Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(21):4058-4071. doi:10.7150/jca.25587 This issue Cite
Research Paper
Development of a Novel Autophagy-related Prognostic Signature for Serous Ovarian Cancer
Yuanyuan An, Fangfang Bi, Yue You, Xinhui Liu, Qing Yang ![]()
Department of Gynecology and Obstetrics, Shengjing Hospital of China Medical University, Shenyang, Liaoning 110004, P.R. China
Received 2018-2-17; Accepted 2018-7-23; Published 2018-10-18
Citation:
An Y, Bi F, You Y, Liu X, Yang Q. Development of a Novel Autophagy-related Prognostic Signature for Serous Ovarian Cancer. J Cancer 2018; 9(21):4058-4071. doi:10.7150/jca.25587. https://www.jcancer.org/v09p4058.htm
Other stylesAbstract
Purpose: Considerable evidence suggests that autophagy plays a crucial role in the biological processes of ovarian cancer. The aim of this study was to develop a novel autophagy-related prognostic signature for serous ovarian cancer.
Methods: A univariate Cox proportional regression model was used to analyze mRNA microarray and clinical data in The Cancer Genome Atlas (TCGA) for the purpose of selecting autophagy-related prognostic genes. A multivariate Cox proportional regression model and the survival analysis were used to develop an eight-gene prognostic signature. The multivariate Cox and stratification analysis suggested that this signature was an independent prognostic factor for serous ovarian cancer patients. Bioinformatics functions were investigated by a principal components analysis and gene set enrichment analysis (GSEA). Finally, the correlation between the prognostic signature and gene mutation status was further analyzed in serous ovarian cancer, and especially with regard to the mutation status of BRCA1 and BRCA2 (BRCA1/2) genes.
Results: Distinctly different autophagy-related gene expression profiles were identified in normal ovarian tissues and serous ovarian cancer tissues. We profiled an autophagy-related gene set and identified eight genes with significant prognostic values for serous ovarian cancer. Subsequently, an autophagy-related ovarian cancer risk signature was constructed, and patients at a high-risk or low-risk for poor prognosis were identified based on their signature. High-risk patients had significantly shorter overall survival (OS) and disease-free survival (DFS) times than low-risk patients. GSEA results suggested an enhanced intensity of autophagy regulation in high-risk patients when compared with low-risk patients. When studied as an independent prognostic factor for serous ovarian cancer, the significant prognostic value of this signature could be seen in the stratified cohorts. For clinical use, we developed a nomogram that included the prognostic classifier and seven clinical risk factors. Additionally, we identified the 10 most frequently mutated genes found in serous ovarian cancer patients, and analyzed them for their differences in high-risk and low-risk patients. Among 293 patients, 62 had BRCA1/2 gene mutations, and this result was significantly correlated with the autophagy-related prognostic signature.
Conclusions: Our findings suggest that the eight-gene autophagy-related signature could serve as an independent prognostic indicator for cases of serous ovarian cancer.
Keywords: Autophagy, Serous ovarian cancer, Prognostic signature, TCGA
Introduction
As the most lethal gynecological cancer world-wide, the incidence of ovarian cancer has increased [1, 2]. Statistics indicate that 85~90% of patients with ovarian cancer have epithelial ovarian cancer, and 75% of those patients have serous ovarian cancer [3, 4]. It is well-known that tumor stage, histological grade, and residual tumor size are significant prognostic factors for ovarian cancer patients [5]. Moreover, recent studies suggest that the mutation status of BRCA1/2 correlates with patient prognosis and the chemosensitivity of ovarian cancer, and especially serous ovarian cancer [6]. Based on our increased knowledge of how molecular heterogeneity affects ovarian cancer progression, it has been suggested that prognostic biomarkers be included in any clinical analysis of ovarian tumors [7].
As a mechanism for maintaining cellular homeostasis, autophagy is a process that degrades cellular components under conditions of stress or nutrient deprivation [8, 9], and therefore plays an important role in numerous biological and pathological processes, including neurodegenerative diseases, cardiomyopathy and cancer [9, 10]. However, the role of autophagy in tumorigenesis remains controversial.
Previous studies have shown that autophagy can either promote or suppress tumor cell survival at different stages of cancer development [11]. During the early stages of cancer, autophagy promotes the degradation of damaged proteins or organelles to alleviate cellular damage and chromosomal instability, and thus suppresses cancer progression [12-14]. However, once the cancer has formed, autophagy allows tumor cells to survive under stressful conditions, and thus promotes tumor progression [15-17].
The relationship between autophagy and ovarian cancer has been previously reported. For instance, autophagy was shown to promote chemoresistance and help maintain the stemness of ovarian cancer stem cells [18]. Notably, previous studies have focused on exploring the relationship between autophagy and ovarian cancer progression; however, large-scale expression data has rarely been used to investigate the role of autophagy in ovarian cancer progression and prognosis.
Because high-throughput expression data is now available, it has become feasible to use global gene expression data for analyzing the relationship between autophagy-related gene expression and the clinical outcomes of ovarian cancer patients. In this study, we detected an autophagy-related risk signature that contains eight autophagy-related genes. This signature can be used to independently and accurately predict the prognostic value for serous ovarian cancer, and is closely correlated with the mutation status of BRCA1/2. Broadly speaking, our study demonstrates that autophagy plays an essential role in cancer, and suggests autophagy-related genes as promising prognostic biomarkers for serous ovarian cancer.
Materials and methods
Autophagy-related gene set
We searched the HADb (Human Autophagy Database) to identify 234 genes that had been described in literature as being involved in the autophagy process [19]. In addition, 394 autophagy-related genes were obtained from the gene set (GO_autophagy, M12441) in the Molecular Signatures Database v4.0 [20].
Patient samples
Firehose was used to generate a normalized gene expression dataset and clinical information pertinent to ovarian serous cystadenocarcinoma patients from The Cancer Genome Atlas (TCGA) database. A full mutation dataset of corresponding patients was obtained from cBioPortal [21, 22]. The nine independent microarray serous ovarian cancer cohorts produced by the HGU133 Plus 2 platform were extracted from the Gene Expression Omnibus (GEO) database (accession number: GSE14407, GSE38666, GSE54388, GSE27651, GSE18520, GSE40595, GSE32062, GSE26193, and GSE51088 [23-31]. The Robust Multi-array Average method was used to normalize raw microarray datasets [32]. The ComBat method was used to remove any batch effects, and thereby eliminate discrepancies between different datasets [33]. When more than one probe matched the same gene ID, the probe with the largest expression value was used for our study. GSE26193 and GSE51088 were selected as our independent validation cohorts because they are the largest datasets containing information on overall survival among the nine different datasets. Overall, 529 samples from microarray of TCGA were used as a discovery cohort, 295 samples from the RNAseq of TCGA were used as an internal validation cohort, 79 samples from GSE26193 and 106 samples from GSE51088 were used as two independent external validation cohorts.
Procedures
The R programming language was used to perform a principle components analysis for the purpose of investigating the distinct gene expression patterns of samples.
An R package survivalROC was used to create receiver operating characteristic (ROC) curves that were used to evaluate the sensitivity and specificity of survival. Next, area under the curve (AUC) values was calculated according to the ROC curves.
GSEA was used to perform the gene set enrichment analysis [20]. A normalized enrichment score (NES) and false discovery rate (FDR) were used to determine statistical significance.
Statistical analysis
A univariate Cox proportional hazard regression analysis was used to evaluate the association between overall survival time and gene expression values obtained from the autophagy-related gene set in the test cohort. The prognostic value of a gene was considered statistically significant when the p-value was < 0.01. Next, a multivariate Cox proportional hazards regression analysis was performed using the pool of candidate prognostic genes. The Akaike information criterion (AIC) was used to select the most appropriate model [34]. By weighting the estimated Cox regression coefficients, the eight-gene risk signature was constructed [35]. Patients were classified into high-risk and low-risk groups according to their risk score, and using the median score as the cutoff point. The Kaplan-Meier method and log-rank test were used to estimate the overall survival (OS), disease-free survival (DFS), and progression-free survival (PFS) times of patients. Independent prognostic factors were identified using the multivariate Cox proportional hazards regression model, and hazard ratios (HR) were calculated using the Cox regression model. The prediction accuracy of this risk model was determined by a time-dependent ROC analysis. A prognostic nomogram was created for OS based on the Cox proportional hazard regression model. A concordance index (C-index) was calculated to determine the performance of the nomogram. The nomogram and calibration plot were created using the rms package of R software. The Chi-square test was used to evaluate the correlation between BRCA1/2 mutation status and the autophagy-related signature. R software (version 3.4.1) was used to perform the bioinformatics analysis. All tests were two-tailed and p-values < 0.05 were considered statistically significant. All data were analyzed using GraphPad Prism 7 software (GraphPad Software Inc., La Jolla, CA, USA). Differences in clinicopathologic parameters between high-risk and low-risk groups were tested by Students t test or the X2 test.
Results
Distinct autophagy-related phenotype expression patterns in normal ovaries and serous ovarian cancer
The general process of our study is described as Fig. 1a. A total of 117 serous ovarian cancer tissues and 52 normal ovarian tissues with mRNA expression data were obtained from 7 GEO datasets. The autophagy-related gene set included 546 genes from the Human Autophagy Database and another gene set (GO_autophagy, M12441) from the Molecular Signatures Database, v4.0. A principal components analysis comparing serous ovarian cancer tissues with normal ovarian tissues based on their autophagy-related genes showed two significantly different distribution patterns. Normal ovarian samples were distributed on the left side, while samples of serous ovarian cancer were distributed on the right side, suggesting distinctly different regulatory roles for autophagy in normal ovaries and serous ovarian cancer (Fig. 1b).
Figure 1
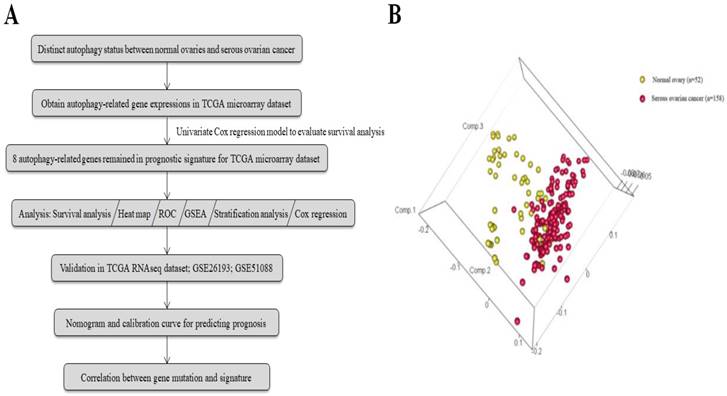
(a) Flowchart for the study; (b) Principle components analysis of autophagy-related genes in normal ovaries and serous ovarian cancer.
Identification of an autophagy-related risk signature for the prognosis of serous ovarian cancer
After reviewing the distinct autophagy-related gene expression pattern found in serous ovarian cancer when compared with the pattern found in normal ovaries, we investigated the role that autophagy might play in serous ovarian cancer, and identified an autophagy-related risk signature that might be useful for establishing the prognosis. A Cox regression analysis was performed to investigate the prognostic value of autophagy-related genes found in 529 serous ovarian cancer patients as listed in the TCGA microarray dataset. Twelve genes (PRKG1, FGF7, WDR45L, NRG4, BLOC1S1, IL24, PEX3, CDKN1B, CD93, SIRT2, PDK4, and SLC22A3) were found to be significantly associated with OS in serous ovarian cancer patients (p < 0.01, Table 1). The AIC was used to select the most appropriate model, which was identified as a model comprised of eight different genes (BLOC1S1, IL24, NRG4, PDK4, PEX3, PRKG1, SIRT2, and WDR45L). Among the eight genes, four genes (BLOC1S1, IL24, NRG4, and WDR45L) were identified as protective factors (HR < 1), while the other four genes (PKD4, PEX3, PRKG1, and SIRT2) were identified as risk factors (HR > 1).
Table 1
Top 12 genes significantly associated with the survival time of patients in the training dataset (N = 529)
| Gene name | HR | P-value |
|---|---|---|
| PRKG1 | 1.425733 | 0.001275 |
| FGF7 | 1.2575 | 0.001398 |
| WDR45L | 0.764353 | 0.002558 |
| NRG4 | 0.858855 | 0.00393 |
| BLOC1S1 | 0.707778 | 0.004328 |
| IL24 | 0.618953 | 0.004421 |
| PEX3 | 1.292997 | 0.00461 |
| CDKN1B | 1.280662 | 0.00528 |
| CD93 | 1.195552 | 0.005346 |
| SIRT2 | 1.377175 | 0.006193 |
| PDK4 | 1.188561 | 0.006719 |
| SLC22A3 | 1.246166 | 0.006826 |
HR: hazard ratio
We then used a multivariate Cox regression model to develop the following autophagy-related risk signature associated with the survival of serous ovarian cancer patients [36].
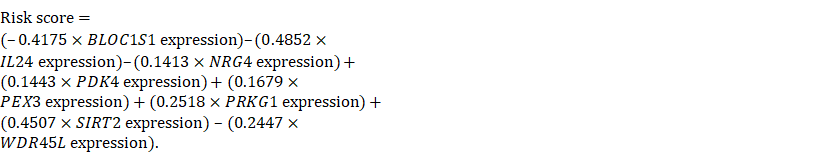
We then calculated the risk score of each patient according to their eight-gene expression signature, and used the median risk value as a cutoff point for classifying patient into a high-risk group (n = 264) and a low-risk group (n = 265), respectively. Patients in the high-risk group had a shorter overall survival time than patients in the low-risk group (median time = 2.84 years vs. 4.72 years, p < 0.001, Fig. 2a). In addition, we also found that patients in the high-risk group had shorter DFS times (median time = 1.15 years vs. 1.77 years, p < 0.001, Fig. 2b).
Serous ovarian cancer cases were divided into high-risk and low-risk groups according to the median eight-gene risk score (Fig. 2c). Protective and risky genes showed distinct expression patterns according to the risk value. High-risk patients expressed higher levels of risky genes (PKD4, PEX3, PRKG1, and SIRT2), while low-risk patients expressed higher levels of protective genes (BLOC1S1, IL24, NRG4, and WDR45L).
ROC curves for 3-year survival were used to reveal the predictive performance of the eight-gene risk signature (Fig. 2d). The 3-year AUC of our signature was 0.703, which was obviously higher than that of age (AUC = 0.626), neoplasm cancer status (AUC = 0.615), residual tumor size (AUC = 0.603), tumor stage (AUC = 0.581), and tumor grade (AUC = 0.544). These results indicated that the ability to predict survival of serous ovarian cancer patients was improved by using the eight-gene risk signature when compared to using clinical factors.
Due to such different prognostic outcomes, we sought to investigate possible differences between the high-risk and low-risk groups by using GSEA, which verified an enhanced autophagy-related signature in the high-risk group (Fig. 2e). Therefore, we propose the existence of an intense regulatory role for autophagy in high-risk serous ovarian patients.
Associations between the autophagy-related risk signature and clinicopathologic features in serous ovarian cancer patients
An analysis was performed to explore associations between various clinical parameters and our risk signature in serous ovarian cancer patients (Table 2). Results showed that the signature was significantly associated with age (p = 0.015), Karnofsky performance status (KPS, p < 0.0001), and neoplasm cancer status (p = 0.0443). Additionally, we found that the signature was only marginally associated with therapy outcome (p = 0.0512) and therapy type (p = 0.0736).
Figure 2
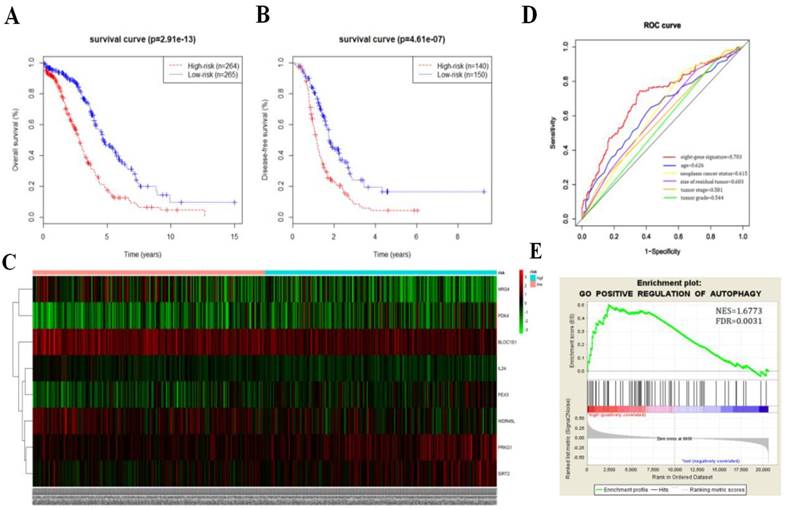
Kaplan-Meier curves for OS (a) and DFS (b) in the high-risk and low-risk groups when stratified by the autophagy-related signature; (c) Hierarchical clustering of high-risk and low-risk patients expressing the eight genes. Red indicates higher expression and green indicates lower expression; (d) ROC analysis at 3-year of overall survival for the eight-gene signature and classical clinicopathologic parameters in the TCGA cohort; (e) A GSEA comparing the autophagy phenotypes in high-risk and low-risk groups.
Table 2
Clinical characteristics of serous ovarian cancer patients by autophagy-related signature.
| Characteristics | High-risk (n = 264) | Low-risk (n = 265) | p-value | |
|---|---|---|---|---|
| Age | 0.0015 | |||
| Mean (years) | 61.20 | 58.02 | ||
| FIGO stage | 0.3899 | |||
| I | 10 (3.8%) | 5 (1.9%) | ||
| II | 11 (4.2%) | 14 (5.3%) | ||
| III | 197 (74.6%) | 211 (79.6%) | ||
| IV | 44 (16.7%) | 34 (12.8%) | ||
| NA | 2 (0.7%) | 1 (0.4%) | ||
| Residual tumor size | 0.3594 | |||
| No macroscopic | 53 (20.1%) | 54 (20.4%) | ||
| 1-10nm | 118 (44.7%) | 114 (43.0%) | ||
| 11-20nm | 14 (5.3%) | 20 (7.5%) | ||
| >20nm | 56 (21.2%) | 44 (16.6%) | ||
| NA | 23 (8.7%) | 33 (12.5%) | ||
| Lymphatic invasion | 0.8329 | |||
| Yes | 67 (25.4%) | 65 (24.5%) | ||
| No | 36 (13.6%) | 41 (15.5%) | ||
| NA | 161 (61%) | 159 (60.6%) | ||
| KPS | <0.0001 | |||
| 40 | 0 (0.0%) | 2 (0.7%) | ||
| 60 | 6 (2.3%) | 9 (3.4%) | ||
| 80 | 6 (2.3%) | 34 (12.8%) | ||
| 100 | 1 (0.3%) | 6 (2.3%) | ||
| NA | 251 (95.1%) | 214 (80.8%) | ||
| Venous invasion | 0.4883 | |||
| Yes | 42 (15.9%) | 43 (16.2%) | ||
| No | 38 (14.4%) | 29 (10.9%) | ||
| NA | 184 (69.7%) | 193 (72.9%) | ||
| Therapy type | 0.0736 | |||
| Chemotherapy | 216 (81.8%) | 237 (89.4%) | ||
| Immunotherapy | 3 (1.1%) | 2 (0.8%) | ||
| Hormone therapy | 3 (1.1%) | 4 (1.5%) | ||
| Targeted molecular therapy | 8 (3.1%) | 2 (0.8%) | ||
| NA | 34 (12.9%) | 20 (7.5%) | ||
| Therapy outcome | 0.0512 | |||
| Complete remission | 132 (50.0%) | 165 (62.3%) | ||
| Partial remission | 35 (13.3%) | 24 (9.1%) | ||
| Progressive disease | 18 (6.8%) | 18 (6.8%) | ||
| Stable disease | 19 (7.2%) | 11 (4.2%) | ||
| NA | 60 (22.7%) | 47 (17.7%) | ||
| Neoplasm cancer status | 0.0443 | |||
| With tumor | 178 (67.4%) | 155 (58.5%) | ||
| Tumor free | 55 (20.8%) | 80 (30.2%) | ||
| NA | 31 (11.8%) | 30 (11.3%) | ||
| New neoplasm event type | 0.7736 | |||
| Recurrence | 128 (48.5%) | 135 (51.0%) | ||
| Progression of disease | 12 (4.5%) | 15 (5.7%) | ||
| Locoregional disease | 3 (1.1%) | 3 (1.1%) | ||
| Metastatic | 1 (0.4%) | 0 (0.0%) | ||
| NA | 120 (45.5%) | 112 (42.2%) | ||
| Radiation therapy | 0.1020 | |||
| Yes | 4 (1.5%) | 1 (0.4%) | ||
| No | 250 (94.7%) | 260 (98.1%) | ||
| NA | 10 (3.8%) | 4 (1.5%) | ||
| Tumor grade | 0.7955 | |||
| G1 | 2 (0.7%) | 4 (1.5%) | ||
| G2 | 33 (12.5%) | 33 (12.5%) | ||
| G3 | 223 (84.5%) | 221 (83.4%) | ||
| G4 | 0 (0.0%) | 1 (0.4%) | ||
| NA | 6 (2.3%) | 6 (2.2%) | ||
| Race | 0.4475 | |||
| White | 227 (86.0%) | 231 (87.2%) | ||
| Black or African American | 12 (4.6%) | 11(4.2%) | ||
| Asian | 8 (3.0%) | 11 (4.2%) | ||
| American Indian | 3 (1.1%) | 0 (0.0%) | ||
| NA | 14 (5.3%) | 12 (4.4%) | ||
KPS: Karnofsky performance score
Table 3
Univariate and multivariate Cox regression analyses of the autophagy-related signature and clinical characteristics predictive of overall survival and disease-free survival.
| Variable | Overall survival | Disease-free survival | ||||||
|---|---|---|---|---|---|---|---|---|
| Univariate | Multivariate | Univariate | Multivariate | |||||
| HR |  -value -value | HR | 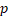 -value -value | HR | 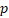 -value -value | HR | 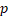 -value -value | |
| Eight-gene signature | 0.414 | <0.001 | 0.463 | <0.001 | 0.488 | <0.001 | 0.599 | 0.007 |
| (low-risk vs high-risk) | ||||||||
| Age (old age vs young age) | 1.019 | 0.001 | 1.016 | 0.033 | 1.012 | 0.058 | ||
| Tumor grade (G3-G4 vs G1-G2) | 1.180 | 0.056 | 1.219 | 0.051 | ||||
| FIGO stage | 1.658 | 0.003 | 1.837 | 0.040 | 1.432 | 0.084 | ||
| (stage III-IV vs stage I-II) | ||||||||
| Residual tumor size | 0.436 | <0.001 | 0.578 | 0.037 | 0.478 | 0.002 | 0.487 | 0.016 |
| (no macroscopic vs visible macroscopic) | ||||||||
| BRCA1/2 mutation stage | 2.021 | 0.001 | 1.874 | 0.004 | 1.752 | 0.020 | 1.602 | 0.065 |
| (no mutation vs mutation) | ||||||||
| Lymphatic invasion | 1.422 | 0.114 | 1.491 | 0.148 | ||||
| (invasion vs no invasion) | ||||||||
| Venous invasion (no invasion vs invasion) | 0.973 | 0.917 | 0.714 | 0.311 | ||||
HR: hazard ratio
Application of the autophagy-related signature in stratified serous ovarian cancer cohorts
In order to investigate the prognostic value of the risk signature in stratified cohorts, patients were classified by residual tumor size, age, and lymphatic invasion status. In all cohorts, the high-risk group had shorter OS (Fig. 3) and DFS times (Fig. 4) than the low-risk group. These results suggest that classification of the autophagy-related risk signature can be used to precisely identify patients with the poor prognosis, without the consideration of clinical parameters.
Figure 3
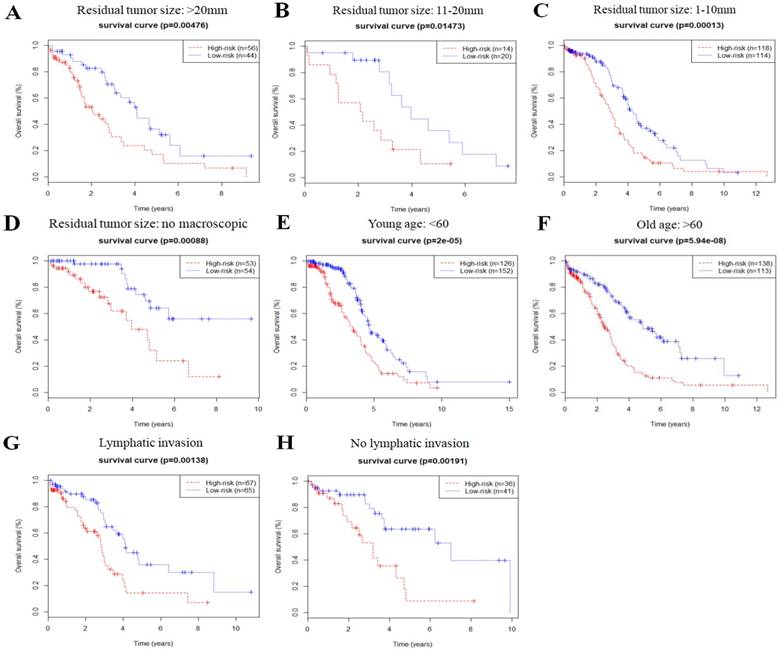
Overall survival of autophagy-related signature in cohorts stratified by residual tumor size (a-d), age (e-f), and lymphatic invasion status (g-h).
The autophagy-related signature is an independent prognostic factor for serous ovarian cancer patients
We performed univariate Cox and multivariate Cox regression analyses of data in the TCGA dataset in order to investigate whether the autophagy-related signature was an independent factor correlated with OS and DFS (Table 3). The univariate Cox analysis showed that the autophagy-related signature, age, tumor stage, residual tumor size, and BRCA1/2 mutation status were all correlated with the overall survival of serous ovarian cancer patients; therefore, those factors were included in a multivariate Cox analysis. Moreover, the risk signature, residual tumor size, and BRCA1/2 mutation status each showed a significant correlation with DFS. Thus our results indicated that the autophagy-related signature might be an independent prognostic factor of OS and DFS when adjusted by those factors.
Validation of the eight-gene autophagy-related prognostic signature using three independent cohorts
To validate the eight-gene risk signature in other datasets, we calculated the risk score for each patient in the TCGA RNAseq dataset as an internal validation, in the GSE26193 and GSE51088 datasets as the independent external validations using same formula. The patients in these three datasets were then divided into a high-risk and a low-risk group based on the median risk score. As expected, the survival curves showed that in the TCGA RNAseq dataset, which including 295 patients, the overall survival time and disease-free survival time were much lower in the high-risk group (OS, median = 2.90 years vs. 4.33 years, p < 0.001, Fig. 5a; DFS, median = 1.22 years vs. 1.98 years, p < 0.001,Fig. 5b). While there was no DFS information in GSE26193, we used the progression-free survival time to create survival curves. Similarly, in dataset GSE26193, which including 79 patients, the high-risk patients had lower overall survival time and progression-free survival time than the low-risk patients (OS, median = 2.42 years vs. 3.90 years, p = 0.026, Fig.5c; PFS, median = 1.66 years vs. 1.87 years, p = 0.042, Fig. 5d). In addition, we also found in dataset GSE51088, which including 106 patients, the overall survival time were much lower in the high-risk group (OS, median = 3.93 years vs. 5.28 years, p = 0.034, Fig. 5e).
We next arrayed the patients according to the risk score in the validation cohorts. The heat map displayed the expression patterns of the eight autophagy-related prognostic genes in the two groups. As expected, in the TCGA RNAseq (Fig. 5h), GSE26193 (Fig.5f) and GSE51088 (Fig. 5g) datasets, the expression levels of the four protective genes were down-regulated in the high-risk group, while the risk factor genes were expressed at high levels. In contrast, those genes showed an opposite expression pattern in the low-risk group.
Consistent with the finding in the test cohort, the autophagy-related signature was further validated by the TCGA RNAseq, GSE26193 and GSE51088 datasets, confirming its power to independently predict the prognosis (Table 4). All these three cohorts validated that this novel autophagy-related signature could work as an independent predictor for the prognosis of serous ovarian cancer.
Figure 4
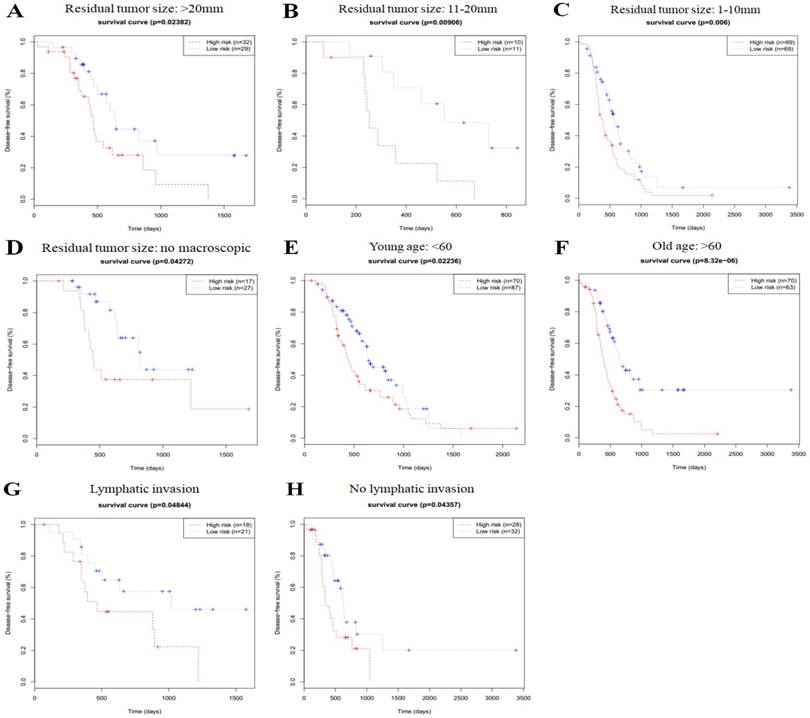
Disease-free survival of autophagy-related signature in cohorts stratified by residual tumor size (a-d), age (e-f), and lymphatic invasion status (g-h).
Figure 5
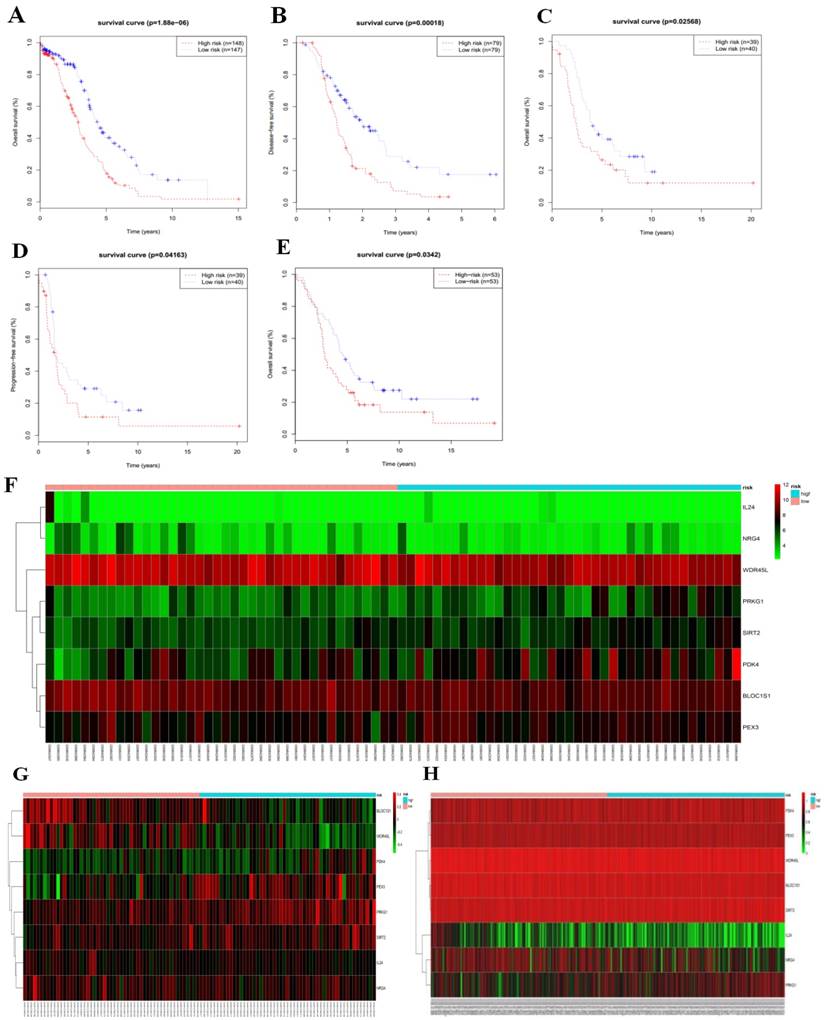
Based on median risk score, the eight-gene risk signature divided patients into high-risk and low-risk groups with distinct prognosis in the TCGA RNAseq cohort (a-b), GSE26193 cohort (c-d), and GSE51088 cohort (e); (f-h) Hierarchical clustering of high-risk and low-risk patients expressing the eight genes in GSE26193 (f), GSE51088 (g), and TCGA RNAseq (h). Red indicates higher expression and green indicates lower expression.
Table 4
Univariate and multivariate Cox regression analyses of the autophagy-related signature and clinical characteristics predictive of OS, DFS and PFS in each validation cohort
| Overall survival | Disease-free survival | Progression-free survival | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Univariate | Multivariate | Univariate | Multivariate | Univariate | Multivariate | |||||||||||||
| HR | p-value | HR | p-value | HR | p-value | HR | p-value | HR | p-value | HR | p-value | |||||||
| TCGA RNAseq cohort (n = 295) | ||||||||||||||||||
| Eight-gene signature (low-risk vs high-risk) | 0.477 | <0.001 | 0.468 | <0.001 | 0.491 | <0.001 | 0.517 | 0.001 | ||||||||||
| Age (old age vs young age) | 1.022 | 0.003 | 1.016 | 0.033 | 1.014 | 0.058 | ||||||||||||
| Tumor grade (G3-G4 vs G1-G2) | 1.663 | 0.038 | 1.777 | 0.120 | 1.337 | 0.051 | ||||||||||||
| FIGO stage (stage III-IV vs stage I-II) | 1.487 | 0.057 | 1.216 | 0.084 | ||||||||||||||
| Residual tumor size (visible vs no macroscopic) | 1.587 | 0.054 | 1.968 | 0.002 | 1.889 | 0.023 | ||||||||||||
| BRCA 1/2 mutation stage (mutation vs no mutation) | 0.613 | 0.033 | 0.758 | 0.237 | 0.584 | 0.020 | ||||||||||||
| Lymphatic invasion (invasion vs no invasion) | 1.319 | 0.359 | 1.109 | 0.148 | ||||||||||||||
| Venous invasion (no invasion vs invasion) | 0.927 | 0.830 | 0.603 | 0.311 | ||||||||||||||
| GSE26193 cohort (n = 79) | ||||||||||||||||||
| Eight-gene signature (low-risk vs high-risk) | 0.564 | 0.028 | 0.552 | 0.023 | 0.599 | 0.044 | 0.553 | 0.021 | ||||||||||
| Tumor grade (G1-G2 vs G3-G4) | 0.877 | 0.351 | 1.054 | 0.719 | ||||||||||||||
| FIGO stage (stage III-IV vs stage I-II) | 1.699 | 0.006 | 1.713 | 0.005 | 1.827 | 0.001 | 1.883 | 0.001 | ||||||||||
| GSE51088 cohort (n = 106) | ||||||||||||||||||
| Eight-gene signature (low-risk vs high-risk) | 0.624 | 0.035 | 0.556 | 0.021 | ||||||||||||||
| Age (old age vs young age) | 1.023 | 0.012 | 1.008 | 0.148 | ||||||||||||||
| Tumor grade (G3-G4 vs G1-G2) | 1.235 | 0.194 | ||||||||||||||||
| FIGO stage (stage III-IV vs stage I-II) | 1.799 | 0.023 | ||||||||||||||||
| Disease status (not free vs free) | 60.734 | <0.001 | 66.097 | <0.001 | ||||||||||||||
HR: hazard ratio
Nomogram development for the prediction of prognostic risk
In order to provide clinicians with a quantitative approach for predicting cancer survival, we assembled a nomogram that integrated both the risk signature and various clinicopathologic risk factors. The nomogram was constructed to estimate 1-, 2-, 3-, 4-, and 5 -year survival probabilities, and it showed that the signature risk score was the most essential factor among the different variables analyzed (Fig. 6a). A C-index was calculated to estimate the performance of this nomogram (Fig. 6b). The C-index of our nomogram was 0.778. A calibration plot showed optimal agreement when compared with an ideal model.
Association between the risk signature and gene mutation status, and especially BRCA1/2 mutation status
The ten genes (BRCA1, BRCA2, CSMD3, FAT3, HMCN1, MUC16, RYR2, TP53, TTN, and USH2A) with the highest mutation rates in 293 patients were selected for further study (Fig. 7a). The association between autophagy-related signature and each of those ten genes is shown in Table 5. The data indicated that mutation of the BRCA1 gene (p = 0.0359) helped to extend the survival of serous ovarian cancer patients, while mutation of the BRCA2 (p = 0.0691) or TTN (p = 0.0884) gene had a marginally favorable effect on survival. The mutation status of BRCA1/2 was recently reported to be an important favorable survival predictor in ovarian cancer [6]. Therefore, we further investigated the effect of combined BRCA1 and BRCA2 gene mutations. As expected, the BRCA1/2 mutation status was significantly correlated with our risk signature (p = 0.0062). Therefore, the patients were arranged according to their risk scores. The genes in our autophagy-related signature showed distinct expression patterns according to the BRCA1/2 mutation status (Fig. 7b).
Discussion
Ovarian cancer is a progressive disease that urgently needs reliable prognostic markers that can aid in its diagnosis and treatment. Computational models have recently been used to explore possible mRNA and non-coding RNA biomarkers for ovarian cancer. Furthermore, numerous studies have focused on the role that autophagy plays in tumorigenesis and the results produced by cancer therapies. While most studies of autophagy-related genes have used cell lines or animal models, our current study used high-throughput expression profiling of autophagy-related genes to investigate the progression and outcomes of serous ovarian cancer patients.
Figure 6
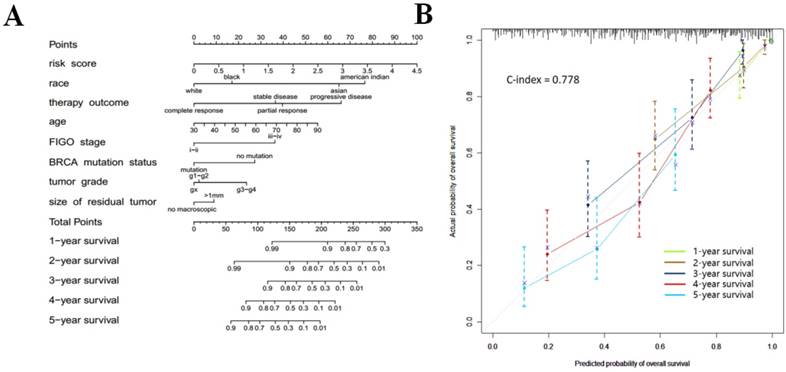
(a) Prognostic nomogram for serous ovarian cancer patients; (b) Calibration curves for the nomogram at different time points.
Figure 7
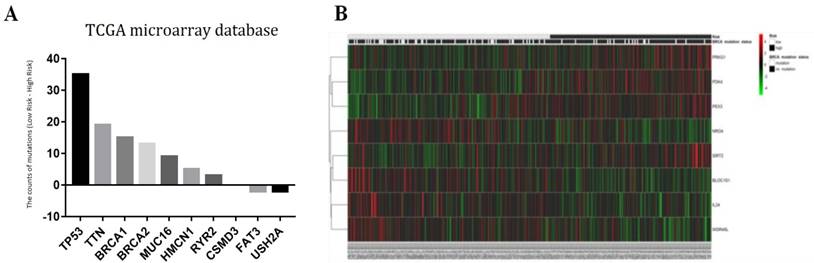
(a) Associations between the risk signature and BRCA1/2 mutation status with its related genes; (b) Top ten genes with highest mutation rates in serous ovarian cancer patients of TCGA microarray database
As a novel therapeutic method, the influence of autophagy on cancer has been discussed for several years. Sui et al. suggested that autophagy might influence how patients respond to chemoradio-therapy [37]. In our study, we first examined differences in the autophagy status of normal ovarian tissues and serous ovarian cancer tissues, and then explored the role that autophagy might play in serous ovarian cancer. Although the controversial and complex role played by autophagy in cancer development has been widely discussed during the past decade, researchers have not reached a conclusion. Therefore, development of a meaningful autophagy-related risk signature should not only benefit the science of disease prognosis, but also provide clinicians with a rationale for using autophagy targeting therapies for treating serous ovarian cancer patients. In this study, we profiled an eight-gene autophagy-related model gene signature that could predict the prognosis of serous ovarian cancer, which was further validated in the GEO database. As described in GSEA, high-risk group showed positive regulation of autophagy, which suggested that the autophagy level might be increased in high-risk group with worse prognostic outcomes. As we all known, cisplatin resistant ovarian cancer cells showed higher autophagy level compared with the cisplatin sensitivity ovarian cancer cells [38], which is in accordance with our results. Previous studies have demonstrated that patients with BRCA1/2 gene mutations had higher chemosensitivity and better prognosis [6], while our results showed that high-risk group correlated with lower possibility of BRCA1/2 gene mutations and higher autophagy level, which is in accordance with the conclusion that chemoresistance of ovarian cancer cells correlated with higher autophagy level. Kwon et al performed a cDNA analysis which showed that among four high-risk genes, PDK4 was greatly overexpressed in ovarian cancer stem cells when compared with parental ovarian cancer cells [39]. Additionally, S Sun et al demonstrated that FAM210B expression loss led to the regulation of PDK4 expression, which is significantly associated with tumor metastasis and decreased survival of ovarian cancer patients [40]. Knockdown of SIRT2 was also found to be associated with ovarian cancer cell proliferation, migration, and invasion [41]. However, with regards to other genes, most studies have focused on their role in autophagy rather than ovarian cancer. Zhao et al. reported that autophagy contributed to the resistance of osteosarcoma cells to doxorubicin [42], while IL24 could suppress autophagy, and thus reverse the multiple drug resistance of osteosarcoma [43]. Moreover, other studies have reported that high levels of PEX3 expression can induce pexophagy, and that ubiquitination of PEX3 is not essential for pexophagy [44].
Table 5
Gene mutation status in serous ovarian cancer according to the autophagy-related signature.
| Characteristics | High-risk (n = 130) | Low-risk (n = 163) | p-value | |
|---|---|---|---|---|
| BRCA1 | 0.0359 | |||
| mutation | 9 (6.9%) | 24 (14.7%) | ||
| no mutation | 121 (93.1%) | 139 (85.3%) | ||
| BRCA2 | 0.0691 | |||
| mutation | 9 (6.9%) | 22 (13.5%) | ||
| no mutation | 121 (93.1%) | 141 (86.5%) | ||
| BRCA1/2 | 0.0062 | |||
| mutation | 18 (13.8%) | 44 (27.0%) | ||
| no mutation | 112 (86.2%) | 119 (73.0%) | ||
| CSMD3 | 0.5995 | |||
| mutation | 10 (7.7%) | 10 (6.1%) | ||
| no mutation | 120 (92.3%) | 153 (93.9%) | ||
| FAT3 | 0.3241 | |||
| mutation | 10 (7.7%) | 8 (4.9%) | ||
| no mutation | 120 (92.3%) | 155 (95.1%) | ||
| HMCN1 | 0.5481 | |||
| mutation | 8 (6.2%) | 13 (8.0%) | ||
| no mutation | 122 (93.8%) | 150 (92.0%) | ||
| MUC16 | 0.1612 | |||
| mutation | 7 (5.4%) | 16 (9.8%) | ||
| no mutation | 123 (94.6%) | 147 (90.2%) | ||
| RYR2 | 0.7849 | |||
| mutation | 7 (5.4%) | 10 (6.1%) | ||
| no mutation | 123 (94.6%) | 153 (93.9%) | ||
| TP53 | 0.2953 | |||
| mutation | 125 (96.2%) | 160 (98.2%) | ||
| no mutation | 5 (3.8%) | 3 (1.8%) | ||
| TTN | 0.0884 | |||
| mutation | 22 (16.9%) | 41 (25.2%) | ||
| no mutation | 108 (83.1%) | 122 (74.8%) | ||
| USH2A | 0.3178 | |||
| mutation | 12 (9.2%) | 10 (6.1%) | ||
| no mutation | 118 (90.8%) | 153(93.9%) | ||
BRCA1/2 mutation status has been well studied as an important molecular marker for ovarian cancer. Ovarian cancer patients with BRCA1/2 mutations have shorter survival times and higher degrees of chemosensitivity [6]. It is known that PARP inhibitors can inhibit the proliferation of cancer cells with BRCA1/2 mutations. Moreover, recent studies have demonstrated that the PARP inhibitor can induce autophagy in BRCA1 or BRCA2 mutated breast cancer cells, and that inhibition of autophagy results in the partial inhibition of PARP inhibitor induced apoptosis [45]. These results suggest that autophagy plays an essential role in regulating BRCA1/2 mutated cancer cell proliferation and apoptosis. Coincidentally, another study showed significant decreases in serum LC3 and Bcl-2 expression levels in breast cancer patients with the 5382insC BRCA1 mutation [46]. In our current study, we found a close correlation between BRCA1/2 mutation status and the risk signature for ovarian cancer, which suggests that autophagy might contribute to the BRCA1/2 mutations found in serous ovarian cancer cells; however, the precise mechanism for this contribution remains unknown. Thus additional studies should be considered to further explore the role played by autophagy in BRCA1/2 mutated serous ovarian cancer.
Further studies showed that our signature was an independent prognostic factor for serous ovarian cancer, which suggests that autophagy status might serve as an accurate prognostic indicator. In this study, patients with higher risk scores significantly showed worse prognosis. Our nomogram demonstrated that the risk score associated with the signature for each patient was the most important variable, indicating its significant meaning when predicting the prognosis of serous ovarian cancer patients. Interestingly, by building this nomogram, we found that American Indian and Asian patients showed poorer prognosis than White patients. As we all known, patients with old ages, visible residual tumor sizes, higher FIGO stages and tumor grades all showed worse prognostic outcomes, which is in accordance with our results. Additionally, patients with BRCA1/2 gene mutations showed higher chemosensitivity and thus caused better prognosis [6]. Thus, this nomogram provided an individualized thorough estimate of survival, rather than using specific covariates. Moreover, perfect agreement was seen between actual observations and predictions in the calibration plot. Thus, the eight-gene signature could precisely predict the prognosis for serous ovarian cancer patients.
The advantage of our study is that we performed a systematic analysis of microarray data and RNAseq data, which provided a robust statistical approach for exploring the role of autophagy in serous ovarian cancer. Although the eight-gene signature allowed for a practical independent prognosis for serous ovarian cancer, our study does have certain limitations. First, only autophagy-related genes were included in the study, and the risk signature does not represent the entire gene transcription profile associated with serous ovarian cancer. Second, our study is restricted because it is retrospective, and our results should be validated in prospective investigations.
In summary, we developed an autophagy-related gene expression model that could independently predict the overall survival of serous ovarian cancer patients. Furthermore, our results suggest that use of a targeted autophagy therapy might be a promising future strategy for treating serous ovarian cancer. Further investigations into the molecular mechanisms of autophagy will demonstrate how autophagy affects survival, and provide new suggestions for treating ovarian cancer patients. Thus, our eight-gene autophagy-related signature may predict the overall survival of serous ovarian cancer patients, and also guide the therapeutic approaches used for those patients.
Abbreviations
AIC: Akaike information criterion; AUC: area under the curve; C-index: concordance index; DFS: disease-free survival; FDR: false discovery rate; GEO: Gene Expression Omnibus; GSEA: gene set enrichment analysis; HADb: Human Autophagy Database; HR: hazard ratio; KPS: Karnofsky performance status; NES: normalized enrichment score; OS: overall survival; PFS: progression-free survival; ROC: receiver operating characteristic; TCGA: The Cancer Genome Atlas.
Acknowledgements
Supported by grant from the Shenyang Science and Technology Bureau (award number: LNCCC-A01-2015)
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kobel M, Kalloger SE, Boyd N, McKinney S, Mehl E, Palmer C. et al. Ovarian carcinoma subtypes are different diseases: implications for biomarker studies. PLoS Med. 2008;5:e232
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5-29
3. Syrios J, Banerjee S, Kaye SB. Advanced epithelial ovarian cancer: from standard chemotherapy to promising molecular pathway targets-where are we now? Anticancer Res. 2014;34:2069-77
4. Marsh DJ, Shah JS, Cole AJ. Histones and their modifications in ovarian cancer - drivers of disease and therapeutic targets. Front Oncol. 2014;4:144
5. Agarwal R, Kaye SB. Prognostic factors in ovarian cancer: how close are we to a complete picture? Ann Oncol. 2005;16:4-6
6. Biglia N, Sgandurra P, Bounous VE, Maggiorotto F, Piva E, Pivetta E. et al. Ovarian cancer in BRCA1 and BRCA2 gene mutation carriers: analysis of prognostic factors and survival. Ecancermedicalscience. 2016;10:639
7. Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet. 2014;384:1376-88
8. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069-75
9. Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463-77
10. Choi HS, Jeong EH, Lee TG, Kim SY, Kim HR, Kim CH. Autophagy Inhibition with Monensin Enhances Cell Cycle Arrest and Apoptosis Induced by mTOR or Epidermal Growth Factor Receptor Inhibitors in Lung Cancer Cells. Tuberc Respir Dis (Seoul). 2013;75:9-17
11. White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308-16
12. White E, Karp C, Strohecker AM, Guo Y, Mathew R. Role of autophagy in suppression of inflammation and cancer. Curr Opin Cell Biol. 2010;22:212-7
13. Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K. et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367-81
14. Jin S, White E. Role of autophagy in cancer: management of metabolic stress. Autophagy. 2007;3:28-31
15. Lorin S, Hamai A, Mehrpour M, Codogno P. Autophagy regulation and its role in cancer. Semin Cancer Biol. 2013;23:361-79
16. Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G. et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51-64
17. Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G. et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460-70
18. Pagotto A, Pilotto G, Mazzoldi EL, Nicoletto MO, Frezzini S, Pasto A. et al. Autophagy inhibition reduces chemoresistance and tumorigenic potential of human ovarian cancer stem cells. Cell Death Dis. 2017;8:e2943
19. Moussay E, Kaoma T, Baginska J, Muller A, Van Moer K, Nicot N. et al. The acquisition of resistance to TNFalpha in breast cancer cells is associated with constitutive activation of autophagy as revealed by a transcriptome analysis using a custom microarray. Autophagy. 2011;7:760-70
20. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545-50
21. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-4
22. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
23. Bowen NJ, Walker LD, Matyunina LV, Logani S, Totten KA, Benigno BB. et al. Gene expression profiling supports the hypothesis that human ovarian surface epithelia are multipotent and capable of serving as ovarian cancer initiating cells. BMC Med Genomics. 2009;2:71
24. Lili LN, Matyunina LV, Walker LD, Benigno BB, McDonald JF. Molecular profiling predicts the existence of two functionally distinct classes of ovarian cancer stroma. Biomed Res Int. 2013;2013:846387
25. Yeung TL, Leung CS, Wong KK, Gutierrez-Hartmann A, Kwong J, Gershenson DM. et al. ELF3 is a negative regulator of epithelial-mesenchymal transition in ovarian cancer cells. Oncotarget. 2017;8:16951-63
26. King ER, Tung CS, Tsang YT, Zu Z, Lok GT, Deavers MT. et al. The anterior gradient homolog 3 (AGR3) gene is associated with differentiation and survival in ovarian cancer. Am J Surg Pathol. 2011;35:904-12
27. Mok SC, Bonome T, Vathipadiekal V, Bell A, Johnson ME, Wong KK. et al. A gene signature predictive for outcome in advanced ovarian cancer identifies a survival factor: microfibril-associated glycoprotein 2. Cancer Cell. 2009;16:521-32
28. Yeung TL, Leung CS, Wong KK, Samimi G, Thompson MS, Liu J. et al. TGF-beta modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. 2013;73:5016-28
29. Yoshihara K, Tsunoda T, Shigemizu D, Fujiwara H, Hatae M, Fujiwara H. et al. High-risk ovarian cancer based on 126-gene expression signature is uniquely characterized by downregulation of antigen presentation pathway. Clin Cancer Res. 2012;18:1374-85
30. Mateescu B, Batista L, Cardon M, Gruosso T, de Feraudy Y, Mariani O. et al. miR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat Med. 2011;17:1627-35
31. Karlan BY, Dering J, Walsh C, Orsulic S, Lester J, Anderson LA. et al. POSTN/TGFBI-associated stromal signature predicts poor prognosis in serous epithelial ovarian cancer. Gynecologic oncology. 2014;132:334-42
32. Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U. et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249-64
33. Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118-27
34. Akaike H. Information Theory and an Extension of the Maximum Likelihood Principle. Parzen E, Tanabe K, Kitagawa G, editors. Selected Papers of Hirotugu Akaike Springer Series in Statistics (Perspectives in Statistics): Springer, New York, NY. 1998
35. Kawaguchi A, Iwadate Y, Komohara Y, Sano M, Kajiwara K, Yajima N. et al. Gene expression signature-based prognostic risk score in patients with primary central nervous system lymphoma. Clin Cancer Res. 2012;18:5672-81
36. Lossos IS, Czerwinski DK, Alizadeh AA, Wechser MA, Tibshirani R, Botstein D. et al. Prediction of survival in diffuse large-B-cell lymphoma based on the expression of six genes. N Engl J Med. 2004;350:1828-37
37. Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M. et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4:e838
38. Zhang XY, Zhang M, Cong Q, Zhang MX, Zhang MY, Lu YY. et al. Hexokinase 2 confers resistance to cisplatin in ovarian cancer cells by enhancing cisplatin-induced autophagy. The international journal of biochemistry & cell biology. 2017;95:9-16
39. Kwon AY, Kim GI, Jeong JY, Song JY, Kwack KB, Lee C. et al. VAV3 Overexpressed in Cancer Stem Cells Is a Poor Prognostic Indicator in Ovarian Cancer Patients. Stem Cells Dev. 2015;24:1521-35
40. Sun S, Liu J, Zhao M, Han Y, Chen P, Mo Q. et al. Loss of the novel mitochondrial protein FAM210B promotes metastasis via PDK4-dependent metabolic reprogramming. Cell Death Dis. 2017;8:e2870
41. Du Y, Wu J, Zhang H, Li S, Sun H. Reduced expression of SIRT2 in serous ovarian carcinoma promotes cell proliferation through disinhibition of CDK4 expression. Mol Med Rep. 2017;15:1638-46
42. Zhao D, Yuan H, Yi F, Meng C, Zhu Q. Autophagy prevents doxorubicininduced apoptosis in osteosarcoma. Mol Med Rep. 2014;9:1975-81
43. Liu Z, Xu L, Yuan H, Zhang Y, Zhang X, Zhao D. Oncolytic adenovirusmediated mda7/IL24 expression suppresses osteosarcoma growth and enhances sensitivity to doxorubicin. Mol Med Rep. 2015;12:6358-64
44. Yamashita S, Abe K, Tatemichi Y, Fujiki Y. The membrane peroxin PEX3 induces peroxisome-ubiquitination-linked pexophagy. Autophagy. 2014;10:1549-64
45. Arun B, Akar U, Gutierrez-Barrera AM, Hortobagyi GN, Ozpolat B. The PARP inhibitor AZD2281 (Olaparib) induces autophagy/mitophagy in BRCA1 and BRCA2 mutant breast cancer cells. Int J Oncol. 2015;47:262-8
46. Abdel-Mohsen MA, Ahmed OA, El-Kerm YM. BRCA1 Gene Mutations and Influence of Chemotherapy on Autophagy and Apoptotic Mechanisms in Egyptian Breast Cancer Patients. Asian Pac J Cancer Prev. 2016;17:1285-92
Author contact
![]() Corresponding author: E-mail: yangqorg
Corresponding author: E-mail: yangqorg
Citation styles
APA
An, Y., Bi, F., You, Y., Liu, X., Yang, Q. (2018). Development of a Novel Autophagy-related Prognostic Signature for Serous Ovarian Cancer. Journal of Cancer, 9(21), 4058-4071. https://doi.org/10.7150/jca.25587.
ACS
An, Y.; Bi, F.; You, Y.; Liu, X.; Yang, Q. Development of a Novel Autophagy-related Prognostic Signature for Serous Ovarian Cancer. J. Cancer 2018, 9 (21), 4058-4071. DOI: 10.7150/jca.25587.
NLM
An Y, Bi F, You Y, Liu X, Yang Q. Development of a Novel Autophagy-related Prognostic Signature for Serous Ovarian Cancer. J Cancer 2018; 9(21):4058-4071. doi:10.7150/jca.25587. https://www.jcancer.org/v09p4058.htm
CSE
An Y, Bi F, You Y, Liu X, Yang Q. 2018. Development of a Novel Autophagy-related Prognostic Signature for Serous Ovarian Cancer. J Cancer. 9(21):4058-4071.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.