Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(22):4108-4116. doi:10.7150/jca.27281 This issue Cite
Research Paper
Comprehensive Analysis of BAP1 Somatic Mutation in Clear Cell Renal Cell Carcinoma to Explore Potential Mechanisms in Silico
Shengming Jin1,2*, Junlong Wu1,2*, Yao Zhu1,2, Weijie Gu1,2, Fangning Wan1,2, Wenjun Xiao1,2, Bo Dai1,2, Hailiang Zhang1,2, Guohai Shi1,2, Yijun Shen1,2, Yiping Zhu1,2 ![]() , Dingwei Ye1,2
, Dingwei Ye1,2 ![]()
1. Department of Urology, Fudan University Shanghai Cancer Center, Shanghai, 200032, China
2. Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, China
*These authors contributed equally to this work.
Received 2018-5-16; Accepted 2018-7-24; Published 2018-10-18
Abstract
Purpose: Aim of this study was to comprehensively analyze BRCA1-associated protein-1 (BAP1) somatic mutation in clear cell renal cell carcinoma (ccRCC) and explore potential therapeutic pathways and molecules.
Patients and methods: In this study, we analyzed 445 ccRCC cases from The Cancer Genome Atlas (TCGA). Comprehensive analysis including survival, transcriptome and methylation between BAP1 mutated and wild-type cases was performed using bioinformatics tools in silico. Pathways and molecules related to BAP1 mutation were analyzed using Database for Annotation, Visualization and Integrated Discovery (DAVID) and protein-protein interaction (PPI) network.
Results: BAP1 mutated ccRCC patients had a worse overall survival (OS) and disease free survival (DFS) than BAP1 wild-type patients. We found 583 up-regulated and 1216 down-regulated different expressed genes (DEGs) in BAP1 mutated tumors. Up-regulated DEGs were enriched in molecular functions and biological processes like protein binding, protein transport and ubiquitin protein ligase binding. Down-regulated DEGs were enriched in pathways like Rap1 signaling pathway, Notch pathway and altered molecular functions like metal ion binding and ubiquitin-protein transferase activity. Furthermore, CAD, TSPO, CTNNB1 and MAPK3 were top hub genes selected using PPI network analysis. Finally, BAP1 mutation had a strong correlation with CpG island methylator phenotype (CIMP).
Conclusion: Our study provides a comprehensive understanding of BAP1 functional somatic mutation in ccRCC patients. Several hub genes like CAD and TSPO may become potential therapeutic targets.
Keywords: clear cell renal cell carcinoma, BAP1, mutation, bioinformatics
Introduction
Renal cell carcinoma (RCC) is one of the most common malignant urologic tumors worldwide. Furthermore, in recent years, RCC has been associated with increased morbidity in China, leading to an estimated 66,800 new cases and 23,400 deaths in 2015 [1]. Clear cell RCC (ccRCC), which accounts for about 70% of all cancers of the kidney [2], is the major subtype. Diagnosis of RCC mainly depends on imaging tests, and when necessary, a renal biopsy is recommended [3]. For the treatment of localized RCC, partial or radical nephrectomy is still the first choice. Immunotherapy and targeted therapy may also be taken into consideration when surgery alone is not enough [3]. However, these treatment measures still have some limitations, and new ways to diagnose and treat RCC are greatly needed.
In recent studies, several mutated genes including PBRM1, SETD2, KDM6A, BAP1, and others, have been identified as having an impact on the outcomes and biological properties of RCC [4-6]. Among these genes, BRCA1-associated protein-1 (BAP1) has been reported to have tumor suppressor activity, which has drawn a lot of interest and may be a target for RCC treatment [7]. BAP1, as a deubiquitinating enzyme, exerts its tumor suppressor activity based on its deubiquitinating activity and nuclear localization, which involves the NH2-terminal ubiquitin COOH-terminal hydrolase (UCH) domain and nuclear localization signal (NLS), respectively (Figure 1A). As previously reported, BAP1-deficient cancer cells were more vulnerable to γ-radiation and more sensitive to olaparib, which indicated that radiotherapy and PARP inhibitors may be more effective in BAP1-mutated cases than in BAP1 wild-type cases [8, 9]. BAP1 loss leads to ubiquitinated H2A accumulation, causing various abnormal transcriptional changes. Histone deacetylase inhibitors may reverse this phenomenon [8]. In addition, a comprehensive understanding of pathway changes caused by BAP1 mutations may also be useful in filtering potential therapeutic targets.
(A) Representative BAP1 protein domains with amino acid sequence. (Numbers on the top indicate amino acid position.) UCH: ubiquittin COOH-terminal hydroaase; NLS: nuclear localization signal. (B) Flow chart of study design and summary of ccRCC cases filtered in TCGA datasets. TCGA: The Cancer Genome Atlas; DEG: different expression gene; CIMP: CpG Island Methylation Phenotype; GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; PPI: protein-protein interaction.
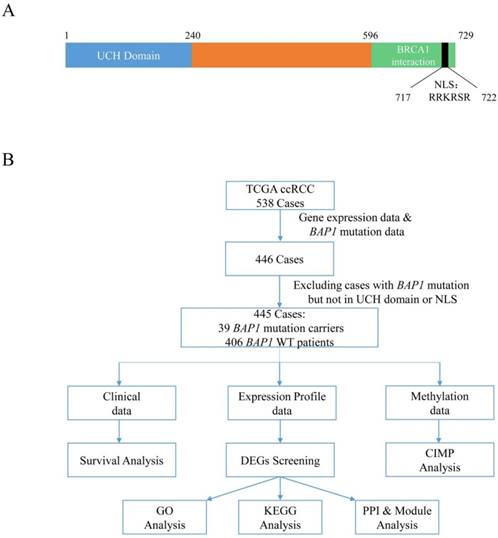
Previous studies revealed that in ccRCC patients, BAP1 has a high mutation rate in somatic cells [7, 10] and BAP1 germline mutations will lead to a hereditary renal carcinoma syndrome. [11, 12] Furthermore, low expression of BAP1 usually predicts a poorer prognosis in ccRCC patients. [13-16] Recently, a phase II trial (NCT03207347) was registered to evaluate the treatment response of the PARP inhibitor, niraparib, in BAP1-mutated cancer patients, including RCC patients, because BAP1 protein is intimately involved in DNA double-strand break repair. This trial may provide a novel therapeutic strategy to improve the prognosis in BAP1-mutated patients.
In this study, we analyzed 445 ccRCC cases using the complete gene expression data and somatic BAP1 mutation data retrieved from The Cancer Genome Atlas (TCGA) database. We performed a comprehensive analysis including survival, transcriptome, and methylation between BAP1-mutated and wild-type cases, and highlighted pathways and molecules related to BAP1 mutations.
Material and methods
Data retrieval
The Cancer Genome Atlas (TCGA) Provisional clinical data, mRNA expression profiles, gene methylation data, and somatic mutation data of ccRCC cases were downloaded from the CbioPortal [17, 18] (http://www.cbioportal.org/. Downloading date: 2017-01-16). The Cancer Genome Atlas (TCGA) is a collaboration between the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI). In the TCGA database, there are multiple types of bioinformation, including transcriptional data, epigenetic data, genomic mutation profiles, and clinical data, across more than 30 cancer types involving >10,000 patients in total. Specifically, the TCGA ccRCC project contained 538 cases. We excluded cases without gene expression data (generated by RNA sequencing and shown in pre-normalized Z-scores) and somatic BAP1 mutation data (generated using genome sequencing). We also excluded one ccRCC case with a BAP1 mutation that was not in the UCH domain or NLS. Finally, 445 ccRCC cases were entered in our analysis, including 39 cases with BAP1 mutations and 406 cases with wild-type BAP1.
Study design
First, we performed a survival analysis using TCGA clinical data. Then, we focused on transcriptome data and analyzed differentially expressed genes (DEGs) between somatic BAP1-mutated and wild-type cases. After DEG screening, we carried out Gene Ontology (GO) functional analysis [19], Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis [20], and protein-protein interaction (PPI) and module analysis (Figure 1B). Additionally, we performed a gene methylation analysis of these cases.
Survival analysis
To test overall survival (OS) and disease-free survival (DFS) differences between cases with or without BAP1 mutations, the Kaplan-Meier method was used to compare survival curves for these two groups. Survival data was censored at five years and P values less than 0.05 were considered statistically significant.
Analysis of DEGs
Gene expression data (mRNA level) were processed using Multi-Experiment Viewer 4.9.0 [21]. The Student's t test was used to examine differences in expression levels between BAP1-mutated and BAP1 wild-type cases. Only genes with an adjusted P value less than 0.01, FDR less than 0.01, and a mean Z score difference larger than 0.5 were considered DEGs.
Functional and pathway enrichment analysis
The Database for Annotation, Visualization and Integrated Discovery (DAVID) is a comprehensive set of functional annotation tools that has been used for systematic and integrative analysis of large gene lists [22]. GO terms are significantly overrepresented in a set of genes from three aspects: the cellular component, molecular function, and the biological process [23]. In our work, the significant GO biological processes, molecular function terms, and KEGG pathway enrichment analyses of the identified DEGs were performed using DAVID, with the threshold of P values less than 0.05 and enrichment gene counts over 5. For module gene analysis, the P value threshold was also set as 0.05 but the gene count threshold was 2.
Protein-protein interaction (PPI) network construction
The Search Tool for the Retrieval of Interacting Genes (STRING) database is a pre-computed global resource for the exploration and analysis of PPI information [24]. In the present study, the STRING 10.5 online tool was used to screen the PPIs of the DEGs. The DEGs with the required confidence level (combined score) greater than 0.4 were selected, and then the PPI network was constructed and visualized using Cytoscape 3.5.1 [25]. Given that most of the networks were scale-free, hub genes were selected with a connectivity degree larger than 10.
Module analysis of the PPI network
Module analysis was performed on the PPI network using Molecular Complex Detection (MCODE), which finds protein complexes and parts of pathways in a network in Cytoscape with a degree cutoff =2, node score cutoff =0.2, k-core =2, and max depth =100 [26]. Then, significant modules with MCODE scores >4 and nodes >6 were selected. Next, GO functional and KEGG pathway enrichment analyses of the most significant modules were performed with a threshold of P value less than 0.05.
Gene methylation analysis
Pre-processed gene methylation data (HM450 platform, shown with β values) downloaded from CBioPortal contained 320 tumor samples, including 260 samples in the 445 selected cases in our study and 160 normal kidney tissues. In these 260 samples, 23 of them were from BAP1-mutated cases.
With regard to CpG island methylator phenotype (CIMP) analysis, we chose genes that were not methylated in the 160 normal samples (mean β value <0.1) and that had a standard deviation of greater than 0.1 (364 genes) in the tumor samples used for clustering. Hierarchical clustering with Wald's method was used to cluster 260 samples, and the clustering dendrogram was cut into two clusters. One of the two exhibited extensive hypermethylation across selected genes and was renamed the CIMP cluster [27].
Ethics statement
The Research Ethics Committee of Shanghai Medical College, Fudan University, China approved this study. For the public TCGA database, we did not require informed consent of patients.
Results
Patient characteristics and survival analysis
Overall, 445 ccRCC cases were divided into two cohorts (39 patients with BAP1 mutations and 406 patients without BAP1 mutations). Demographics and clinical characteristics of these patients were analyzed using the Student's t test and Chi-square test (Table 1). The two cohorts were similar in terms of age, laterality, pN stage, and Fuhrman Grade. However, they differed in gender (P=0.003), pT stage (P=0.028), pM stage (P=0.025), and tumor stage (P=0.001).
Clear-cell renal cell carcinoma patient demographics and clinical characteristics in TCGA.
Characteristics | TCGA ccRCC cohort | P value | Total cohort (N=445) | |
|---|---|---|---|---|
| BAP1 mutation carriers (N=39) | Non-BAP1 mutation carriers (N=406) | |||
| Age, median (range) | 58 (32 - 85) | 61 (26 - 90) | 0.872 a | 60 (26 - 90) |
| Number (%) | ||||
| Gender | 0.003 b | |||
| Male | 17 (43.6) | 273 (67.2) | 290 (65.2) | |
| Female | 22 (56.4) | 133 (32.8) | 155 (34.8) | |
| Lateral | 0.417 b | |||
| Left | 21 (53.8) | 191 (47.0) | 212 (47.6) | |
| Right | 18 (46.2) | 215 (53.0) | 233 (52.4) | |
| pT stage | 0.028 b | |||
| T1 & T2 | 18 (46.2) | 260 (64.0) | 278 (62.5) | |
| T3 & T4 | 21 (53.8) | 146 (36.0) | 167 (37.5) | |
| pN stage | 0.069 c | |||
| N0 | 24 (61.5) | 180 (44.3) | 204 (45.8) | |
| N1 | 4 (10.3) | 10 (2.5) | 14 (3.1) | |
| Nx | 11 (28.2) | 216 (53.2) | 227 (51.0) | |
| pM stage | 0.025 b | |||
| M0 | 28 (71.8) | 347 (85.5) | 375 (84.3) | |
| M1 | 11 (28.2) | 59 (14.5) | 70 (15.7) | |
| Tumor stage | 0.001 b | |||
| I & II | 13 (33.3) | 249 (61.3) | 262 (58.9) | |
| III & IV | 26 (66.7) | 157 (39.7) | 183 (41.1) | |
| Fuhrman Grade | 0.087 c | |||
| I & II | 12 (30.8) | 185 (45.6) | 197 (44.3) | |
| III & IV | 26 (66.6) | 217 (53.4) | 243 (54.6) | |
| Unclear | 1 (2.6) | 4 (1.0) | 5 (1.1) | |
| *CIMP | ||||
| CIMP | 14 (60.9) | 67 (28.3) | 0.001 b | 81 (31.2) |
| Non-CIMP | 9 (39.1) | 170 (71.7) | 179 (68.8) | |
a. Student's t test; b. Chi-square test; c. Chi-square test, exclude unclear cases.
* Numbers do not sum to total because of missing values.
BAP1 mutations in these patients are shown in Table 2, and the total mutation frequency was 8.76% (39/445). Among 538 ccRCC cases in TCGA, 40 patients carried a somatic BAP1 mutation. Most of the BAP1 mutations altered the UCH domain or NLS (39/40), and only 1 out of 40 mutation sites did not involve these two regions, which suggested that mutations in the UCH domain or NLS may play a role in tumorigenesis.
BAP1 mutation summary in TCGA ccRCC cohort.
| Characteristics | BAP1 mutation carriers in TCGA ccRCC cohort (N=39) |
|---|---|
| Mutation frequency, (%) | 39/445 (8.76) |
| Number of mutation carriers, (%) | |
| Mutation type | |
| Missense mutation | 12 (30.77) |
| Inframe-shift InDels | 0 |
| Truncating mutation | 28 (71.79) a |
| Mutation site | |
| Altering UCH domain | 11 (28.21) |
| Altering NLS | 16 (41.03) |
| Altering both UCH domain and NLS | 12 (30.77) |
a. one ccRCC patient had a missense mutation and a truncating mutation both. One patient had two different truncating mutations. UCH domain: ubiquitin COOH-terminal hydrolase domain; NLS: nuclear-localization signals.
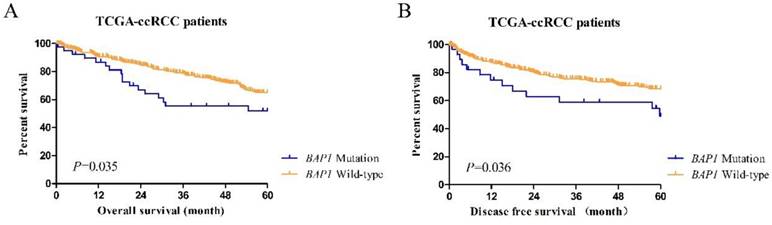
Cases in the BAP1-mutated and BAP1 wild-type groups showed different prognoses (Figure 2). Kaplan-Meier curves of 5-year OS and DFS between mutated and wild-type groups indicated that ccRCC patients with somatic BAP1 functional mutations had a significantly shorter OS (P=0.035) and DFS (P=0.036).
(A) The Kaplan-Meier plot of overall survival (censored at 60 months) in TCGA ccRCC patients (BAP1 mutation cases versus BAP1 wild-type cases). (B) The Kaplan-Meier plot of disease-free survival (censored at 60 months) in TCGA ccRCC patients (BAP1 mutation cases versus BAP1 wild-type cases).
Identification of DEGs
By processing gene expression data (mRNA level), we found a total of 1,799 DEGs between BAP1-mutated and BAP1 wild-type cases. Among them, 583 were upregulated DEGs, while 1,216 were downregulated DEGs.
GO functional and pathway enrichment analysis
The significant GO biological processes and molecular function term analysis of the identified DEGs were carried out using DAVID. In all, upregulated DEGs were mainly enriched in 25 GO functions and downregulated DEGs were mainly enriched in 96 GO functions. The most significant 10 GO terms for these two groups of DEGs are listed in Figures 3 A & B. Protein binding was the most significant GO term for upregulated DEGs and metal ion binding was the most significant GO term for downregulated DEGs.
(A) Top 10 significant GO terms (biological process or molecular function) in up-regulated DEGs in BAP1 mutated cases. (B) Top 10 significant GO terms (biological process or molecular function) in down-regulated DEGs in BAP1 mutated cases. (C) Top 10 significant KEGG pathways in up-regulated DEGs in BAP1 mutated cases. (D) Top 10 significant KEGG pathways in down-regulated DEGs in BAP1 mutated cases.
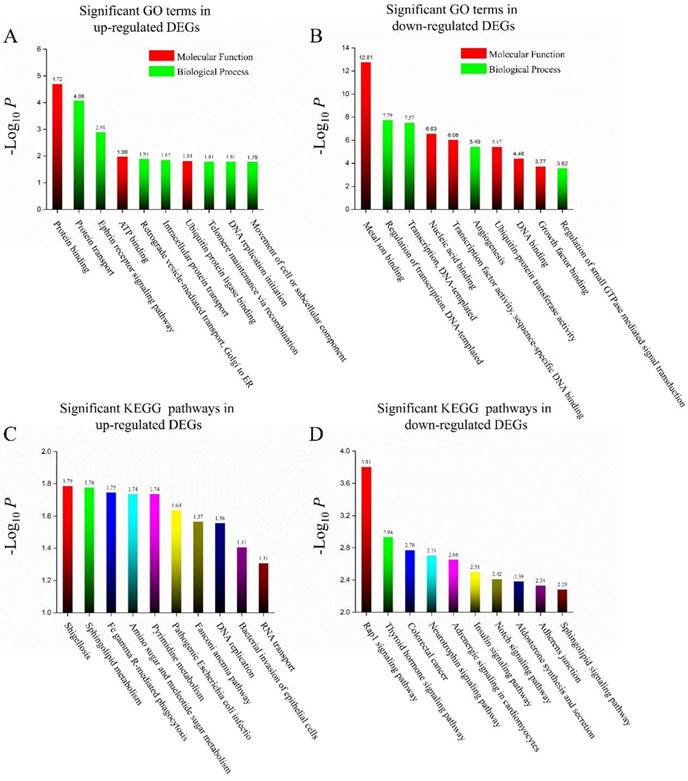
In addition, KEGG pathway enrichment analysis was also performed. Ten pathways for upregulated and 28 pathways for downregulated DEGs were found. The 10 most significant pathways are listed for both groups, as well (Figures 3 C & D).
PPI network, hub genes, and module analysis
The STRING database was used to build up the PPI network of DEGs. Furthermore, 36 hub proteins in the upregulated PPI network and 158 in the downregulated PPI network were discovered. The top 20 hub proteins in each network are shown in Table 3.
The hub proteins in the up-regulated and down-regulated protein-protein interaction network. (Top 20 in each)
| Protein Symbol | Degree | Protein Symbol | Degree | Protein Symbol | Degree | Protein Symbol | Degree |
|---|---|---|---|---|---|---|---|
| Up-regulated | |||||||
| TP53 | 61 | DNM2 | 22 | CCT3 | 18 | ORC1 | 15 |
| CAD | 49 | GRB2 | 22 | CDK16 | 17 | PABPC1 | 15 |
| TSPO | 48 | SMARCA4 | 22 | BIRC5 | 15 | RFC5 | 15 |
| SRC | 42 | ALDH18A1 | 20 | CFL1 | 15 | SEC61A1 | 15 |
| RAD51 | 33 | DECR1 | 20 | NME2 | 15 | AP2M1 | 13 |
| Down-regulated | |||||||
| CTNNB1 | 76 | MAPK8 | 61 | SIRT1 | 45 | PRKACB | 37 |
| MAPK3 | 69 | KDR | 56 | INSR | 42 | EDN1 | 36 |
| FYN | 68 | RHOB | 48 | EGR1 | 39 | KALRN | 36 |
| HDAC1 | 64 | ITGA2 | 47 | SMARCA2 | 39 | SMAD2 | 36 |
| BCL2 | 63 | KAT2B | 47 | FLT1 | 37 | SMAD4 | 36 |
In the upregulated PPI, two modules were chosen with MCODE scores >4 and nodes >6: Module-Up-A with 21 nodes (MCODE Score=4.50) and Module-Up-B with 9 nodes (MCODE Score=4.25). At the same time, three modules were chosen in the downregulated PPI network: Module-Down-A with 31nodes (MCODE Score=7.67), Module-Down-B with 21 nodes (MCODE Score=6.10), and Module-Down-C with 30 nodes (MCODE Score=4.48) (Table 4). Proteins involved in Module-Up-A and Module-Down-A are shown in Supplementary Figure 1.
Module Analysis of up- and down- regulated DEGs between BAP1 WT/Mutated cases.
| Modules | No. of DEGs | No. of connections | MCODE Score |
|---|---|---|---|
| Up-regulated | |||
| Module-Up-A | 21 | 45 | 4.50 |
| Module-Up-B | 9 | 17 | 4.25 |
| Down-regulated | |||
| Module-Down-A | 31 | 115 | 7.67 |
| Module-Down-B | 21 | 61 | 6.10 |
| Module-Down-C | 30 | 65 | 4.48 |
GO functional and KEGG pathway analyses of DEGs in Module-Up-A and Module-Down-A were implemented. The results were as follows: In Module-Up-A, DEGs were enriched in GO functions like antigen processing and presentation of exogenous peptide antigen via MHC class II and intracellular protein transport. Regarding the KEGG pathway, it included endocrine and other factor-regulated calcium reabsorption, Huntington's disease, and synaptic vesicle cycle pathways. Regarding Module-Down-A, DEGs were enriched in GO functions such as negative regulation of transcription from RNA polymerase II promoter and rRNA transcription. For the KEGG pathway, DEGs tended to be enriched in calcium signaling pathways, cGMP-PKG signaling pathways, and Chagas disease (American trypanosomiasis) pathways (Tables S1 & S2).
Gene methylation analysis
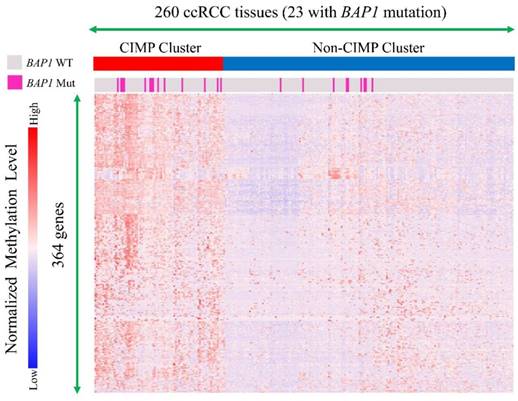
Gene methylation data of 260 ccRCC tumor tissues (23 with BAP1 mutations) were clustered and cases were divided into a CIMP cluster and non-CIMP cluster based on the clustering results (Figure 4). It indicated that BAP1-mutated cases compared with BAP1 wild-type cases had a significantly higher probability of being in a CIMP cluster (60.9% vs. 28.3%, P=0.001).
A subgroup of clear cell renal cell carcinoma manifests a CpG island methylator Phenotype (CIMP). Molecular subtyping by means of TCGA DNA methylation platform revealed two subtypes of ccRCC, one of which showed widespread DNA hypermethylation patterns characteristic of CIMP-associated tumors. BAP1 mutation cases had a significantly higher probability to obtain CIMP than BAP1 wild type cases
Discussion
Combined analysis including genome, transcriptome, proteome, and clinical data has been widely used in the field of cancer research. In our study, we comprehensively analyzed ccRCC cases extracted from the TCGA database, focusing on BAP1 functional somatic mutations. We highlighted several pathways or molecules altered dramatically between BAP1-mutated and wild-type cases, which may indicate new diagnostic biomarkers and targets for novel therapy development.
By analyzing hub genes in the upregulated PPI network, we found that TP53, CAD, and translocator protein (TSPO) had the highest connective degree. The TP53 gene encodes a tumor suppressor protein, which plays a vital role in DNA repair, the cell cycle, and apoptosis. Mutations in TP53 drives carcinogenesis in various cancers [28]. One study indicated that TP53 overexpression was an independent adverse prognostic factor in laryngeal squamous cell carcinoma [29]. TP53 protein levels can be determined using a commercial reverse phase protein array (RPPA). In addition, we examined TP53 protein levels using RPPA data deposited in TCGA and found that there was no significant difference between the BAP1-mutated and BAP1 wild-type groups (Figure S2 A). This indicated that although TP53 mRNA levels were higher in BAP1-mutated cases, a post-transcriptional mechanism was inhibiting mRNA translation. This phenomenon emphasized the importance of performing a comprehensive analysis when using bioinformatics to avoid misinterpretation of the data.
Regarding CAD, it is a multifunctional enzyme, composed of carbamoyl-phosphate synthetase II, aspartate transcarbamylase, and dihydroorotase [30]. Previous studies demonstrated that CAD had the potential to be developed as an antitumor target [31]. The de novo pyrimidine synthesis pathway is essential for cancer development, and CAD controls the first three steps in the pathway. Activity of CAD is further modulated by phosphorylation through the ERK-MAP kinase, cAMP-dependent protein kinase, and mTOR signaling cascade. Thus, the mTOR inhibitor everolimus and temsirolimus may be used to inhibit mTOR signaling and thereby inhibit CAD activity to block cancer cell proliferation. Furthermore, the structure of CAD has been determined recently. The structural data suggests that targeting the dihydroorotase domain of the human CAD protein may have antitumoral potential. In accordance with the upregulation of CAD in ccRCC, one study also revealed that CAD expression was increased in invasive and relapsing androgen-dependent tumors [32].
Regarding TSPO, it is a cholesterol- and drug-binding protein primarily located in the outer mitochondrial membrane. TSPO not only plays an important role in steroidogenesis, but also has a direct or indirect link with multiple other cellular functions including apoptosis, cell proliferation, differentiation, anion transport, porphyrin transport, heme synthesis, and regulation of mitochondrial function [33]. Based on these characteristics, TSPO-binding chemicals have exhibited an inspiring effect in PET imaging and anticancer therapies[34]. TSPO-binding drugs have also been demonstrated to cause death of several cancer cells, and TSPO has already been viewed as a novel target for cancer chemotherapy [35].
In evaluating the downregulated PPI network, we found CTNNB1 and MAPK3 to have the highest degree of connectivity. CTNNB1 is responsible for encoding β-catenin, which is a multifunctional protein that plays a significant role in maintaining physiological homeostasis. β-catenin not only maintains the integrity of epithelial tissues but also directs transcription of various genes on extracellular instigations [36]. In RCC patients, low expression of β-catenin was related to venous growth inside the tumor, extratumoral venous growth, and perineural growth. Furthermore, downregulated membranous expression intensity of β-catenin was predictive of a shorter recurrence-free survival (RFS), indicating that β-catenin could become a biomarker of aggressiveness in RCCs [37]. We also examined CTNNB1 protein levels between groups, and the differential expression observed was statistically significant (Figure S2 B).
Mitogen-activated protein kinase (MAPK3) plays an important role in one of the MAPK pathways, which regulate cell proliferation, differentiation, migration, and apoptosis. Several studies revealed that positive expression of both MAPK3 and AMPK were associated with a better prognosis in several cancers [38, 39]. These findings suggested that low expression of MAPK3 showed potential as a worse prognostic indicator.
Recently, BAP1 was reported to be mutated in up to 14% of sporadic ccRCCs and was associated with more aggressive tumors and poorer patient outcomes [9, 40]. In addition, ccRCC with a pathogenic germline BAP1 mutation has already been defined as a tumor predisposition syndrome [41]. It is unlikely that biopsy and genetic profiling of BAP1-mutated cases could change the treatment principle in localized kidney cancer patients; however, for metastatic or locally advanced patients with a BAP1 functional mutation, this might alter the therapeutic strategy. More clinical trials of new therapies like PARP inhibitors and immunotherapy, for patients with BAP1 mutations, will appear in the future. Treatment for BAP1-mutated cases, especially advanced cases, will become more personalized.
Our study had some limitations. First, all data analyzed in this study were derived from the TCGA database, not from us. Although TCGA is a huge data repository with different dimensions of data, most patients are Caucasian, African, or of Afro-Caribbean descent, and there are few Asian people. Thus, comprehensive analysis of BAP1 mutation patterns in Asian ccRCC patients is still needed in the future. Second, results of this study were analyzed, clustered, and predicted in silico, and not verified using molecular biology experiments. Third, there were only a limited number (39 patients) of BAP1-mutated cases included in this study. Therefore, as a next step, experimental studies based on our findings should be performed.
Conclusion
Our study provides a comprehensive understanding of BAP1 functional somatic-mutated ccRCC patients, and lists several pathways, biological processes, and molecules that may be involved in the progression and development of BAP1-mutated tumors. Furthermore, several top hub genes like CAD and TSPO may be potential therapeutic targets in BAP1-mutated ccRCC.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The authors acknowledge the contribution of TCGA database and would like to thank all the stuff in the department of urology at Shanghai cancer center.
Funding
This study was funded by the National Natural Science Foundation of China (Grant No.81370073 & Grant No.81772706) and Shanghai Rising Star Program (Grant No.16QA1401100).
Author contributions
Shengming Jin and Junlong Wu analyzed the data and drafted the manuscript. Weijie Gu, Fangning Wan and Wenjun Xiao helped interpreted the data. Bo Dai, Hailiang Zhang and Guohai Shi prepared all figures, Yijun Shen and Yao Zhu edited all tables. Yi-Ping Zhu and Dingwei Ye designed the study. All authors have reviewed and approved the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. CA: a cancer journal for clinicians. 2016;66:115-32
2. Jonasch E, Gao J, Rathmell WK. Renal cell carcinoma. BMJ (Clinical research ed). 2014;349:g4797
3. Ljungberg B, Bensalah K, Canfield S, Dabestani S, Hofmann F, Hora M. et al. EAU guidelines on renal cell carcinoma: 2014 update. European urology. 2015;67:913-24
4. Hakimi AA, Ostrovnaya I, Reva B, Schultz N, Chen YB, Gonen M. et al. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: a report by MSKCC and the KIRC TCGA research network. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:3259-67
5. Kapur P, Pena-Llopis S, Christie A, Zhrebker L, Pavia-Jimenez A, Rathmell WK. et al. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: a retrospective analysis with independent validation. The Lancet Oncology. 2013;14:159-67
6. Gossage L, Murtaza M, Slatter AF, Lichtenstein CP, Warren A, Haynes B. et al. Clinical and pathological impact of VHL, PBRM1, BAP1, SETD2, KDM6A, and JARID1c in clear cell renal cell carcinoma. Genes, chromosomes & cancer. 2014;53:38-51
7. Brugarolas J. PBRM1 and BAP1 as novel targets for renal cell carcinoma. Cancer journal (Sudbury, Mass). 2013;19:324-32
8. Piva F, Santoni M, Matrana MR, Satti S, Giulietti M, Occhipinti G. et al. BAP1, PBRM1 and SETD2 in clear-cell renal cell carcinoma: molecular diagnostics and possible targets for personalized therapies. Expert review of molecular diagnostics. 2015;15:1201-10
9. Pena-Llopis S, Vega-Rubin-de-Celis S, Liao A, Leng N, Pavia-Jimenez A, Wang S. et al. BAP1 loss defines a new class of renal cell carcinoma. Nature genetics. 2012;44:751-9
10. Brugarolas J. Molecular genetics of clear-cell renal cell carcinoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2014;32:1968-76
11. Farley MN, Schmidt LS, Mester JL, Pena-Llopis S, Pavia-Jimenez A, Christie A. et al. A novel germline mutation in BAP1 predisposes to familial clear-cell renal cell carcinoma. Molecular cancer research: MCR. 2013;11:1061-71
12. Popova T, Hebert L, Jacquemin V, Gad S, Caux-Moncoutier V, Dubois-d'Enghien C. et al. Germline BAP1 mutations predispose to renal cell carcinomas. American journal of human genetics. 2013;92:974-80
13. Joseph RW, Kapur P, Serie DJ, Eckel-Passow JE, Parasramka M, Ho T. et al. Loss of BAP1 protein expression is an independent marker of poor prognosis in patients with low-risk clear cell renal cell carcinoma. Cancer. 2014;120:1059-67
14. Joseph RW, Kapur P, Serie DJ, Parasramka M, Ho TH, Cheville JC. et al. Clear Cell Renal Cell Carcinoma Subtypes Identified by BAP1 and PBRM1 Expression. The Journal of urology. 2016;195:180-7
15. Kapur P, Christie A, Raman JD, Then MT, Nuhn P, Buchner A. et al. BAP1 immunohistochemistry predicts outcomes in a multi-institutional cohort with clear cell renal cell carcinoma. The Journal of urology. 2014;191:603-10
16. Minardi D, Lucarini G, Milanese G, Di Primio R, Montironi R, Muzzonigro G. Loss of nuclear BAP1 protein expression is a marker of poor prognosis in patients with clear cell renal cell carcinoma. Urologic oncology. 2016;34:338.e11-8
17. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2:401-4
18. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1
19. Gene Ontology Consortium. going forward. Nucleic acids research. 2015;43:D1049-56
20. Tanabe M, Kanehisa M. Using the KEGG database resource. Current protocols in bioinformatics. 2012;38(1.12):1-43
21. Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N. et al. TM4: a free, open-source system for microarray data management and analysis. BioTechniques. 2003;34:374-8
22. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44-57
23. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics. 2000;25:25-9
24. von Mering C, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B. STRING: a database of predicted functional associations between proteins. Nucleic acids research. 2003;31:258-61
25. Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics (Oxford, England). 2011;27:431-2
26. Rhrissorrakrai K, Gunsalus KC. MINE: Module Identification in Networks. BMC bioinformatics. 2011;12:192
27. Cancer Genome Atlas Research N, Linehan WM, Spellman PT, Ricketts CJ, Creighton CJ, Fei SS. et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. The New England journal of medicine. 2016;374:135-45
28. Leroy B, Anderson M, Soussi T. TP53 mutations in human cancer: database reassessment and prospects for the next decade. Human mutation. 2014;35:672-88
29. Qiu X, You Y, Huang J, Wang X, Zhu H, Wang Z. LAMP3 and TP53 overexpression predicts poor outcome in laryngeal squamous cell carcinoma. International journal of clinical and experimental pathology. 2015;8:5519-27
30. Evans DR, Bein K, Guy HI, Liu X, Molina JA, Zimmermann BH. CAD gene sequence and the domain structure of the mammalian multifunctional protein CAD. Biochemical Society transactions. 1993;21:186-91
31. Grande-Garcia A, Lallous N, Diaz-Tejada C, Ramon-Maiques S. Structure, functional characterization, and evolution of the dihydroorotase domain of human CAD. Structure (London, England: 1993). 2014;22:185-98
32. Morin A, Fritsch L, Mathieu JR, Gilbert C, Guarmit B, Firlej V. et al. Identification of CAD as an androgen receptor interactant and an early marker of prostate tumor recurrence. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2012;26:460-7
33. Batarseh A, Papadopoulos V. Regulation of translocator protein 18 kDa (TSPO) expression in health and disease states. Molecular and cellular endocrinology. 2010;327:1-12
34. Selvaraj V, Stocco DM. The changing landscape in translocator protein (TSPO) function. Trends in endocrinology and metabolism: TEM. 2015;26:341-8
35. Austin CJ, Kahlert J, Kassiou M, Rendina LM. The translocator protein (TSPO): a novel target for cancer chemotherapy. The international journal of biochemistry & cell biology. 2013;45:1212-6
36. Thakur R, Mishra DP. Pharmacological modulation of beta-catenin and its applications in cancer therapy. Journal of cellular and molecular medicine. 2013;17:449-56
37. Aaltomaa S, Lipponen P, Karja V, Lundstedt S, Lappi J, Kosma VM. The expression and prognostic value of alpha-, beta- and gamma-catenins in renal cell carcinoma. Anticancer research. 2004;24:2407-13
38. Baba Y, Nosho K, Shima K, Meyerhardt JA, Chan AT, Engelman JA. et al. Prognostic significance of AMP-activated protein kinase expression and modifying effect of MAPK3/1 in colorectal cancer. British journal of cancer. 2010;103:1025-33
39. Kim JG, Lee SJ, Chae YS, Kang BW, Lee YJ, Oh SY. et al. Association between phosphorylated AMP-activated protein kinase and MAPK3/1 expression and prognosis for patients with gastric cancer. Oncology. 2013;85:78-85
40. The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43-9
41. Schmidt LS, Linehan WM. Genetic predisposition to kidney cancer. Seminars in oncology. 2016;43:566-74
Author contact
![]() Corresponding authors: Prof. Ding-Wei Ye, M.D. (dwyelicom) and Dr. Yi-Ping Zhu, M.D. (qdzhuyipingcom), Department of Urology, Fudan University, Shanghai Cancer Center, No. 270 Dong'an Road, Shanghai 200032, People's Republic of China. Tel: 86-21-64175590-2807; Fax: 86-21-64434556.
Corresponding authors: Prof. Ding-Wei Ye, M.D. (dwyelicom) and Dr. Yi-Ping Zhu, M.D. (qdzhuyipingcom), Department of Urology, Fudan University, Shanghai Cancer Center, No. 270 Dong'an Road, Shanghai 200032, People's Republic of China. Tel: 86-21-64175590-2807; Fax: 86-21-64434556.