Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(23):4314-4324. doi:10.7150/jca.27943 This issue Cite
Research Paper
Autophagy Plays a Critical Role in Insulin Resistance- Mediated Chemoresistance in Hepatocellular Carcinoma Cells by Regulating the ER Stress
Linjing Li1,3*, Xinyue Liu1*, Liye Zhou3, Wei Wang4, Zhuan Liu1, Yan Cheng5, Jing Li1, Hulai Wei2 ![]()
1. Department of Clinical Laboratory Center, The Second Hospital of Lanzhou University, Lanzhou 730000, P.R. China;
2. Key Laboratory of Preclinical Study for New Drugs of Gansu Province, School of Basic Medical Sciences, Lanzhou University, Lanzhou 730000, P.R. China;
3. Dana-Farber cancer institute, Harvard Medical School, 450 Brookline Avenue, Boston, MA, 02215-5450, USA;
4. Marlene and Stewart Greenbaum Comprehensive Cancer Center, University of Maryland School of Medicine, Baltimore, MD, 21201, USA;
5. Northwest University for Nationalities, Lanzhou 730000, P.R. China.
*contributed equally to this article
Received 2018-6-17; Accepted 2018-9-14; Published 2018-10-21
Citation:
Li L, Liu X, Zhou L, Wang W, Liu Z, Cheng Y, Li J, Wei H. Autophagy Plays a Critical Role in Insulin Resistance- Mediated Chemoresistance in Hepatocellular Carcinoma Cells by Regulating the ER Stress. J Cancer 2018; 9(23):4314-4324. doi:10.7150/jca.27943. https://www.jcancer.org/v09p4314.htm
Other stylesAbstract
The high mortality of hepatocellular carcinoma (HCC) patients is associated with several independent risk factors including type 2 diabetes mellitus (T2DM) and insulin resistance (IR), which could be caused by various pathological processes such as tumorigenesis and inflammation in the liver. In previous report, we declared that IR contributes to multidrug resistance in HCC by activation of endoplasmic reticulum (ER) stress. Here, our study revealed that the enhanced autophagy induced by IR significantly prompts the chemotherapeutic drug resistance in hepatoma cells, which was validated by stimulation and inhibition of the autophagy respectively. A potential reason is that autophagy acts as a regulator of ER stress in the IR-mediated chemoresistance in HCC. In conclusion, autophagy facilitates the HCC survival in chemotherapeutic drug treatment by maintaining the homeostasis in the ER indicating the regulatory role of autophagy in ER stress contributes to IR-mediated chemoresistance in hepatocellular carcinoma cells. Collectively, these data implied inhibition of autophagy is a potential treatment of inherent IR-mediated chemoresistance in HCC.
Keywords: Insulin resistance, Hepatocellular carcinoma, Autophagy, Endoplasmic reticulum, Chemoresistance
Introduction
Hepatocellular carcinoma (HCC) ranks as the fifth most common cancer type and the third leading cause of cancer death worldwide[1]. Most of HCC patients are diagnosed at the terminal stage and are not amenable to potentially curative therapies including surgeries and radiofrequency ablation, thus chemotherapy becomes the main approach of HCC treatment. However, conventional chemotherapeutics cannot produce satisfactory clinical outcomes mainly due to the inherent multiple drug resistance (MDR)[2]. Emerging evidences demonstrated that the poor outcome of HCC patients is associated with T2DM and IR caused by tumorigenesis and inflammation in the liver [3-5]. Our previous studies also showed that IR enhances the tolerance of hepatoma cells to multiple chemotherapeutics[6,7] indicating that IR is involved in the inherent MDR of HCC.
It has been reported that IR can trigger unfolded protein response (UPR) of ER stress in liver cells to restore ER homeostasis by upregulation the expression of specific proteins such as Glucose regulated protein 78 (GRP78/Bip), autophosphorylation of protein kinase-R-like ER kinase (p-PERK) and X-box binding protein 1 (XBP-1) to halt protein synthesis, enhance protein degradation, control damage, restore normal ER functions, and promote cell survival[8-10]. In turn, severe and chronic ER stress could also tip the balance in favor of pro-apoptotic UPR response resulting in cell death[11]. In previous report, we demonstrated that IR contributes to multidrug resistance in HCC via PERK signaling pathway, which is involved in activation of UPR[7]. However, it is still poorly understood how UPR activation maintains the homeostasis of ER to enhance the IR-mediated chemotherapy resistance in hepatoma cells.
Increasing number of researchers declared that UPR acts as the driver to promote the intracellular autophagy which could in turn supplement the protein degradation during ER stress to decrease the accumulation of misfolded protein[12-14]. Macro-autophagy (conventional autophagy) is an intracellular lysosomal degradation process mediated by the formation of autophagosomes. The process is used by normal and cancer cells as mechanisms of recycling building blocks and conserving energy, as well as eliminating toxic materials for homeostasis and survival[15]. Autophagy can function as a tumor suppressor in normal cells. Dysregulation of autophagy may lead to malignant transformation and carcinogenesis. In tumor, autophagy promotes tumor growth and progression by helping tumor cells to survive in metabolically-challenged harsh tumor microenvironments[16]. Recent study suggested that the impairment of autophagic degradation promotes apoptotic cell death in multidrug-resistant liver cancer cells[17]. However, the role of autophagy in IR-mediated chemotherapy resistance in hepatocellular carcinoma cells is still unclear.
In current report, we investigated the role of autophagy in IR-mediated chemo-resistance in HCC using a stable insulin resistant hepatoma cell model (HepG2/IR) [7]. We observed a remarkable enhancement of autophagic flux in HepG2/IR cells compared to that in parental HepG2 cells. The enhanced autophagy significantly contributed to the chemotherapeutic drug resistance in HepG2/IR cells. In addition, we also found that the ER stress response can be enhanced by inhibiting autophagy or be impaired by reinforcing autophagy in HCC. In conclusion, autophagy not only facilitates the HCC survive in severe environment such as chemotherapeutic drug treatment, but also helps to maintain the homeostasis of ER. Overall, our present study indicates that the regulatory role of autophagy in ER stress in contributing to IR- mediated chemoresistance in hepatocellular carcinoma cells, which could be a potential mechanism of inherent chemoresistance in HCC.
Materials and Methods
Reagents and antibodies
Insulin, 3-(4,5)-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), Monodansylcadaverine (MDC), Triton X-100, Tween-20, Cisplatin (DDP) ,3-Methyladenine (3-MA), Rapamycin(RAPA) and Dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Pioglitazone hydrochloride (PH) was purchased from Santa Cruz Biotechnology (CA, USA). Annexin V/PI assay kit was purchased from Invitrogen/ Thermo Fisher Scientific (MA, USA). BCA protein assay kit, RIPA lysis buffer and pre-stained color protein marker were purchased from Beyotime (Beijing, China). Antibodies against cleaved Caspase-3, Bcl-2, PERK, GRP78, Beclin-1, LC3B, P62 and β-actin were obtained from Cell signaling Technology (Boston, MA, USA). Antibodies against p-PERK and CHOP were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). DMEM medium, fetal bovine serum (FBS) and penicillin-streptomycin were purchased from Gibco (Grand Island, NY, USA).
Cell culture and induction for insulin resistance
Human hepatocellular carcinoma HepG2 cells were purchased from American Type Culture Collection (ATCC HB-8065, Rockville, MD, USA) and maintained in DMEM supplemented with 10% FBS at 37°C with 5% CO2. insulin resistance (IR) was induced in HepG2 cells according to the previously described method[7]. Briefly, cells were incubated in serum-free DMEM for 6 hr then treated with insulin at a final concentration of 0.5 μM for 72 hr. The resultant cells were named as HepG2/IR cells. HepG2/IR/PH cells were obtained by treating HepG2/IR cells with pioglitazone hydrochloride (PH) (10 mM) for 24 hr to reverse IR. Authentication of the cells was performed by short tandem repeat (STR) analysis. Cells were monitored for mycoplasma contamination by MycoAlert® Mycoplasma Detection Kit (Lonza, Rockland, MA, USA).
Cell viability assay
Briefly, the cells were harvested and plated into 96-well plates for 12 hr. at the density of 1×105 cells/mL in 200μl complete medium, then exposed to 1.00-256.00 mg/L Cisplatin (DDP; Sigma-Aldrich) for various durations with or without pre-treatment by RAPA (2μmol/L) or 3-MA(2mmol/L) for 4 hr. Each treatment was repeated in six separate wells. 20μl of MTT reagent (5mg/ml) in phosphate-buffered saline (PBS) was added to each well and incubated the plates at 37°C with 5% CO2 for 4 hr. The formazan crystals were dissolved in 150μl of dimethyl sulfoxide and added into each well. The optical density (OD) was recorded at a wavelength of 490 nm using a Powerwave X plate reader (Bio-Tek, USA). Cell proliferation inhibition rates were calculated using the following formula: cell proliferation inhibition rate = [(OD control - OD experiment)/OD experiment] × 100%. The half-maximal inhibitory concentration (IC50) was also calculated as reported previously [7].
Flow cytometric analysis (FCM)
An annexin V/propidium iodide (PI) double staining assay (Invitrogen, USA) was used to determine cell apoptosis. 1×106 cells were collected and suspended in 400μl binding buffer (400μl) and incubated with Annexin V-FITC and propidium iodide (PI) for 0.5 hr. and then suspended in binding buffer. The samples were analyzed by a Coulter Epics XL flow cytometer (Beckman Coulter, USA). The early apoptotic index was determined by the percentage of Annexin V-positive and PI-negative cells. The later period index was determined by the percentage of Annexin V- positive and PI-positive cells[7].
Ultrastructure
The morphological features were assessed using an electron microscopy[7]. Briefly, Cells were immobilized in 3% glutaraldehyde, and further processed for dehydration, infiltration and embeding into LX-112 resin with serial changes into Ethanol and LX112 solutions. Finally, the samples were transferred in pyramid-tip-mold and polymerized at 60°C for 72 h. Semi-thin sections were cut using an ultra-microtome (Leica EM UC7) after attaching pyramid on mounting cylinders and stained with toluidine blue to identify the position of cells. Ultra-thin-sections (70-100nm) were cut and collected on 200 mesh grids. The grids were stained with 1% uranyl acetate at RT for 10 min and followed by Reynolds lead citrate at RT for 5 min. The ultrastructure was visualized using a JEM 1230 transmission electron microscopy (JEOL, Japan).
Detection of autophagic vacuoles by Monodansylcadaverine (MDC) staining
MDC has been proposed as a special tracer for autophagic vacuoles[18] The autophagic vacuoles were incubated with 0.05 mmol/L MDC in PBS in the dark for 30 min at 37°C after treated by drugs. After the incubation, cells were washed with PBS and immediately observed using a AX80 fluorescence microscope (Olympus, Japan).
Western blotting
Briefly, cells under different treatments were lysed and the protein concentrations were assessed using the Bradford assay (Roche). The proteins were separated by SDS-PAGE electrophoresis and then transferred to PVDF membranes. The membranes were blocked with non-fat milk and probed overnight with primary antibodies (anti-Beclin-1, anti-LC3-II, anti-P62, anti-bcl-2, anti-GRP78, anti-PERK, anti-p-PERK, anti-cleaved Caspases-3, or anti-β-actin) followed by incubating with IRDye800CW or IRDye700DX-conjugated secondary antibodies (LI-COR, USA) for 50 mins. Protein bands were visualized using an Odyssey double-color infrared-laser imaging system (LI-COR). Relative expression was then plotted to reflect true protein expression (average infrared fluorescence intensity of the target protein / β-actin).
Statistical analysis
Data are expressed as means ± SD. Statistical analysis was performed using Student's t-test with SPSS 15.0 (SPSS Inc. Chicago, IL). P values less than 0.05 were considered as statistically significant difference. The data were representative of three independent experiments performed in triplicate.
Results
Autophagy is activated in insulin resistant hepatoma cells
Our previous studies demonstrated that UPR in ER stress was involved in chemotherapy resistance in IR hepatoma cells[6,7]. ER stress has also been reported to be able to promote the intracellular autophagy[12-14]. Therefore, we first asked whether autophagy is activated by IR in hepatoma cells by the use of a stable insulin resistant hepatoma cells model (HepG2/IR) established previously [7]. Protein expression levels of autophagic flux markers including Beclin-1, LC3-II, and autophagic substrate P62 were determined using immunoblotting[19]. The results displayed that expression levels of Beclin-1 and LC3-II were elevated; while P62 was impaired in HepG2/IR cells comparing to control HepG2 cells (Figure 1B). In contrast, reversion of IR by insulin sensitizer Pioglitazone Hydrochloride (PH)[6] significantly suppressed the upregulation of Beclin-1 and LC3-II and promoted the accumulation of P62 in HepG2/IR cells (Figure 1B).
Furthermore, transmission electron microscopy (TEM) were used to visualize ultrastructure changes in cells. Compared with the parental HepG2 cells, HepG2/IR cells showed an increasing number of double-membrane vesicles fusing with lysosomes accompanied with evident ER expansion. These morphological changes were reversed by the treatment of PH (Figure 1A, upper plane). In concert with ultrastructure, we observed increased accumulation of autophagic vacuoles in HepG2/IR cells by MDC staining, which can also be reversed by PH treatment (Figure 1A, lower plane). These data indicate that autophagy is enhanced in insulin resistant hepatoma cells.
Enhanced autophagy promotes IR-associated chemotherapeutic drug resistance in hepatoma cells
Autophagy activated by ER stress is an important regulatory mechanism in maintaining the cellular homeostasis[20,21]. We hypothesized that the activation of autophagic flux is a key factor of IR-mediated chemotherapy drug resistance in HCC. To test this, we pretreated HepG2 and HepG2/IR cells with autophagy inducer rapamycin (RAPA) and then treated them with cisplatin (DDP), a widely used chemotherapy drug for solid tumors. Firstly, the protein expression levels of autophagic flux markers and the cell survival in DDP treated HepG2/IR and HepG2 cells were evaluated. The autophagic flux in HepG2 and HepG2/IR cells were modestly activated by DDP treatment alone. Notably, with treatment of DDP, HepG2/IR cells showed dramatically higher levels of LC3-II and Beclin-1, and a significantly lower level of autophagic substrate P62 than that in HepG2 cells (Figure 2A-D). In comparison with HepG2 cells, a significant lower apoptosis rate was induced by DDP treatment in HepG2/IR cells (Figure 2E, F), which was further confirmed by the higher expression level of anti-apoptotic Bcl-2 and lower expression level of pro-apoptotic cleaved-caspase-3 in HepG2/IR cells (Figure 2G-I). Consistent with these results, more intracellular autophagic vacuoles formation were observed by TEM and FLM in HepG2/IR cells upon treatment with DDP. In addition, less apoptotic morphological changes were detected by TEM in HepG2/IR cells, indicated by vacuolization of cytoplasm, condensed and fragmented nucleus and formation of apoptotic bodies (Figure 2J, K). These data demonstrated that higher autophagic flux activated in HepG2/IR cells is involved in IR-mediated chemotherapy drug resistance.
Figure 1
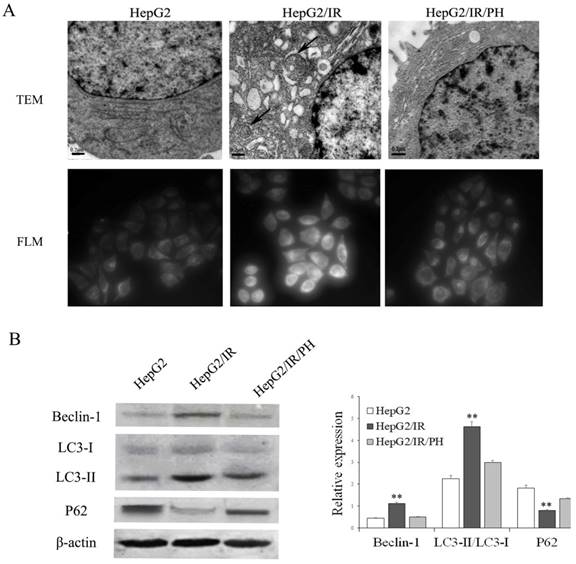
Analysis of autophagy activation in HepG2/IR cells. (A) Ultrastructure detected by transmission electron microscopy (TEM) (original magnification, ×20,000). The fluorescence intensity of MDC was observed by fluorescence light microscopy (FLM) (original magnification, ×1,000). (B) HepG2 cells were treated with 0.5 μmol/L insulin for 72 hr to induce HepG2/IR cells, and then treated with 10mmol/L PH for 24 hours to reverse IR. Cell lysates were used for autophagy markers detection by immunoblotting. The quantitative data are the Beclin-1/β-actin, LC3-II/LC3-I, P62/β-actin. All experiments were repeated three times, and data were presented as mean ± SD of triplicate experiments. ** P<0.01 vs HepG2 cells.
Figure 2
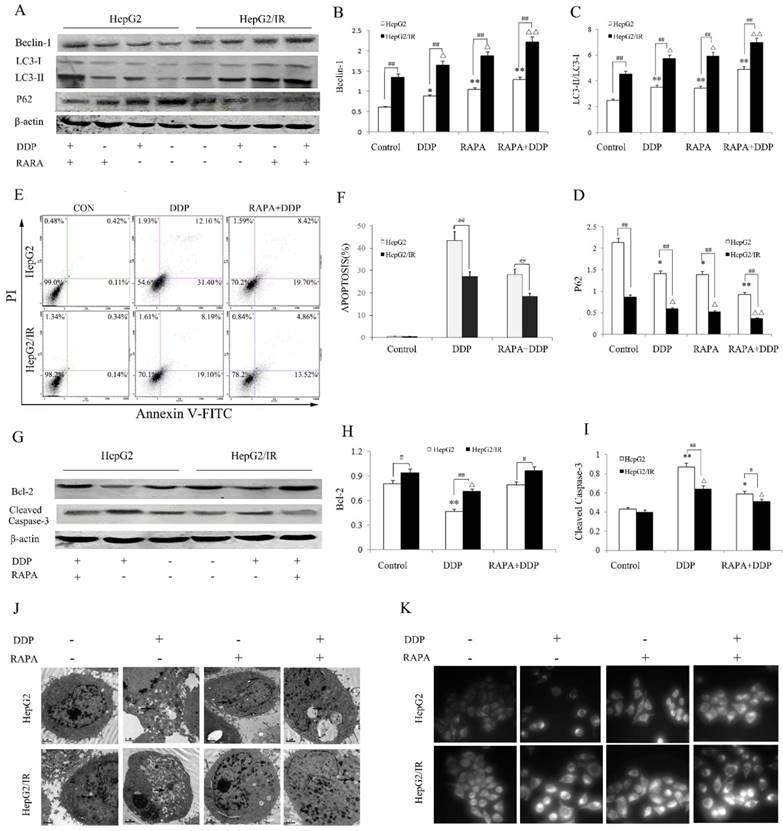
Autophagic enhancement promotes IR-mediated chemotherapeutic drug resistance in hepatoma cells. (A-D) Autophagy markers were detected by immunoblotting at 48 hr following treatment with 16mg/L DDP with or without pretreated by 2 μmol/L RAPA for 4 hr in both HepG2 and HepG2/IR cells. The quantitative data are the Beclin-1/β-actin (B), LC3-II/LC3-I (C) and P62/β-actin (D). (E-F). HepG2 and HepG2/IR cells were exposed to 16mg/L DDP for 48 hr with or without pretreated by 2μmol/L RAPA for 4 hr and then harvested. Apoptotic cell rates were detected with Annexin V-FITC/PI double staining assay and then acquired by flow cytometry. The apoptotic cell rate is shown (F). (G-I) The analysis of cleaved Caspase-3 and Bcl-2 at 48 hr following treatment with 16mg/L DDP with or without pretreated by 2μmol/L RAPA for 4 hr by Western blot. The quantitative data are the Bcl-2/β-actin (H) and Cleaved Caspase-3/β-actin (I). (J) FLM, original magnification×1,000. (K) TEM, original magnification×2,000. All experiments were repeated three times, and data were presented as mean ± SD of triplicate experiments. * P<0.1, ** P<0.01, vs control group of HepG2 cells; △ P<0.1, △△ P<0.01, vs control group of HepG2/IR cells; # P<0.1, ## P<0.01. vs HepG2 cells in the same treatment group.
To further determine whether autophagy can enhance the IR-mediated chemotherapy drug resistance in hepatoma cells, we treated the HepG2/IR and HepG2 cells with DDP following treatment by autophagy inducer RAPA. A significant stronger autophagic flux was induced by combinational treatment of RAPA and DDP in HepG2/IR than in HepG2 cells, displayed with higher levels of LC3-II and Beclin-1 and a lower expression of P62 in HepG2/IR cells. Compared with the DDP treated group, the extent of autophagy enhancement was shown as Beclin-1 increased by 44.60% and 33.90%, LC3-II increased by 39.31% and 21.62% while P62 decreased by 34.12% and 37.05% respectively in HepG2 and HepG2/IR cells. As expected, both HepG2 and HepG2/IR cells exhibited slightly higher autophagic flux by RAPA treatment alone comparing to their corresponding control group (Figure 2A-D). Although DDP treatment could induce apoptosis in both cell lines, RAPA pre-treatment markedly induced a stronger resistance to DDP in HepG2/IR cells than in HepG2 cells as determined by the MTT (Table 1) and Annexin V and PI double staining assay. The apoptosis rate decreased by 35.36% (HepG2) and 32.65% (HepG2/IR), respectively (Figure 2E, F). These results were further confirmed by the observation that RAPA up-regulated the expression of anti-apoptotic Bcl-2 and down-regulated pro-apoptotic cleaved-caspase-3 protein levels in both HepG2/IR and HepG2 cells (Figure 2G-I). Moreover, morphological alterations in HepG2/IR and HepG2 cells induced by DDP following the RAPA pretreatment showed elevated autophagic vacuoles and less apoptotic bodies in HepG2/IR cells than that in HepG2 cells (Figure 2J, K). These results further suggest that enhancement of autophagy is involved in IR-mediated chemotherapeutic resistance in hepatoma cells.
Table 1
Enhancement of autophagic flux promoted the DDP resistance
| Treated time | 50% inhibitory conc of DDP (mg/L) | |||
|---|---|---|---|---|
| HepG2 | RAPA+ HepG2 | HepG2/IR | RAPA+ HepG2/IR | |
| 24 h | 150.4±0.31 | 359.72±0.25** | 274.4±0.22 | 513.23±0.21△△ |
| 48 h | 10.27±0.24 | 22.01±0.17** | 17.04±0.17 | 26.11±0.15△△ |
| 72 h | 7.36±0.14 | 14.57±0.22** | 11.69±0.19 | 18.93±0.21△△ |
** P<0.01 vs HepG2 cells, △△ vs HepG2/IR cells
Depressed autophagy impaired IR-mediated chemotherapeutic drug resistance in hepatoma cells
We further explored the effect of autophagy inhibition on DDP induced cytotoxicity in hepatoma cells. HepG2 and HepG2/IR cells were treated by DDP with or without 3-MA, an autophagy inhibitor. Compared to the control group, the activity of autophagy in HepG2 and HepG2/IR cells was significantly suppressed by 3-MA, which was shown by decreased LC3-II and Beclin-1 accompanied with increased autophagic substrate P62. 3-MA pre-treatment also impaired the activation of autophagy induced by DDP, which was shown as Beclin-1 decreased by 54.90% and 32.90%, LC3-II decreased by 51.60% and 31.5%, while P62 increased by 69.54% and 148.41% respectively in HepG2 and HepG2/IR cells. Overall, a lower activity of autophagy was observed in HepG2 cells upon combinational treatment of 3-MA and DDP compared with that in HepG2/IR cells (Figure 3A-D).
Compared to the DDP treatment alone, 3-MA pretreatment dramatically enhanced drug sensitivity of HepG2 and HepG2/IR cells as determined by the MTT (Table 2), and Annexin V/PI double staining assay which was revealed by obviously increased DDP-induced apoptosis in HepG2 (59.51%) and HepG2/IR cells (71.73%). The enhanced drug sensitivity also led to massive cell death upon DDP treatment for 72 hr. In addition, a significantly higher apoptosis rate was observed in 3-MA and DDP treated HepG2 cells than in HepG2/IR cells (Figure 3E, F). The autophagy inhibition induced drug sensitivity was further confirmed by evidently decreased Bcl-2 and increased cleaved caspase 3 expression levels in HepG2 than that in HepG2/IR cells following 3-MA and DDP treatment (Figure 3G-I).
Table 2
Inhibition of autophagic flux promoted the DDP sensitivity
| Treated time | 50% inhibitory conc of DDP (mg/L) | |||
|---|---|---|---|---|
| HepG2 | 3-MA+ HepG2 | HepG2/IR | 3-MA+ HepG2/IR | |
| 24 h | 150.4±0.31 | 55.89±0.25** | 274.4±0.22 | 90.14±0.19△△ |
| 48 h | 10.27±0.24 | 4.34±0.09** | 17.04±0.17 | 5.45±0.13△△ |
** P<0.01 vs HepG2 cells, △△ vs HepG2/IR cells
Furthermore, we observed that 3-MA pretreatment significantly attenuated DDP activated autophagic vacuoles accumulation within the cytoplasm in HepG2 and HepG2/IR cells (Figure 3J). Consistent with the MDC staining results, 3-MA and DDP combinational treatment induced a significant depletion of autophagic vacuoles with notable apoptotic changes including cell shrinkage, condensation and margination of nuclear chromatin, and nuclear fragmentation in both HepG2 and HepG2/IR cells, which was detected by TEM. Interestingly, less autophagic vacuoles and more apoptotic changes appeared in HepG2 cells than that in HepG2/IR cells (Figure 3K). Collectively, these data demonstrated that inhibition of autophagic flux by 3-MA promotes chemotherapeutic drug-induced apoptotic cell death, suggesting that enhanced autophagy contributes to the chemotherapeutic drug resistance in hepatocellular carcinoma.
Autophagy maintains the homeostasis in ER to promote survival of insulin resistant hepatoma cells
Autophagy is an essential survival pathway for many types of cancer. The inhibition of autophagy may severely impair the cellular stress response and result in cell death [20]. To investigate the influence of autophagy on ER stress in hepatoma cells, we detected morphological changes of HepG2 and HepG2/IR cells treated with RAPA and 3-MA, respectively. Our results showed that RAPA treatment resulted in less ER expansion, degranulation, and mitochondrial swelling accompanied with more autophagic vacuoles accumulation; while 3-MA induced severe ER expansion, degranulation, and mitochondrial swelling coupled with less autophagosomes (Figure 4A). These morphological changes appeared in both HepG2 and in HepG2/IR cells, suggesting that enhancement of autophagy may attenuate ER stress; while inhibition of autophagy may aggravate ER stress.
Figure 3
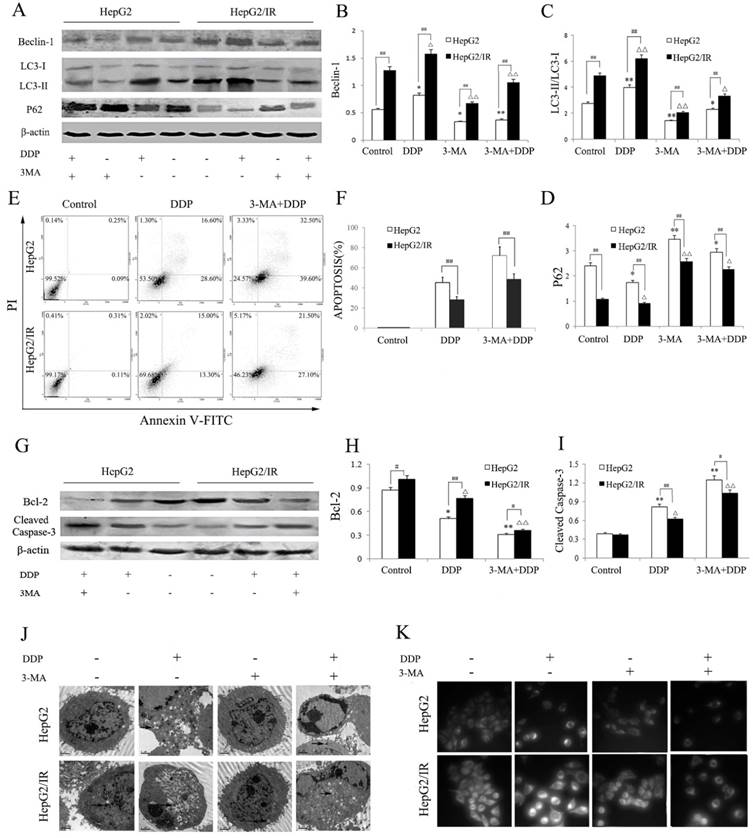
Autophagy inhibition impaired IR-mediated chemotherapeutic drug resistance in hepatoma cells. (A-D) Autophagy markers were detected by immunoblotting at 48 hr following treatment with 16mg/L DDP with or without pretreated by 2 mmol/L 3-MA for 4 hr in both HepG2 and HepG2/IR cells. The quantitative data are the Beclin-1/β-actin (B), LC3-II/LC3-I (C) and P62/β-actin (D). (E-F) HepG2 and HepG2/IR cells were exposed to 16mg/L DDP for 48 h with or without pretreated by 2mmol/L 3-MA for 4 hr and then harvested. Apoptotic cell rates were detected with Annexin V-FITC/PI double staining assay followed by flow cytometry. The apoptotic cell rate is shown (F). (G-I) The analysis of cleaved Caspase-3 and Bcl-2 at 48 hr following treatment with 16mg/L DDP with or without pretreated by 2mmol/L 3-MA for 4 hr by Western blot. The quantitative data are the Bcl-2/β-actin (H) and Cleaved Caspase-3/β-actin (I). (J) FLM, original magnification×1,000. (K) TEM, original magnification×2,000. All experiments were repeated three times, and data were presented as mean ± SD of triplicate experiments. * P<0.1, ** P<0.01, vs control group of HepG2 cells. △ P<0.1, △△ P<0.01, vs control group of HepG2/IR cells. # P<0.1, ## P<0.01 vs HepG2 cells in the same treatment group.
Figure 4
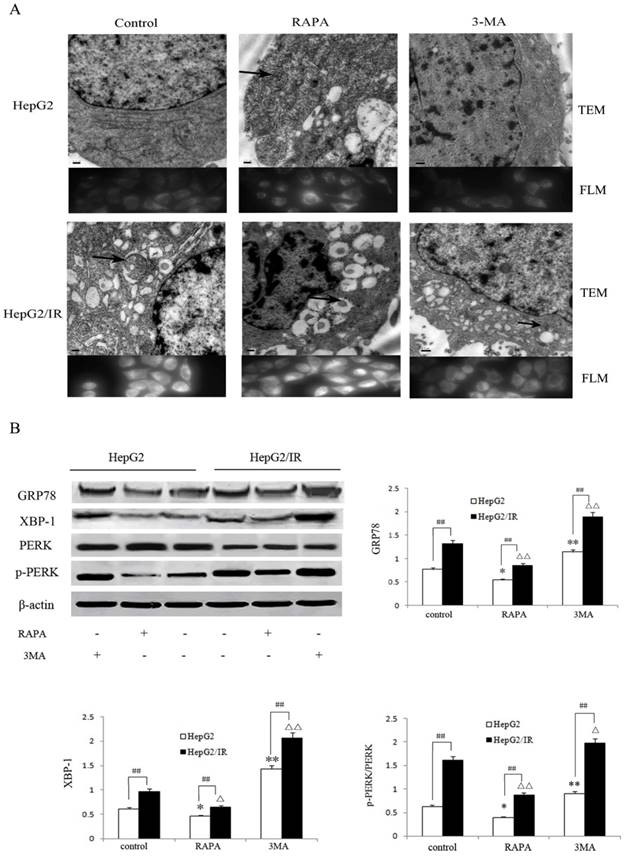
Autophagy regulates the ER stress response in HepG2/IR cells. (A) FLM, original magnification×1,000 and TEM, original magnification×2,000. (B) Western blot analysis of GRP78, XBP-1, PERK and p-PERK following 4 hr treatment by 2μmol/L RAPA or 2mmol/L 3-MA. The quantitative data are the GRP78/β-actin, XBP-1/β-actin and p-PERK/PERK. All experiments were repeated three times, and data were presented as mean ± SD of triplicate experiments. * P<0.1, ** P<0.01, vs control group of HepG2 cells. △ P<0.1, △△ P<0.01, vs control group of HepG2/IR cells. # P<0.1, ## P<0.01 vs HepG2 cells in the same treatment group.
Moreover, we evaluated the markers of ER stress including Glucose regulated protein 78 (GRP78), phosphorylation-protein kinase-like endoplasmic reticulum kinase (p-PERK)/ protein kinase-like endoplasmic reticulum kinase (PERK) and X-box binding protein 1 (XBP-1)[22] following treatment with RAPA and 3-MA respectively. The extent of down-regulation of GRP78 (35.6%), p-PERK/PERK (33.0%) and XBP-1(46.0%) in HepG2/IR cells were significantly greater than that in HepG2 cells under RAPA treatment (Figure 4B). In contrast, inhibition of autophagy notably upregulated the expression of GRP78, p-PERK/PERK and XBP-1 by 48.1%, 134.4%, and 42.9% in HepG2 cells than that in HepG2/IR cells (Figure 4B). Collectively, these results indicate that autophagy plays a protective role by modulating the ER stress to maintain the ER homeostasis and protect hepatocellular carcinoma cells from apoptotic death, especially in insulin resistant hepatoma cells with enhanced autophagy.
Discussion
HCC is a highly malignant cancer with a significant recurrence rate and a poor prognosis [23]. As most of pharmacological strategies used in HCC treatment are poorly effective[24], it is critical to elucidate the refractory mechanism of chemotherapy for developing novel strategies to overcome drug resistance. Insulin resistance (IR) in hepatocytes is a common consequence of inflammation and tumorigenesis in the liver, when insulin signaling pathways are blocked [3,25]. Our previous studies suggested that IR is involved in the development of multiple chemotherapeutic drugs tolerance in HCC [6,7]. In this study, we further revealed the role of autophagy in protecting the HCC from apoptotic cell death under the chemotherapy treatment by maintaining the ER homeostasis in insulin resistant HCC.
Firstly, a higher basal level of autophagy was observed in stable insulin resistant HCC (Fig 1), which supports the notion that IR can elevate the autophagic flux and autophagosome completion in hepatoma cells. However, our observation is different from some reports which demonstrated that hepatic autophagy is suppressed by IR in obese type II diabetes mice model[26,27]. A possible reason could be that the role of autophagy in hepatoma cells is dissimilar from that in normal liver cells. A basal level of autophagy is maintained in normal liver cells to eliminate or repair damaged organelles[15], while more importantly hepatic autophagy is highly activated to respond to the starvation when the level of nutrients and insulin in plasma is low[28]. Thus, it is reasonable to speculate that a severe autophagy deficiency in normal liver cells could be induced in the presence of IR or hyperinsulinemia caused by chronic exposure to high energy and nutrient. In fact, hepatoma cells are usually under the condition of nutrient deficiency due to sustaining proliferation[29]. Autophagy plays a key role in survival-promoting capability of tumor cells by providing energy and building blocks required for supporting the proliferative and metabolic demands in nutrient deficiency conditions[16]. However, excessive autophagy could also lead to tumor cell death[30,31]. Although IR can suppress the autophagic flux[26,27,32], the ER stress triggered by IR can also boost autophagy in tumor cells[12-14]. Thus, the overall cellular autophagy level in IR hepatoma cells would be determined by both IR and ER stress and maintained at a slightly higher level. It would be plausible that tumorigenesis-induced IR, which is different from obesity-induced IR in hepatocytes, maintains a mildly enhancement of autophagy to protect hepatoma cells from autophagic death.
We declared here the higher level of autophagy is a contributor to the IR-mediated chemoresistance in hepatoma cells. It is supported by the observation that the enhanced autophagy (Fig 2 A-D) is accompanied with a stronger drug resistance (Fig 2 E-I) in HCC with stable insulin resistance (HepG2/IR). The protective role of autophagy in IR-mediated survival of HCC was further confirmed by the observation that an increased apoptosis rate of HCC was induced by the autophagic inhibitor (Fig 3). It was reported that a modest, but not excessive, level of autophagy plays a critical role in tumor cell survival[33]. A mild increase of autophagic flux was also promoted by IR in HCC as demonstrated by a moderate alteration of Beclin-1 (an autophagic regulator) and LC3-II (a contributor to form a stable membrane of autophagosomes) in HepG2/IR cells following the treatment with chemotherapeutics. The autophagy still maintained a moderate enhancement in HepG2/IR cells treated with autophagic inducer (Fig 2 A-D), suggesting a stability of autophagy in IR hepatoma cells. Hence, it would be possible that IR enabled hepatoma cells to maintain the autophagic homeostasis at a mildly elevated level, which contributes to IR-mediated chemoresistance. However, the autophagic substrate P62 changed dramatically in HepG2/IR cells following the combinational treatment of chemotherapy drug and autophagic inducer. A possible reason could be that P62 plays central functions in processes such as autophagy, apoptosis, inflammation, cell survival, cells death, and signal transduction via its multiple binding domains[34]. Future studies are necessary to address the exact role of P62 in chemoresistance of insulin resistant hepatoma cells.
Moreover, we demonstrate the potential role of autophagy in maintaining ER homeostasis in insulin resistant hepatoma cells. Similar to autophagy, ER stress plays a dual role in cell fate determination depending on the intensity of ER stress[11,35]. Our previous study showed that the PERK signaling pathway in ER stress is activated by IR and plays a protective role in the IR-mediated multidrug resistance in hepatoma cells[7]. Several recent studies have demonstrated that PERK pathway can induce protective autophagy activation and eventually lead to tumor cell survival[36-38]. However, highly activated PERK also switches autophagy to C/-EBP homologous protein (CHOP)-mediated cell apoptosis[39]. Little difference between expression levels of CHOP in HepG2/IR and HepG2 cells suggests that ER stress in insulin resistant hepatoma cells are maintained at a steady state[7]. In fact, autophagy is an essential survival mechanism for tumor cells to transport misfolded proteins for degradation, lower cellular stress[20,35], and decrease the accumulation of misfolded protein during ER stress[12-14]. In the present study, we discovered that the autophagy serves as an adaptive response to regulate the ER stress, maintain the ER homeostasis, and promote survival in IR hepatoma cells. Collectively, we infer that IR could regulate the autophagy triggered by ER stress, which is activated in IR hepatoma cells under chemotherapy. In turn, the moderately activated autophagy can also regulate the ER stress in hepatoma cells with IR. Hence, autophagy plays a critical role in IR-mediated chemoresistance in hepatocellular carcinoma cells by regulating the ER stress.
In summary, our findings provide a new insight regarding the role of autophagy in IR-mediated chemoresistance in hepatoma cells and suggest an innovative strategy to overcome inherent drug resistance in liver cancer by inhibiting autophagic flux. In vivo studies are needed to further investigate the possible reasons of chemoresistance in insulin resistant liver cancer cells.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81602622), Scientific Research Project of Gansu Medical and Health Industry (GSWSKY2016-14), Internationally Technological Cooperation Project of Gansu Province (18YF1WA117), Cuiying Scientific and Technological Innovation Program of Lanzhou University Second Hospital and Scientific Research Initiation Funds for Doctor of Lanzhou University Second Hospital.
Author Contributions
L.J., X.Y., and H.L. conceived and designed the experiments. L.J., Z.L. and Y.C. performed the experiments. L.J., X.Y., L.Y., W.W. and J.L. analyzed the data. L.J., L.Y., and W.W. wrote the paper. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistic. CA Cancer J Clin. 2015;65:25-29
2. Chenivesse X, Franco D, Brechot C. MDR1 (multidrug resistance) gene expression in human primary liver cancer and cirrhosis. Hepatol. 1993;18(2):168-172
3. Leclercq IA, Morais ADS, Schroyen B. et al. Insulin resistance in hepatocytes and sinusoidal liver cells: Mechanisms and consequences. Hepatol. 2007;47:142-156
4. Feng YH, Lin CY, Huang WT. et al. Diabets mellitus impairs the response to intra-arterial chemotherapy in hepatocellular carcinoma. J Med Oncol. 2011;28:1080-1088
5. Mantovani A, Targher G. Type 2 diabetes mellitus and risk of hepatocellular carcinoma: spotlight on nonalcoholic fatty liver disease. Ann Transl Med. 2017;5(13):270
6. Li L, Li G, Wei H. et al. The endoplasmic reticulum stress response is associated with insulin resistance- mediated drug resistance in HepG2 cells. Neoplasma. 2015;62(2):180-190
7. Liu X, Li L, Li J. et al. Insulin resistance contributes to multidrug resistance in HepG2 cells via activation of the PERK signaling pathway and upregulation of Bcl-2 and P-gp. Oncology Reports. 2016;35(5):251-258
8. Quan X, Wang J, Liang C. et al. Melatonin inhibits tunicamycin-induced endoplasmic reticulum stress and insulin resistance in skeletal muscle cells. Biochem Biophys Res Commun. 2015;12:30123-30126
9. Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci. 2001;26:504-510
10. Calfon M, Zeng H, Urano F. et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92-96
11. Demay Y, Perochon J, Szuplewski S. et al. The PERK pathway independently triggers apoptosis and a Rac1/Slpr/JNK/Dilp8 signaling favoring tissue homeostasis in a chronic ER stress Drosophila model. Cell Death Dis. 2014;9(5):1452
12. Rouschop K, Beucken T, Dubois L. et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. 2010;120:127-141
13. Chir W, Maurin A, Carraro V. et al. The eIF2 alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41:7683-7699
14. Rzymski T, Milani M, Pike L. et al. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene. 2010;29:4424-4435
15. Rubinsztein DC, Shpilka T, Elazar Z. Mechanisms of autophagosome biogenesis. Curr Biol. 2012;22(1):R29-R34
16. Bulent O, Doris MB. Targeting autophagy in cancer management strategies and developments. Cancer Management and Research. 2015;11(7):291-299
17. Sun H, Huang M, Yao N. et al. The cycloartane triterpenoid ADCX impairs autophagic degradation through Akt overactivation and promotes apoptotic cell death in multidrug-resistant HepG2/ADM cells. Biochem Pharmacol. 2017;146:87-100
18. Biederbick A, Kern HF, Elsasser HP. Monodansylcadaverine (MDC) is a specific in vivo markerfor autophagic vacuoles. Eur J Cell Biol. 2005;171:603-614
19. Klionsky DJ, Abdelmohsen K, Abe A. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1-222
20. Jin S, White E. Role of autophagy in cancer: management of metabolic stress. Autophagy. 2007;3:28-31
21. Yu Cai, Jyothi Arikkath, Lu Yang. et al. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy. 2016;12(2):225-244
22. Minakshi R, Rahman S, Jan AT. et al. Implications of aging and the endoplasmic reticulum unfolded protein response on the molecular modality of breast cancer. Exp Mol Med. 2017;49(11):e389
23. El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142:1264-1273
24. Marin JJ, Romero MR, Briz O. Molecular bases of liver cancer refractoriness to pharmacological treatment. Curr Med Chem. 2010;17:709-740
25. Fabiani S, Fallahi P, Ferrari SM. et al. Hepatitis C virus infection and development of type 2 diabetes mellitus: Systematic review and meta-analysis of the literature. Rev Endocr Metab Disord. 2018 doi: 10.1007/s11154-017-9440-1. [Epub ahead of print]
26. Liu HY, Han J, Cao SY. et al. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J Biol Chem. 2009;284(45):31484-92
27. Yang L, Li P, Fu S. et al. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11(6):467-78
28. Mortimore GE, Hutson NJ, Surmacz CA. Quantitative correlation between proteolysis and macro- and microautophagy in mouse hepatocytes during starvation and refeeding. Proc Natl Acad Sci. 1983;80:2179-2183
29. Huang D, Li T, Wang L. et al. Hepatocellular carcinoma redirects to ketolysis for progression under nutrition deprivation stress. Cell Res. 2016;26(10):1112-1130
30. Jayanto D, Eric H, Guido K. Does autophagy contribute to cell death? Autophagy. 2005;1:66-74
31. Ding WX, Ni HM, Gao W. et al. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem. 2007;282:4702-4710
32. Ost A, Svensson K, Ruishalme I. et al. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol Med. 2010;16:235-246
33. KS Choi. Autophagy and cancer. Experimental &Molecular Medicine. 2012;44:109-120
34. Islam MA, Sooro MA, Zhang P. Autophagic Regulation of p62 is Critical for Cancer Therapy. Int J Mol Sci. 2018:19 (5)
35. Avril T, Vauléon E, Chevet E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis. 2017;6(8):e373
36. Wang J, Huang S, Tian R. et al. The protective autophagy activated by GANT-61 in MYCN amplified neuroblastoma cells is mediated by PERK. Oncotarget. 2018;9(18):14413-14427
37. Wang H, Zhang G. ER stress-mediated autophagy protects against β,β-dimethylacrylshikonin-induced apoptosis in lung adenocarcinoma cells. Cancer Sci. 2018 DOI 10.1111/cas.13616
38. Storniolo A, Alfano V, Carbotta S. et al. IRE1α deficiency promotes tumor cell death and eIF2α degradation through PERK dependent autophagy. Cell Death Discov. 2018;4:3. DOI 10.1038/s41420-017-0002-9
39. Rozpedek W, Pytel D, Mucha B. et al. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr Mol Med. 2016;16(6):533-44
Author contact
![]() Corresponding author: Hulai Wei, Key Laboratory of Preclinical Study for New Drugs of Gansu Province, School of Basic Medical Sciences, Lanzhou University, 199 Donggang West Road, Lanzhou,Gansu 730000, P.R. China. E-mail: weihulaiedu.cn
Corresponding author: Hulai Wei, Key Laboratory of Preclinical Study for New Drugs of Gansu Province, School of Basic Medical Sciences, Lanzhou University, 199 Donggang West Road, Lanzhou,Gansu 730000, P.R. China. E-mail: weihulaiedu.cn
Citation styles
APA
Li, L., Liu, X., Zhou, L., Wang, W., Liu, Z., Cheng, Y., Li, J., Wei, H. (2018). Autophagy Plays a Critical Role in Insulin Resistance- Mediated Chemoresistance in Hepatocellular Carcinoma Cells by Regulating the ER Stress. Journal of Cancer, 9(23), 4314-4324. https://doi.org/10.7150/jca.27943.
ACS
Li, L.; Liu, X.; Zhou, L.; Wang, W.; Liu, Z.; Cheng, Y.; Li, J.; Wei, H. Autophagy Plays a Critical Role in Insulin Resistance- Mediated Chemoresistance in Hepatocellular Carcinoma Cells by Regulating the ER Stress. J. Cancer 2018, 9 (23), 4314-4324. DOI: 10.7150/jca.27943.
NLM
Li L, Liu X, Zhou L, Wang W, Liu Z, Cheng Y, Li J, Wei H. Autophagy Plays a Critical Role in Insulin Resistance- Mediated Chemoresistance in Hepatocellular Carcinoma Cells by Regulating the ER Stress. J Cancer 2018; 9(23):4314-4324. doi:10.7150/jca.27943. https://www.jcancer.org/v09p4314.htm
CSE
Li L, Liu X, Zhou L, Wang W, Liu Z, Cheng Y, Li J, Wei H. 2018. Autophagy Plays a Critical Role in Insulin Resistance- Mediated Chemoresistance in Hepatocellular Carcinoma Cells by Regulating the ER Stress. J Cancer. 9(23):4314-4324.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.