v09p4029.htm
Other stylesAbstract
CIP2A is a well-known oncoprotein whose expression is elevated in multiple human solid tumor types. However, its role in renal cell carcinoma (RCC) development is poorly understood. Thus, in our present study, we used the renal cancer cell lines 786-O, A498 and CAKI-1 and the renal epithelial cell line HK-2 to clarify the function of CIP2A in RCC. We found that CIP2A expression is much higher in the RCC cells than in the normal renal epithelial cell. Lentivirus covered coding region CIP2A cDNA sequence and CIP2A siRNA were used to up and down regulate CIP2A expression in vitro. We found that overexpression of CIP2A promoted G1/S transition and cell proliferation. In addition, up-regulation of CIP2A significantly enhanced the invasion and migration capabilities of the cells. Furthermore, CIP2A promoted epithelial-mesenchymal transformation (EMT) and chemoresistance to cisplatin in RCC cells. Taken together, our findings demonstrate that CIP2A plays an important role in proliferation, invasion and chemoresistance to cisplatin in RCC cells. CIP2A may serve as an ideal molecular target for RCC therapeutics.
Keywords: CIP2A, renal cell carcinoma, cisplatin resistance, cell proliferation, cell invasion.
Introduction
Renal cell carcinoma (RCC) refers to cancer that originates from the renal epithelium. RCC accounts for more than 90% of cancers of the kidney. Clear cell RCC (ccRCC) is the most common form of RCC and accounts for most cancer-related deaths [1]. Surgery remains the primary treatment approach for localized RCC. Approximately 30% of patients eventually develop metastases after nephrectomy for localized RCC [2]. However, routine treatments for RCC, namely, chemotherapy, are ineffective. Although cisplatin has proven efficient in treating many types of tumors [3], RCC is not sensitive to chemotherapy. Response rates for chemotherapy alone are low (approximately 4% to 6%)[4]. Thus, there is an urgent need for novel RCC treatment targets, further explorations of the mechanistic basis of RCC at the molecular level are needed.
Cancerous Inhibitor of PP2A (CIP2A) is a newly discovered oncoprotein. It can directly interact with the oncogenic transcription factor c-myc, inhibits PP2A activity towards c-myc serine 62 (S62), and thereby prevents c-myc proteolytic degradation [5]. In several human cancers, including gastric cancer and hepatocellular carcinoma, CIP2A expression is much higher in the tumor tissue than in the surrounding normal tissue [6, 7]. In our previous study[8], we also found that CIP2A was also overexpressed in RCC.CIP2A plays an important role in tumor growth[5].In addition, several studies also demonstrated that CIP2A knockdown can enhance the sensitivity of certain types of cancer cells to chemotherapeutic drugs [9, 10]. However, the molecular biological function and the underlying mechanisms of CIP2A in RCC remain unclear.
In this study, we found that the expression of CIP2A is positive correlated with proliferation, invasion and chemoresistance to cisplatin in RCC cells.
Materials and methods
Materials and Cell culture
The 786-O, A498, CAKI-1 and HK-2 cell lines were obtained from American Type Culture Collection (Manassas, VA, USA). The 786-O cells were cultured in RPMI1640 (HyClone, Logan, UT, USA) containing 10% fetal bovine serum (FBS) (Gibco, Grand Island, NY), and the A498 cells were cultured in MEM with Earle's Balanced Salts and 1% NEAA containing 10% FBS. CAKI-1 and HK-2 cells were cultured in Dulbecco's Modified Eagle Medium: nutrient mixture F-12 (DMEM/F12; HyClone) containing 10% FBS (Gibco). PI3K inhibitor LY294002 (Sigma, St Louis, MO, USA) was used to block PI3K-AKT signal pathway. Cells were treated for 24 hours with LY294002 at a concentration of 40 μM.
Small interfering RNA (siRNA) transfection
The CIP2A-targeting siRNA and control siRNA were purchased from Genepharma (Genepharma, Shanghai, China). The sequence of the CIP2A siRNA was 5'-GACAACUGUCAAGUGUACCACUCUU-3'. The sequence of the siRNA for negative control was 5'-UUCUCCGAACGUGUCACGUTT-3'. Cells were transfected with either the CIP2A siRNA or control siRNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions.
Lentivirus transfection
A lentivirus (Genechem, Shanghai, China) covered coding region CIP2A cDNA sequence was used to establish RCC cell lines with overexpression of CIP2A.The GV492 vector contained the elements Ubi-MCS-3FLAG-CBh-gcGFP-IRES-puromycin.RCC cells was transfected with Enhanced Infection Solution (Genechem) and polybrene(Genechem) following the manufacturer's instructions. After 8 h of incubation, the transfection medium should be change to complete medium.
Chemicals
After siRNA and lentivirus transfection, RCC cells were treated with cisplatin (Sigma, St Louis, MO, USA) respectively. Considering RCC cell lines were differently sensitive to cisplatin [11, 12], RCC cell lines were treated with different concentrations cisplatin before performed CCK-8 assay and western blotting analysis.
Immunofluorescence
Cells were cultured on glass slides in 24-well plates. According to the protocol, the cells were washed with PBS and fixed in 4% formaldehyde for 30 mins. The cells on the coverslips were permeabilized with 0.5% Triton X-100 and blocked with normal serum. The cells were then incubated with the anti-CIP2A antibody (1:100) overnight at 4°C and incubated with the FITC-conjugated goat anti-rabbit secondary antibody for 1 h at 37°C. Nuclei were stained with DAPI, which is shown in blue.
Western blotting analysis
Cells were harvested and lysed in RIPA buffer containing PMSF (Phenylmethanesulfonyl fluoride, Beyotime). After centrifugation at 10,000×g for 15 min at 4°C, the protein concentrations of the supernatants were determined using the BCA method, and the supernatants were stored at -80°C. Antibodies against CIP2A, Akt, P-Akt (Abcam, Cambridge, UK), and β-actin, Tubulin, Cyclin E, P-Rb, Cyclin D1, E-cadherin, and Vimentin (CST Danvers, MA, US) were used to detect the expression of corresponding proteins. Protein (30 μg) was separated by SDS-PAGE and transferred onto a PVDF membrane (Millipore, Bedford, Massachusetts, USA) using a semi-dry blotting apparatus (Bio-Rad, Hercules, CA, USA). After blocking with 5% non-fat milk at room temperature for 1 h, the membrane was incubated with primary antibodies overnight at 4 °C. After washing the membrane in TBST three times, secondary antibodies conjugated with HRP were applied for 2 h at room temperature. Protein bands were visualized by enhanced chemiluminescence (Millipore).
EdU and colony formation assays
A Cell-Light EdU Apollo567 In Vitro Kit (RiBoBio, Guangzhou, China) was used for the EdU assays. In brief, cells (1×104) were seeded into 96-well plates and treated with lentivirus or siRNA. After incubation with 50 μM EdU for 2 h at 37 °C, the cells were fixed in 4% formaldehyde for 30 min and permeabilized with 0.5% TritonX-100 for 10 min at room temperature. Then washed the cells with PBS, 1× ApolloR reaction cocktail (100 μl) was added to react with the EdU for 30 min. Subsequently, Hoechest33342 (100 μl) was added for 30 min to visualize the nuclei. The prepared samples were then examined by fluorescence microscopy. For the colony formation assays, 1000 cells treated with siRNA or lentivirus were seeded in 6-well plates. After 10 days, the cells were stained with 0.1% crystal violet and photographed and we counted the number of cell clones (consist of 50 cells or more).
Cell cycle analysis by flow cytometry
To determine the effect of CIP2A on the cell cycle, both 786-O and A498 cells were treated with either CIP2A siRNA or control siRNA. After the treatment, the cells were harvested, washed twice with ice-cold PBS, and fixed with ice-cold 75% ethanol. Cells were incubated in a solution containing propidium iodide (PI) for 1 h at 4°C. The cell cycle distribution was analysed by flow cytometry.
Transwell assay
Invasion chambers were obtained from Corning(Corning Costar, Lowell, MA, USA). Each well was coated with 10 µl of basement membrane (Matrigel, BD Biosciences, NJ, USA) and mixed with 50 µl of serum-free medium; the wells were then used for in vitro invasion assays. Cells (1×105) were suspended in 100 µl of serum-free medium and added to the upper chamber; 500 µl of medium containing 20% FBS was added to the lower chamber. The invasion chambers were then incubated at 37°C for 24 h. After incubation, the inserts and cells on the upper side of the filter were removed. The filters were fixed and stained in accordance with the manufacturer's instructions. Cells that had invaded the underside of the filter were counted. Each experiment was repeated three times. Migration assays is similar to invasion assay except upper chambers without basement membrane. After incubation at 37°C for 8 h, the upper chambers were used in migration assays. The rest of assay was performed as the invasion assay.
CCK-8 assay
Cell vitality was estimated via a CCK-8 assay that used cells in the logarithmic growth phase. Cell suspensions (4000 cells/well) were added to 96-well plates at a volume of 200 µl/well. After 1 day, samples were treated with various concentrations of cisplatin. For each group, four parallel wells were prepared and incubated at 37°C and 5% CO2 for 24 h. At the end of the culture period, 10 µl CCK-8 was added to each well. After incubation for 2 h, absorbance was measured at 450 nm using a microplate reader. Inhibition of cell growth was calculated using the formula supplied in the assay instructions. Each group was tested to determine cell vitality at different times.
Statistical analysis
Statistical analyses were performed with SPSS 20.0 (IBM, USA) and GraphPad Prism 7 (GraphPad Software, USA). A two-tailed Student's t-test was used to determine statistically significant differences between treatment and control values. Two-way anova was used for analysis of CCK-8 assay results. (*P<0.05, **P<0.01). All data are presented as the mean±SD of three independent experiments.
Results
The effects of CIP2A on proliferation of HK-2 cells and RCC cells
We visualized CIP2A expression by immune-fluorescence. Although CIP2A expression was observed in both cell lines (Figure S1), CIP2A expression in HK-2 is much weaker compared to RCC cells. By western blot analysis, it is also confirmed that the expression of CIP2A is dramatically upregulated in RCC cell lines (786-O, A498 and CAKI-1) compared to the normal renal epithelial cell line HK-2(Figure 1A and 1B).The RCC cell groups with the CIP2A siRNA showed decreased CIP2A protein levels by Western blotting (Figure. 1C).After transfection with lentivirus, over-expression of CIP2A in HK-2 was confirmed by Western blotting (Figure. 1C).Our previous study indicated that the high CIP2A expression level was correlated with a poor prognosis [8]. To investigate the relationship between CIP2A and renal cancer cell proliferation, both the EdU and colony formation assays were performed. The EdU assay was considered a sensitive and specific evaluation method for the assessment of proliferation. We used CIP2A siRNA to perform a loss-of-function assay. As shown in Figure 2A, the rate of proliferative cells in the CIP2A siRNA-treated groups was clearly decreased compared with the control siRNA treated group. To further confirm the function of CIP2A in proliferation, we then performed a gain-of-function assay in HK-2 cell line by transfecting lentivirus. The results indicated that upregulation of CIP2A promoted the proliferation of HK-2 cells.In colony formation assays, both loss-of-function and gain-of-function assays also revealed that CIP2A promote proliferation in renal cell lines in vitro (Figure. 2B).
Association between the expression of CIP2A and cell cycle in HK-2 cell line and RCC cell lines
Flow cytometry were performed to test whether the CIP2A could affect the cell cycles of the renal cancer cell lines. The percentage of cells in the G1 phase was increased and proportion of cells in the S phase was decreased with the CIP2A knockdown. Meanwhile, up-regulation of CIP2A promoted G1/S transition in HK-2 cells. This indicated that CIP2A might regulate cell cycle at the G1/S phase in RCC cells and HK-2 cells (Figure. 3A, 3B). However, the percentage of cells in the G2 phase was not apparently changed on flow cytometry. During the G1/S transition, retinoblastoma protein (pRb) could be phosphorylated by complex contains cyclin D1 or E and cyclin-dependent kinases (cdks) [13]. Both cyclin E-cdk2 complexes phosphorylation and cyclin D-cdk4/6 complexes phosphorylation are indispensable for activation of pRb. The pRb acts to constrain the G1/S transition in mammalian cells. [14]. By western blotting test, we found that expression of CIP2A was positively correlated with the expression of Cyclin D1, Cyclin E and pRb (Figure. 3C).Taken together, CIP2A might regulated cell cycle at G1-S phase.
Figure 1
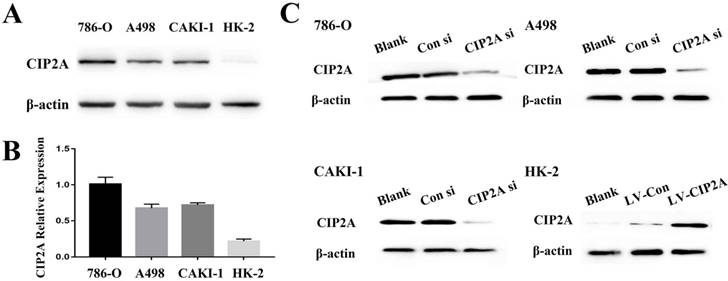
Expression of CIP2A in renal cell line. (A), (B): Expression of CIP2A protein in renal cells. (C). Representative Western blotting showing changes of CIP2A in the protein levels after siRNA or lentivirus transfection.
Figure 2
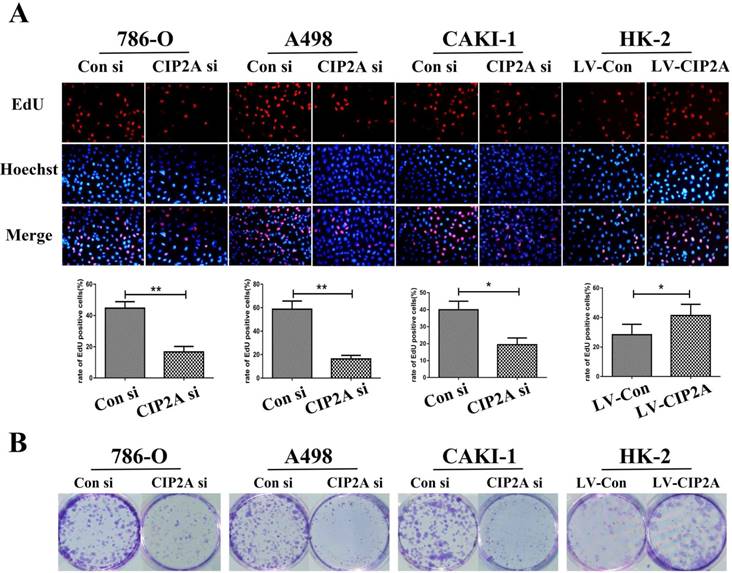
Depletion of CIP2A inhibits cell growth in RCC cells, whereas CIP2A overexpression demonstrates promotion of cell proliferation in HK-2 cells by EdU cell proliferation analysis and colony formation assays. (A): Representative profiles of Edu cell growth in renal cells after CIP2A knockdown or CIP2A up-regulation. Rate of EdU-positive cells in S phase. (B): Effects of CIP2A alteration on the colony formation of renal cells. The data expressed as the mean ± SD. (*p < 0.05, **P<0.01).
Figure 3
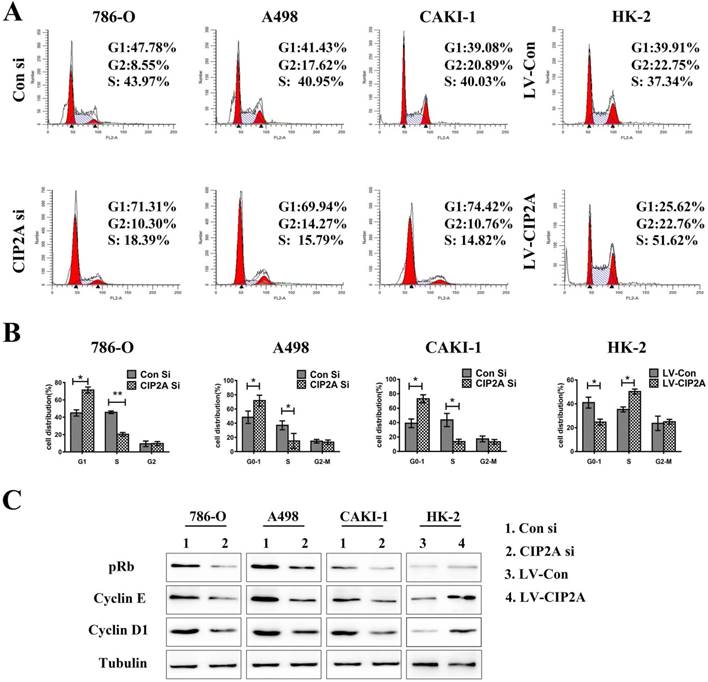
CIP2A regulates cell cycle progression at the G1-S phase transition. (A), (B): Cell cycle flow cytometry identified G1/S arrest when CIP2A was knockdown in RCC cells. Up-regulation of CIP2A promoted G1/S transition in HK-2 cells. (C). Western blotting of G1/S-related proteins verified that CIP2A regulates the G1/S phase transition of renal cells. (*p < 0.05, **P<0.01).
CIP2A promotes invasive and migratory capabilities in renal cell lines
The transwell assay was then performed to test the invasive and migratory capabilities of renal cell lines (Figure. 4A). The results demonstrated that high level of CIP2A could promotes invasion and migration capabilities of RCC cells. Knockdown of CIP2A could impair both cell migration and invasion of RCC cells.Although depletion of CIP2A didn't significantly inhibit the migration and invasion of HK-2 cells, up-regulation of CIP2A could markedly enhance the invasive and migratory capabilities of HK-2 cells.
CIP2A was linked to the sensitivity to cisplatin and EMT progression in RCC cells
Since CIP2A knockdown can enhance the sensitivity of certain types of cancer cells to chemotherapeutic drugs [9, 10], we speculated that CIP2A might play a role in resistance to cisplatin in RCC. We treated RCC cells with different concentrations of cisplatin, and investigated the inhibition rates in different cell lines (Figure. 5). We found that knockdown of CIP2A made cisplatin chemotherapy more effective in RCC cells, while overexpression of CIP2A significantly enhanced chemoresistance to cisplatin in RCC cells. In western blotting assays, we found CIP2A levels increased in a dose-dependent manner in both types of cells when treated with cisplatin (Figure. 6A). By contrast, more mesenchymal-like phenotype exhibited when RCC cells treated with higher dose of cisplatin (Figure. 6A), therefore our results indicate that CIP2A might relate to EMT progression and sensitivity to cisplatin in RCC cells.
Figure 4
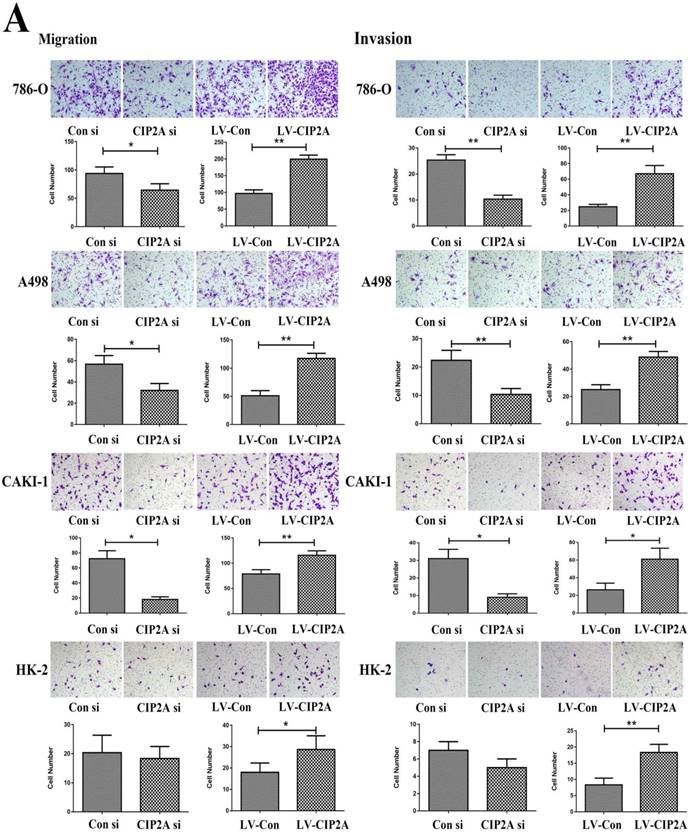
CIP2A regulates the invasion and migration ability of renal cancer cells. (A): Invasion and migration of RCC cells was inhibited after treated with CIP2A siRNA. Up-regulation of CIP2A can enhance the invasive and migratory ability of renal cells. (*p < 0.05, **P<0.01).
CIP2A affected the progression of EMT and phosphorylation level of Akt in renal cancer cell lines
Lentivirus covered coding region CIP2A cDNA sequence was used to establish cell lines with CIP2A overexpression, and the overexpression efficiency was determined by western blotting. To clarify relationship between CIP2A expressions and EMT, we evaluated the expressions of EMT related proteins after CIP2A up-regulated or down-regulated in RCC cell lines (Figure. 6B). The results showed that the EMT progression was blocked by knockdown of CIP2A. The overexpression of CIP2A could effectively enhance the progression of EMT. The results of western blotting showed that either LY294002 or the CIP2A siRNA could suppress the expression of P-Akt (Figure. 6C). Besides, when both LY294002 and the CIP2A siRNA were used, the inhibitory effect was significantly enhanced, implying that CIP2A could regulate the phosphorylation levels of Akt independent of PI3K-AKT pathway in renal cancer cells.
Figure 5
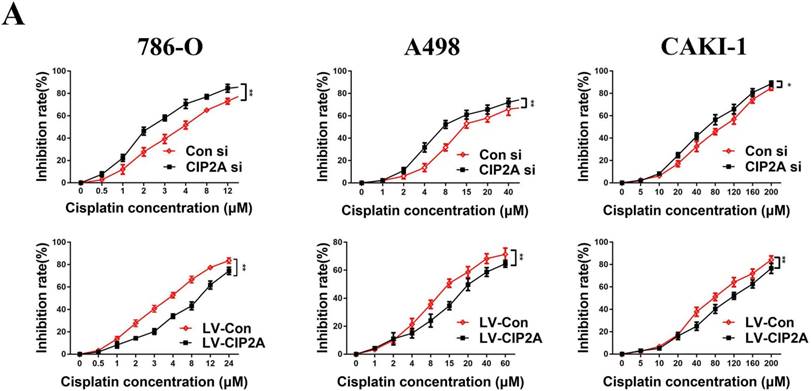
CIP2A promoted chemoresistance to cisplatin in RCC cells. Chemoresistance impaired by CIP2A knockdown. (*p < 0.05, **P<0.01).
Discussion
CIP2A is a known oncoprotein in human malignancies. In our previous study, we found that the CIP2A immunostaining level was positively correlated with the primary tumor stage, lymph node metastasis, distant metastasis, TNM stage and histological grade[8]. However, the mechanism of action of CIP2A in renal cancer cells remains unknown. In the present study, we found that expression of CIP2A was positively correlated with proliferation, invasion and chemoresistance to cisplatin in RCC cells.
EdU and colony formation were performed to evaluate function of CIP2A in regulating RCC cells proliferation. Our study revealed that knockdown of CIP2A could impair the proliferation of RCC cells. Moreover, cells with overexpression CIP2A exhibited more growth ability than control group. All the results indicate that CIP2A could mediate renal cancer cell growth. Similar to our data, several literatures reported that CIP2A had a closer relationship with proliferation in different cancer cells [15, 16]. Cell cycle disorganization could lead to an uncontrolled cellular proliferation[17]. In this study, we found that CIP2A depletion caused a significant triggered G1-S arrest in RCC cell lines, whereas overexpression of CIP2A in HK-2 cell line markedly promoted G1-S transition. Similar with our find, the G1/S arrest after CIP2A depletion has also been noted in cervical cancer [18]. DNA synthesis during S phase is required for cell proliferation. The transition from G0/G1 to S-phase could restart proliferation in quiescent mammalian cells. Cyclin D1 was proved to be a critical target of proliferative signals in G1[19]. Cyclin D1 and E exhibit different roles at the pRb node [13]. The pRb play a key role in transition of G1-S phase in mammalian cells. In this present study, the change of CIP2A expression and G1-S phase related protein (Cyclin D1, Cyclin E and pRb) levels have a positive correlation. Taken together, this could indicate that CIP2A might regulate cellproliferation by controlling G1-S phase transition ofthe cell cycle.
A wide range of studies reported that the AKT signaling pathway is frequently aberrantly activated in many human cancers [20, 21]. The AKT signaling pathway also plays critical roles in regulating cellular growth and proliferation [22]. In western blotting assay (Figure 6B), we found that over-expression of CIP2A promoted the expression of P-Akt. To evaluate how CIP2A affect P-Akt, LY294002 was used to block PI3 kinase-dependent Akt phosphorylation.LY294002 is a highly selective inhibitor of phosphatidylinositol 3 (PI3) kinase. In this study, when both LY294002 and the CIP2A siRNA were used (Figure 6C), the inhibitory effect was significantly enhanced, indicating that CIP2A could affect expression of P-Akt independent PI3K/AKT signaling pathway. Similar to the results of our study, a research by Chen KF et al [6]demonstrated that CIP2A could increase the expression of P-Akt by negatively regulating Akt related PP2A activity in hepatocellular carcinoma cells. Considering all above results, CIP2A could also exhibits its proliferative effects by regulating phosphorylation levels of Akt in renal cancer cells.
Figure 6
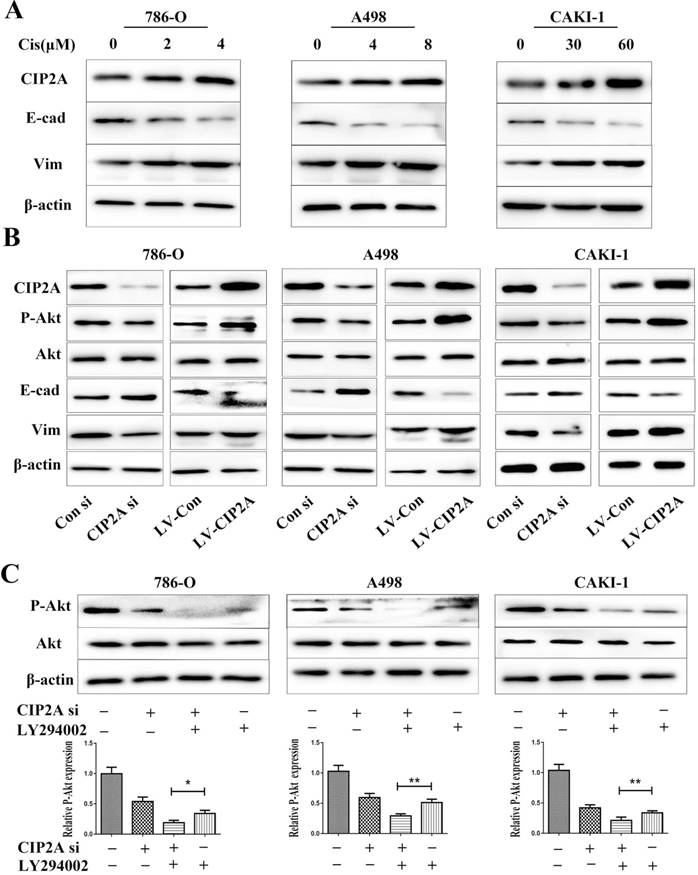
CIP2A affected the progression of EMT and phosphorylation level of Akt in RCC cell lines. (A). Impact of different concentrations cisplatin on CIP2A expression and EMT marker expression. (B). Cells with CIP2A overexpression exhibited more mesenchymal-like phenotype. Changes of CIP2A accompanied with variation of P-Akt expressions. (C).After cells were treated with CIP2A siRNA and/or LY294002 for 72 h, the levels of P-Akt were analysed by western blotting with β-actin as a control. (*p < 0.05, **P<0.01).
Our results demonstrated that CIP2A plays an important role in regulating the invasion and migration capabilities of renal cell lines. The EMT progression was considered as an important factor in invasion and subsequent metastasis in many cancers [23, 24]. EMT progression also is an irreplaceable part in metastasis of renal cancer[25]. Some studies showed clearly that the activation of the Akt pathway contributes to EMT progression and cancer cell motility [26, 27]. We have already found that CIP2A positively regulated P-AKT level. In addition, we also found that down-regulation of CIP2A could inhibit the progression of EMT in renal cancer cells, whereas up-regulation of CIP2A could induce EMT. Thus, we speculate that CIP2A promotes RCC cell migration and invasion, probably through modulating P-AKT level and EMT progression. However, the underlying interaction between proteins still needs further study.
Most RCCs are refractory at the start of chemotherapy[28]. For this reason, chemotherapy is not recommended as the first-line therapy for RCC. Evidence has suggested that CIP2A is associated with drug sensibility in multiple tumors [29, 30]. However, there has remained a lack of research regarding the relationship between CIP2A and cisplatin in renal cancer. In this work, CIP2A levels were founded increased in a dose-dependent manner in both types of cells when treated with cisplatin. Furthermore, upon treatment with cisplatin, greater cisplatin sensibility was observed in cells with CIP2A down-regulation than in control cells. RCC cells lines with CIP2A overexpression manifested greater resistance to cisplatin than the corresponding control cells. A wide range of researches have demonstrated that inhibition of P-Akt probably is an ideal strategy to treat variety types of cancer which resistance to cisplatin. [31-33]. Furthermore, recent studies have indicated that EMT accompanies platinum drug resistance in various cancer types[34]. In this study, we found that RCC cells treated with high dose of cisplatin expressed more mesenchymal marker protein than cells treated with low dose of cisplatin. These suggest that EMT pathway might involve in cisplatin resistance. C-myc was also been proven to play an important role in chemoresistance to cisplatin [35, 36]. Oncoprotein CIP2A could affect EMT pathway and expression of P-Akt by virtue of its control on PP2A and MYC stabilization in many cancer cells [8, 37]. Considering all the related research and our study, we concluded that CIP2A might promote resistance to cisplatin in RCC cells by regulating EMT progression, the level of P-Akt or c-myc. The underlying mechanism of this chemoresistance still needs to be investigated.
Conclusion
Taken together, our study firstly provided a functional analysis of CIP2A in renal cancer cells. For the first time, we revealed that CIP2A enhanced resistance to cisplatin in RCC cells. Moreover, CIP2A regulates cell proliferation and cell cycle of RCC cells. In addition, CIP2A promotes RCC cell migration and invasion probably through modulating P-AKT expression and EMT progression. In conclusion, CIP2A may serve as an ideal molecular target for RCC therapeutics. However, further studies should be done to unravel the efficacy of CIP2A as a therapeutic target in vivo.
Abbreviations
CIP2A, Cancerous Inhibitor of PP2A; ccRCC, clear cell Renal Cell Carcinoma; EMT, Epithelial-mesenchymal transition.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was supported by grants from Natural Science Foundation of China (81372335). Natural Science Foundation of Shandong (No. 2015ZRE27283).
Competing Interests
All authors declare that they have no conflict of interest.
References
1. Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M. et al. Renal cell carcinoma. Nature reviews Disease primers. 2017;3:17009
2. Rabinovitch RA, Zelefsky MJ, Gaynor JJ, Fuks Z. Patterns of failure following surgical resection of renal cell carcinoma: implications for adjuvant local and systemic therapy. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 1994;12:206-12
3. Rancoule C, Guy JB, Vallard A, Ben Mrad M, Rehailia A, Magne N. [50th anniversary of cisplatin]. Bulletin du cancer. 2017;104:167-76
4. Yagoda A, Abi-Rached B, Petrylak D. Chemotherapy for advanced renal-cell carcinoma: 1983-1993. Seminars in oncology. 1995;22:42-60
5. Junttila MR, Puustinen P, Niemela M, Ahola R, Arnold H, Bottzauw T. et al. CIP2A inhibits PP2A in human malignancies. Cell. 2007;130:51-62
6. Chen KF, Liu CY, Lin YC, Yu HC, Liu TH, Hou DR. et al. CIP2A mediates effects of bortezomib on phospho-Akt and apoptosis in hepatocellular carcinoma cells. Oncogene. 2010;29:6257-66
7. Li W, Ge Z, Liu C, Liu Z, Bjorkholm M, Jia J. et al. CIP2A is overexpressed in gastric cancer and its depletion leads to impaired clonogenicity, senescence, or differentiation of tumor cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:3722-8
8. Ren J, Li W, Yan L, Jiao W, Tian S, Li D. et al. Expression of CIP2A in renal cell carcinomas correlates with tumour invasion, metastasis and patients' survival. British journal of cancer. 2011;105:1905-11
9. Xu P, Yao J, He J, Zhao L, Wang X, Li Z. et al. CIP2A down regulation enhances the sensitivity of pancreatic cancer cells to gemcitabine. Oncotarget. 2016;7:14831-40
10. Zhang X, Xu B, Sun C, Wang L, Miao X. Knockdown of CIP2A sensitizes ovarian cancer cells to cisplatin: an in vitro study. International journal of clinical and experimental medicine. 2015;8:16941-7
11. Li G, Bell T, Merino EJ. Oxidatively activated DNA-modifying agents for selective cytotoxicity. ChemMedChem. 2011;6:869-75
12. Harker WG, Tom C, McGregor JR, Slade L, Samlowski WE. Human tumor cell line resistance to chemotherapeutic agents does not predict resistance to natural killer or lymphokine-activated killer cell-mediated cytolysis. Cancer research. 1990;50:5931-6
13. Loden M, Stighall M, Nielsen NH, Roos G, Emdin SO, Ostlund H. et al. The cyclin D1 high and cyclin E high subgroups of breast cancer: separate pathways in tumorogenesis based on pattern of genetic aberrations and inactivation of the pRb node. Oncogene. 2002;21:4680-90
14. Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Molecular and cellular biology. 1998;18:753-61
15. Liu X, Peng B, Li Y, Lei N, Li W, Zhang JY. p90/CIP2A mediates breast cancer cell proliferation and apoptosis. Molecular biology reports. 2014;41:7471-8
16. Chen XD, Tang SX, Zhang JH, Zhang LT, Wang YW. CIP2A, an oncoprotein, is associated with cell proliferation, invasion and migration in laryngeal carcinoma cells. Oncology reports. 2017;38:1005-12
17. Golias CH, Charalabopoulos A, Charalabopoulos K. Cell proliferation and cell cycle control: a mini review. International journal of clinical practice. 2004;58:1134-41
18. Zhang W, Chen H, Chen Y, Liu J, Wang X, Yu X. et al. Cancerous inhibitor of protein phosphatase 2A contributes to human papillomavirus oncoprotein E7-induced cell proliferation via E2F1. Oncotarget. 2015;6:5253-62
19. Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes & development. 1993;7:812-21
20. Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, Gonzalez-Baron M. PI3K/Akt signalling pathway and cancer. Cancer treatment reviews. 2004;30:193-204
21. Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R. Landscape of Phosphatidylinositol-3-Kinase Pathway Alterations Across 19784 Diverse Solid Tumors. JAMA oncology. 2016;2:1565-73
22. Spangle JM, Roberts TM, Zhao JJ. The emerging role of PI3K/AKT-mediated epigenetic regulation in cancer. Biochimica et biophysica acta. 2017;1868:123-31
23. Felipe Lima J, Nofech-Mozes S, Bayani J, Bartlett JM. EMT in Breast Carcinoma-A Review. Journal of clinical medicine. 2016:5
24. Song W, Gobe GC. Understanding Molecular Pathways and Targets of Brachyury in Epithelial-mesenchymal Transition (EMT) in Human Cancers. Current cancer drug targets. 2016;16:586-93
25. He H, Magi-Galluzzi C. Epithelial-to-mesenchymal transition in renal neoplasms. Advances in anatomic pathology. 2014;21:174-80
26. Grille SJ, Bellacosa A, Upson J, Klein-Szanto AJ, van Roy F, Lee-Kwon W. et al. The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer research. 2003;63:2172-8
27. Xu W, Yang Z, Lu N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell adhesion & migration. 2015;9:317-24
28. Kakehi Y, Segawa T, Hashimura T, Yoshida O. [Circumvention of the intrinsic multidrug-resistance in renal cell carcinoma]. Hinyokika kiyo Acta urologica Japonica. 1992;38:1319-24
29. Choi YA, Park JS, Park MY, Oh KS, Lee MS, Lim JS. et al. Increase in CIP2A expression is associated with doxorubicin resistance. FEBS letters. 2011;585:755-60
30. Cai F, Zhang L, Xiao X, Duan C, Huang Q, Fan C. et al. Cucurbitacin B reverses multidrug resistance by targeting CIP2A to reactivate protein phosphatase 2A in MCF-7/adriamycin cells. Oncology reports. 2016;36:1180-6
31. Cheng C, Qin Y, Zhi Q, Wang J, Qin C. Knockdown of long non-coding RNA HOTAIR inhibits cisplatin resistance of gastric cancer cells through inhibiting the PI3K/Akt and Wnt/beta-catenin signaling pathways by up-regulating miR-34a. International journal of biological macromolecules. 2017
32. Zhang G, Li M, Zhu X, Bai Y, Yang C. Knockdown of Akt sensitizes osteosarcoma cells to apoptosis induced by cisplatin treatment. International journal of molecular sciences. 2011;12:2994-3005
33. Amini-Farsani Z, Sangtarash MH, Shamsara M, Teimori H. MiR-221/222 promote chemoresistance to cisplatin in ovarian cancer cells by targeting PTEN/PI3K/AKT signaling pathway. Cytotechnology. 2017
34. Brozovic A. The relationship between platinum drug resistance and epithelial-mesenchymal transition. Archives of toxicology. 2017;91:605-19
35. Wang L, Arras J, Katsha A, Hamdan S, Belkhiri A, Ecsedy J. et al. Cisplatin-resistant cancer cells are sensitive to Aurora kinase A inhibition by alisertib. Molecular oncology. 2017;11:981-95
36. Li QQ, Xie YK, Wu Y, Li LL, Liu Y, Miao XB. et al. Sulforaphane inhibits cancer stem-like cell properties and cisplatin resistance through miR-214-mediated downregulation of c-MYC in non-small cell lung cancer. Oncotarget. 2017;8:12067-80
37. De P, Carlson J, Leyland-Jones B, Dey N. Oncogenic nexus of cancerous inhibitor of protein phosphatase 2A (CIP2A): an oncoprotein with many hands. Oncotarget. 2014;5:4581-602
Author contact
![]() Corresponding author: Zhonghua Xu: Tel: +86 18560086000; E-mail: xuzhonghua1963com; Juchao Ren: Tel: +86 18560083898; E-mail: rjchao1983com
Corresponding author: Zhonghua Xu: Tel: +86 18560086000; E-mail: xuzhonghua1963com; Juchao Ren: Tel: +86 18560083898; E-mail: rjchao1983com
Citation styles
APA
Zhang, Y., Fang, L., Zang, Y., Ren, J., Xu, Z. (2018). CIP2A Promotes Proliferation, Invasion and Chemoresistance to Cisplatin in Renal Cell Carcinoma. Journal of Cancer, 9(21), 4029-4038. https://doi.org/10.7150/jca.25005.
ACS
Zhang, Y.; Fang, L.; Zang, Y.; Ren, J.; Xu, Z. CIP2A Promotes Proliferation, Invasion and Chemoresistance to Cisplatin in Renal Cell Carcinoma. J. Cancer 2018, 9 (21), 4029-4038. DOI: 10.7150/jca.25005.
NLM
Zhang Y, Fang L, Zang Y, Ren J, Xu Z. CIP2A Promotes Proliferation, Invasion and Chemoresistance to Cisplatin in Renal Cell Carcinoma. J Cancer 2018; 9(21):4029-4038. doi:10.7150/jca.25005. https://www.jcancer.org/v09p4029.htm
CSE
Zhang Y, Fang L, Zang Y, Ren J, Xu Z. 2018. CIP2A Promotes Proliferation, Invasion and Chemoresistance to Cisplatin in Renal Cell Carcinoma. J Cancer. 9(21):4029-4038.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.