Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(2):367-377. doi:10.7150/jca.27947 This issue Cite
Research Paper
Alteration of Adaptive Immunity in a Colorectal Peritoneal Carcinomatosis Model
Rongchen Shi1*, Wei Xiang1*, Xia Kang1, Lili Zhang2, Jinping Wang3, Hongming Miao1 ![]() , Fengtian He1
, Fengtian He1 ![]()
1. Department of Biochemistry and Molecular Biology, Third Military Medical University (Army Medical University), Chongqing 400038, China
2. Department of Military Psychology, School of Psychology, Third Military Medical University (Army Medical University), Chongqing 400038, China
3. Department of Neurology, Chongqing Emergency Medical Center, Chongqing 400014, China
*Co-first authors
Received 2018-6-18; Accepted 2018-10-3; Published 2019-1-1
Citation:
Shi R, Xiang W, Kang X, Zhang L, Wang J, Miao H, He F. Alteration of Adaptive Immunity in a Colorectal Peritoneal Carcinomatosis Model. J Cancer 2019; 10(2):367-377. doi:10.7150/jca.27947. https://www.jcancer.org/v10p0367.htm
Other stylesAbstract
Colorectal cancer (CRC) usually gives rise to transcoelomic spread and ultimately causes peritoneal carcinomatosis (PC). However, mechanism studies, especially the immunological basis of colorectal PC, are rarely revealed due to lack of a suitable PC model. Here we selected a mouse colorectal cancer cell line MC-38 for intraperitoneal inoculation in the C57BL/6 mice to mimic the development of colorectal PC. We demonstrated that the injected CRC cells preferentially and rapidly migrated and colonized in the visceral fat tissues, but not in other visceral organs. With flow cytometric analysis, we found the proportions of spleen T cells and B cells were not affected by PC progression, while the ratios of blood CD4+ and CD8+ T cells were largely influenced. Especially, the quantity or activity of CD4+ and CD8+ T cells in visceral fats were intimately regulated by PC development. Taken together, we successfully constructed a colorectal PC model in immune-competent mice and revealed the alteration of adaptive immunity in PC development. Our study might potentiate the research and therapy strategies of colorectal PC.
Keywords: peritoneal carcinomatosis, adaptive immunity, colorectal cancer
Introduction
Colorectal cancer (CRC) is the third most common malignancy, with a major cause of mortality by metastasis1-2. Besides the hematogenous and lymphatic routes, CRC usually gives rise to transcoelomic spread and ultimately causes peritoneal carcinomatosis (PC)3-5. PC is encountered in around 7% of patients at primary surgery, in 4% to 19% of patients during follow-up after curative surgery, in up to 40% to 80% of patients who succumb to CRC4.
In the past, the CRC patients with PC are considered as being in the terminal phase of their disease, and receive only supportive treatments, including the palliative surgery and systemic chemotherapy, achieving a survival rate not exceeding 7 months with 5-FU and Leucovorin6, reaching up to 23.4 month survival with Oxaliplatine and Irinotecan7. In the last decade, PC is considered as an initial loco-regional disease and renewed approaches for this disease are contributed by many research groups. The complete cytoreduction of macroscopic disease combined with hyperthermic intraperitoneal chemotherapy (HIPEC) has shown curative benefits on colorectal PC8-9, achieving overall survival rates up to 46 months and five-year survival rates up to 51%7.
The occurrence of colorectal PC relies on two important steps10. Firstly, the primary CRC cells need spread into peritoneal cavity. The primary invasive CRC could break through the bowel all and the cancer cells might fell off11. This case is encountered at primary surgery, with an incident rated around 7%12. The most frequent route for peritoneal dissemination comes from surgery-resulted rupture of bowel and vascular system4. The invading cancer cells in the lymphatic and blood vessels disseminate into the peritoneal cavity and targets a more adaptable microenvironment for malignant development. It's reported that this kind of PC cases account for up to 19% of postoperative patients4. Secondly, the disseminated cancer cells need target and colonize in a peritoneal tissue or organ, which could provide a pro-tumor microenvironment.
It's reported that the PC of gastrointestinal cancers and ovarian cancers prefers to grow in omentum majus. Some groups demonstrate, in a ovarian PC model, that fat tissues support tumor growth by providing the cancer cells with fatty acids13-14. However, few studies mentioned the immune status in PC progression, perhaps due to the lack of in vivo PC model with competent immunity. Subsequently, the exploration of immunotherapy for PC was restrained, although the T cell-based immunotherapy had obtained a great achievement in many terminal cancers15. In the present study, we set up a colorectal PC model in immune-competent mice and revealed the alteration of adaptive immunity in PC development. Our study might potentiate the research and therapy strategies of colorectal PC.
Materials and Methods
Cell lines
Mouse colorectal cancer cell lines MC-38 cells were provided by JENNIO Biological Technology (Guangzhou, China) and MC-38G (GFP-tagged MC-38 cells) were maintained in our research group. Those cells were maintained in Dulbecco's modified Eagle's medium (DMEM) high glucose, supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/ml) and streptomycin (100 ug/ml), in a 5% CO2 atmosphere at 37 °C.
Animal model
All the animal experiments were approved by the Institutional Animal Care and Use Committee at Third Military Medical University (TMMU), and all the experiments were performed in accordance with the “Guide for the care and use of laboratory animals” published by the US NationalInstitutes of Health (publication no. 85-23, revised 1996). All the mice were housed in a pathogen-free facility with a 12-h light, 12-h dark cycle in TMMU. All the mice were provided with food and purified water ad libitum. Each cage contained no more than 5 mice. MC-38 cells and MC-38G cells (1 × 106 cells in 0.1 ml PBS) were intraperitoneally injected into abdominal cavity of 6 week-old male C57BL/6 mice. Then the tumor-inoculated mice were observed dynamically. The pathological observation of PC development including tumor infiltration and systemic parameters were performed on day 0, 5, 10, 15 and 20. The alteration of T cells and B cells was evaluated on day 0, 3, 5, 7 and 9. The production of IFN-γ in T cells was measured on day 0, 5, 10 and 15.
HE staining
Organs and tissue were harvested, formalin-fixed, and processed into paraffin blocks, and then sections were cut and stained with hematoxylin and eosin (C0105, Beyotime, China).
Immunofluorescence staining
All tissue samples were fixed and processed into paraffin blocks. After dewaxing, antigen repair (10 mmol/L sodium citrate for 5 min in a pressure cooker) and removing endogenous enzymes in Hydrogen Peroxide Block, the sections were incubated with the primary antibodies anti-GFP (ab13970, Abcam, UK, the dilution ratio was 1:500) and anti-Perilipin (ab3526, Abcam, UK, the dilution ratio was 1:200) at 4ºC overnight. Then the samples were incubated in secondary antibody (703-545-155, Jackson ImmunoResearch, USA; ab150074, Abcam, UK, the dilution ratio was 1:500) for 1-2 h at room temperature in the dark. After rinsing three times in 1X PBS for 5 min each, the slides were counterstained with Hoechst (C1026, Beyotime, China, the dilution ratio was 1:500) to reveal cell nuclei. Finally, fluorescence microscopy was used to evaluate specimen.
Immunostaining and fluorescence-activated cell sorting (FACS)
Mice were sacrificed by CO2. The spleen was dissected and gently rubbed between the two rough sides of frosted slides (#FSL006, Beyotime, Shanghai, China). Splenic samples were isolated by gently lapping spleens over a 75-μm filter (F513442, Sangon Biotech, China), and then washing in FACS buffer (PBS with 0.5% endotoxin-free FBS, 2 mM EDTA, and 25 mM HEPES). After spinning down for 5 min at 500g, erythrocytes were removed by ACK lysis buffer (R1010, Solarbio, China) for 5 minutes. Then the reaction was stopped by adding equivoluminal PBS buffer and splenocytes were collected by centrifugation. Finally, cell pellet were resuspended with FACS buffer.
Circulating blood samples were collected using blood collection tubes (171001, KANG JIAN, China) to prevent clotting. After using mouse peripheral blood lymphatic separation kit (#P8620, Solarbio, China), cells were collected and lysed in ACK lysis buffer as described above. In the same way, blood samples were resuspended in FACS buffer.
Adipose tissue were resected from mice and mechanically dissociated with ophthalmic forceps into about one cubic millimeter and then digested with 25 ml Roswell Park Memorial Institute (RPMI) 1640 medium, supplemented with 3% FBS and 1mg/ml collagenase IV ( A004186, Sangon Biotech, China) for 45 min at 37 °C. Meanwhile the solution was vibrated by magnetic stirrer. After dissociation, adipose tissue suspensions were filtered through a 75-μm filter, and were spun down for 10 min at 500g. Then adipose tissue samples were lysed with ACK buffer as above, and then resuspended in FACS buffer.
At last, after staining with antibodies at 4 °C for 45 min followed by 2 washes in FACS buffer, the cells were resuspended in FACS buffer for FACS analysis (FACSVerse C6, BD Biosciences). The antibodies included APC anti-mouse CD45 antibody (#103112, Biolegend), PE anti-mouse CD90.2 antibody (#105308, Biolegend), PerCP/Cy5.5 anti-mouse CD8a antibody (#100734, Biolegend), and APC/Cy7 anti-mouse CD4 (#105308, Biolegend).
Treatment of antibodies in vivo
The antibody treatment was performed as described previously16. Briefly, MC-38 cells (1 × 106 cells in 0.1 ml PBS) were intraperitoneally injected into abdominal cavity of 6 week-old male C57BL/6 mice. Then, the MC-38 tumor-bearing mice were treated with monoclonal antibodies anti-CD8 (MAB116, Novus Biologicals) or IgG (MAB002, Novus Biologicals) as control. Tumor cells were inoculated on day 1. Each mouse was treated 3 times (days 4, 7 and 10) via the tail vein at a dose of 100 ug/day. The tumor nodes in visceral fats were checked on day 15.
Statistical analysis
All analyses were performed using GraphPad Prism 5. All data were expressed as the mean± s.e.m., and were analyzed using two-tailed unpaired T-test. For all statistical analyses, * indicates P< 0.05, ** indicates P<0.01, and *** indicates P<0.005.
Results
CRC cells mainly target the visceral fat tissues
To mimic the disseminated CRC cells in peritoneal cavity, a mouse colorectal cancer cell line MC-38 was employed for intraperitoneal inoculation in C57BL/6 mice. The PC progression was observed dynamically. We demonstrated that the PC nodes were visible 15 days post inoculation in our experimental system (Figure 1A-E). Interestingly, the PC nodes mainly located in visceral fats, such as omentum majus (Figure 1B), mesenterium (Figure 1C) and epididymal fats (Figure 1E). After 20 days of inoculation, the PC nodes could be observed in almost all the visceral fat tissues, even in perirenal fats (Figure 1D). Meanwhile, the body weight of tumor bearing mice was significantly lost in comparison with the tumor-free mice (Table 1). However, the weight of organs like spleen, kidney, lung and liver didn't differ notably between the PC and control group (Table 1). It should be pointed out that the tumor-bearing mice usually died around 25 days post tumor injection (data not shown).
Table 1
The weight of visceral organs in the PC model
| Parameters | PBS | MC-38 |
|---|---|---|
| BW (g) | 20.66±0.34 | 17.20±0.75** |
| Spleen weight (g) | 0.13±0.0260 | 0.13±0.0118 |
| Spleen to BW ratio (%) | 0.61±0.0013 | 0.74±0.0007 |
| Kidney weight (g) | 0.30±0.0215 | 0.33±0.0196 |
| Kidney to BW ratio (%) | 1.45±0.0010 | 1.89±0.0011** |
| Lung weight (g) | 0.14±0.0090 | 0.15±0.0046 |
| Lung to BW ratio (%) | 0.69±0.0004 | 0.86±0.0003** |
| Liver weight (g) | 1.26±0.1081 | 1.45±0.0828 |
| Liver to BW ratio (%) | 6.10±0.0052 | 8.42±0.0048** |
Six-week-old mice were intraperitoneally inoculated with MC-38 cells (1×106 cells in 100 ul PBS for each mouse) or PBS as control. 20 days later, the visceral organs were dissected and weighed. Data are mean± s.e.m. (n=5, **P<0.01, ***P<0.005, student's t-test)
To further confirm the target of CRC cells in PC development, we dissected the visceral organs, such as guts, livers, lungs, kidneys and spleens. No tumor nodes were observed in gross anatomy of those organs 20 days post of tumor inoculation, except the mesenterium attached to guts (Figure 2A). We also didn't find any tumor lesions in spleens, kidneys, lungs or livers from tumor bearing mice with pathological observation (Figure 2B).
CRC cells quickly invaded into the visceral fats
In the present PC model, CRC cells mainly migrated to the visceral fat tissues. As shown in Figure 3A, the epididymal fats were isolated and easy to investigate. Thus we choose this fat tissue to dynamically assess the progression of colorectal PC. In consistence, the PC nodes in epididymal fats were visible 15 days post tumor injection (Figure 3A). With HE staining, we could find CRC cell infiltration on day 10 after setup of the present PC model (Figure 3B). To further characterize the detailed metastasis in epididymal fat, the GFP-tagged MC-38 cells were used to set up the PC model. We demonstrated, on day 5 post PC model setup, that the MC-38 tumor cells were infiltrated in the epididymal fat (Figure 3C). This result indicated that the inoculated CRC cells could rapidly migrate into the visceral fat tissues.
The status of systemic adaptive immunity in the colorectal PC model
Adaptive immunity plays a crucial role in the development of tumors, and T cell-based immunotherapy is increasingly being approved for clinical use15, 17. The CD8+ T cells, also known as cytotoxic T lymphocyte, can directly attack foreigners such as bacteria, viruses, and cancer cells once they become activated18. By contrast, CD4+ T cells (mature T-helper cells) mainly organize an immune response through indirectly recruiting other immune cells19. Therefore, we further measured the adaptive immune alteration in spleen and blood with immunostaining and FACS. We found the ratios of T cells and B cells in spleens were not altered significantly in response to tumor injection (Figure 4A-B). Further, the spleen CD4 and CD8 subsets of T cells were also assed and no obvious differences were observed after PC setup (Figure 4C).
Noteworthily, numbers of blood T cells underwent a quick decrease with PC development, while blood B cells didn't alter notably (Figure 5A-B). In details, the ratios of blood CD4+ T cells decreased slightly over time after tumor inoculation, whereas the ratios of CD8+ T cells increased significantly (Figure 5C). In comparison with CD8, the amount of blood CD4+ T cells continuously decreased with PC progression (Figure 5B-C).
Figure 1
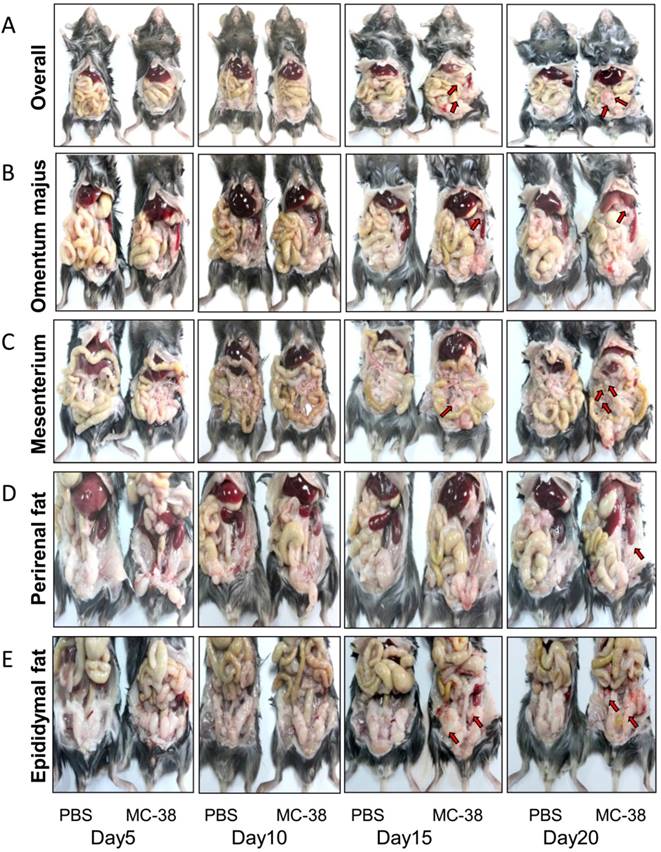
Peritoneal CRC cells mainly target the visceral fat tissues. (A) The overall status of colorectal PC development in mice. Six-week-old mice were intraperitoneally inoculated with MC-38 cells (1×106 cells in 100 ul PBS for each mouse) or PBS as control, and PC progression was observed dynamically. The red arrows indicated tumor nodes. Representative images were displayed. (n=5) (B-E) PC progression in the omentum majus (B), mesenterium (C), perirenal fat (D) and epididymal fat (E) of the mice described in (A). The red arrows indicated tumor nodes. Representative images were displayed. (n=5)
Figure 2
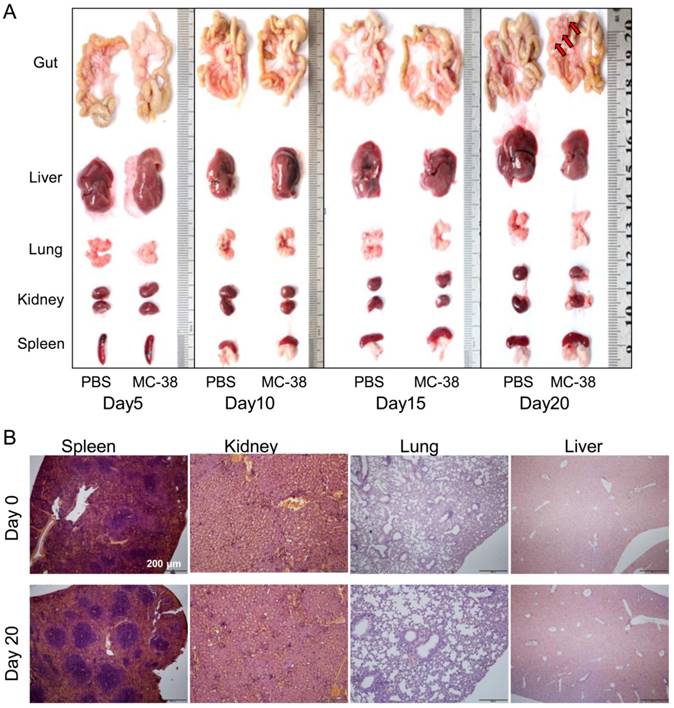
Peritoneal CRC cells doesn't target visceral organs. (A) The gross anatomy of visceral organs from MC-38 PC models as indicated above in (Figure 1A). Representative images of the fresh organs were displayed. (n=5) (B) H&E staining of the visceral organs from PC models described above in (A). Representative images were displayed. Scale bars, 200 μm. (n=5)
The status of local adaptive immunity in the colorectal PC model
Comparing with the systemic immunity, the local immunity in tumor microenvironment appears more effective. Therefore, we further investigated the adaptive immunity alteration in visceral fats and the epididymal fat was selected as a representative. With FACS analysis, we demonstrated that the amount of T cells was continuously increased over time after tumor inoculation, while the B cells altered modestly (Figure 6A-B). Further, we found the ratio of CD4+ T cells increased on day 5 after tumor injection, while declined to basal levels quickly (Figure 6C). More interestingly, the ratio of fat CD8+ T cells was decreased on day 5 after tumor inoculation and quickly restored and increased over time (Figure 6C). Combining with the alteration of total T cells in fats, we concluded that the amount of fat CD8+ T cells was continuously increased in the process of PC development.
It's reported that the tumoricidal T cells would be exhausted with cancer progression. The exhausted T cells expressed less IFN-γ than the functional T cells20. Thus we further detected the IFN-γ levels in fat T cells dynamically in the PC model. We found that the levels of IFN-γ in CD4+ T cells increased by around 2 fold on day 10 after tumor injection and then began to drop, as compared to tumor-free mice (Figure 7A-B). However, the IFN-γ levels in fat CD8+ T cells fluctuated modestly over time (Figure 7C). It's interesting that the levels of IFN-γ in CD8+ T cells were much higher than that in CD4+ T cells (Figure 7B-C).
According to the alteration of T cell quantity and quality in the fat tissues, we presumed that the T cells were not exhausted at least before day 10 and that CD8+ T cell might be a major regulator of tumor immunity in our colorectal PC model. Next, we used CD8 antibodies to eliminate CD8+ T cells to investigate the role of CD8+ T cells in colorectal PC progression. We demonstrated that treatment of CD8 antibodies largely potentiated the development of MC-38 cell-based PC (Figure 8A-B). Those results indicated that PC development could be modulated by immunotherapy.
Figure 3
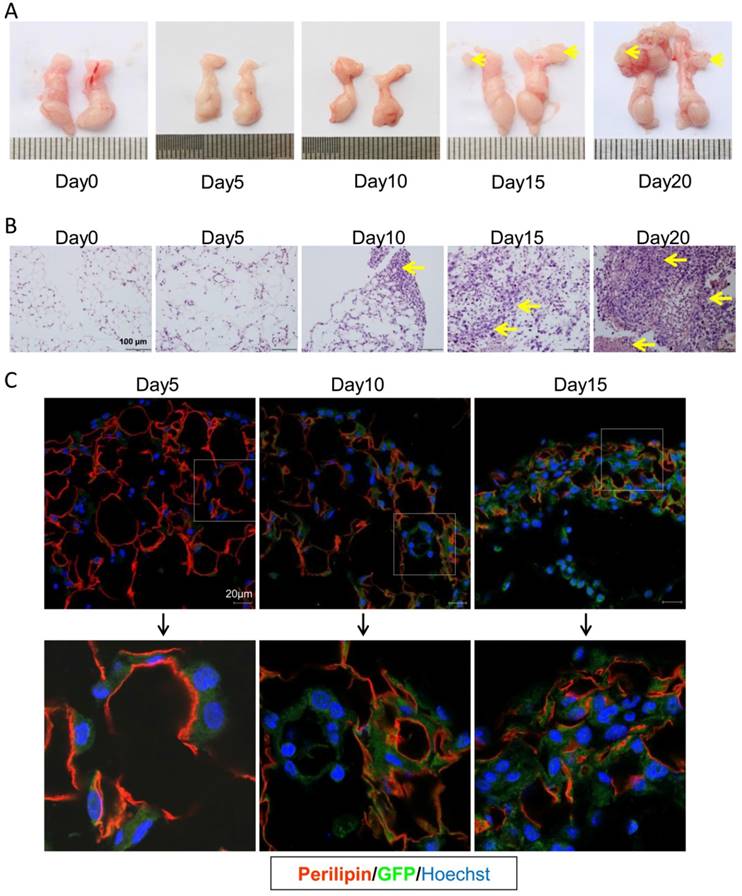
CRC cells quickly invade into the visceral fats. (A) The gross anatomy of the epididymal fats in MC-38 PC models as described above in (Figure 1A). Representative images were displayed. The arrows indicated the tumor nodes. (n=5) (B) H&E staining of the epididymal fat tissues described in (A).The arrows indicated tumor lesions. Shown were representative images. Scale bars, 100 μm. (n=5) (C) The rapid infiltration of CRC cells in the epididymal fat. Six-week-old mice were intraperitoneally injected with GFP-tagged MC-38 cells (1×106 cells in 100 ul PBS for each mouse) or PBS as control. The epididymal fat was collected dynamically and stained with GFP (Green) or perilipin (red) antibodies by immunofluorescence analysis. The nucleus was visualized by hoechst staining (Blue). Representative images were shown. Scale bars, 20 μm. (n=5)
Figure 4
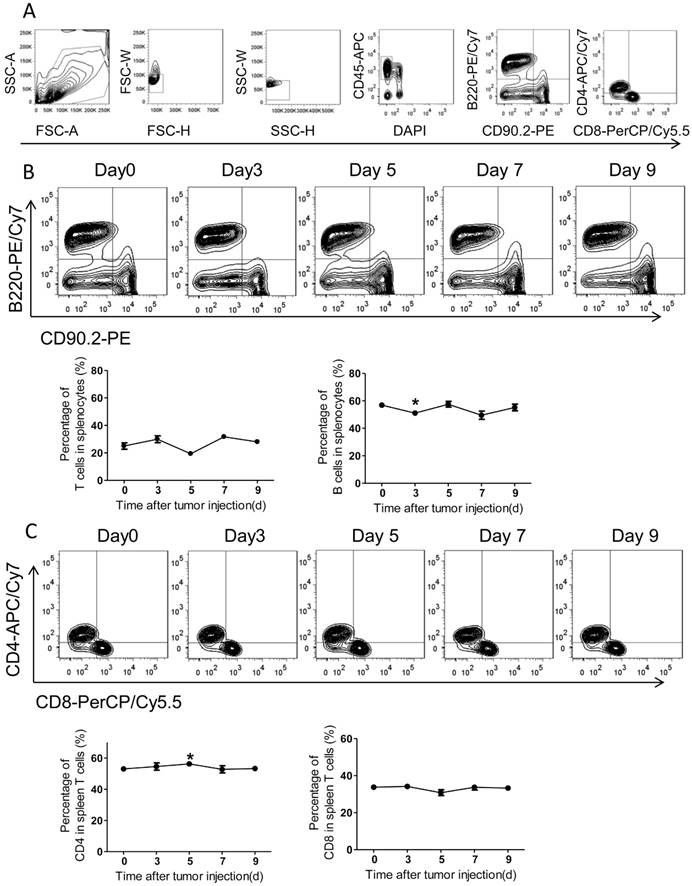
The adaptive immunity in spleens of the colorectal PC model. (A) FACS gating strategy for spleen T/B cells. Debris and doublets were removed, and then the spleen T/B cells were assessed as DAPI-CD45+CD90.2+/B220+. The levels of T/B cells and CD8+/CD4+ T cells were also measured. (B-C) The levels of spleen T/B cells at different time points. Six-week-old mice were intraperitoneally injected with MC-38 cells. Then the splenocytes were isolated for flow cytometric analysis of T/B cells (B) and CD8+/CD4+ T cells (C). Shown are representative images. Data are mean± s.e.m. (n=3, *P< 0.05, student's t-test)
Discussion
By intraperitoneal inoculation of MC-38 cells, we set up a colorectal PC model in mice. We demonstrated that the CRC cells mainly migrated and colonized in the visceral fat tissues. With the analysis of adaptive immunity, we found that T cells were intimately altered with the development of PC. Thus we provided an ideal colorectal PC model for cancer and immunology research.
The present PC model can mimic the characteristics of colorectal PC in clinics. Besides, gastrointestinal and ovarian cancer PC could also be mimicked by intraperitoneal inoculation of corresponding cancer cells. It's reported that ovarian cancer cells were also predominantly colonized in visceral fats such as omentum majus, in mouse PC models13. It's really interesting that visceral fats are the major targets of PC in different types of cancers. The mechanisms for the attractive and supportive effects of fat tissues on peritoneal cancer cells need further to be explored. After that, potential targets for preventing colorectal PC might be revealed.
It should be pointed out that a number of PC models have been established by a number of previous and recent studies with different purposes. For general research on cancer cell biology, the PC models are mainly constructed in the nude mice by intraperitoneal inoculation of any types of cancer cells regardless of species21-22. Especially, some human colorectal cancer cell lines could be implanted in nude mice to test drug effects21-22. Besides, the murine MC-38 or CT-26 cell-based colorectal PC models were also established in the immune competent mice in the present or other studies23-25. More interestingly, the cancer PC models were also reported in rats26 and rabbits27. However, few studies investigated the basic immunological changes with the PC development.
Figure 5
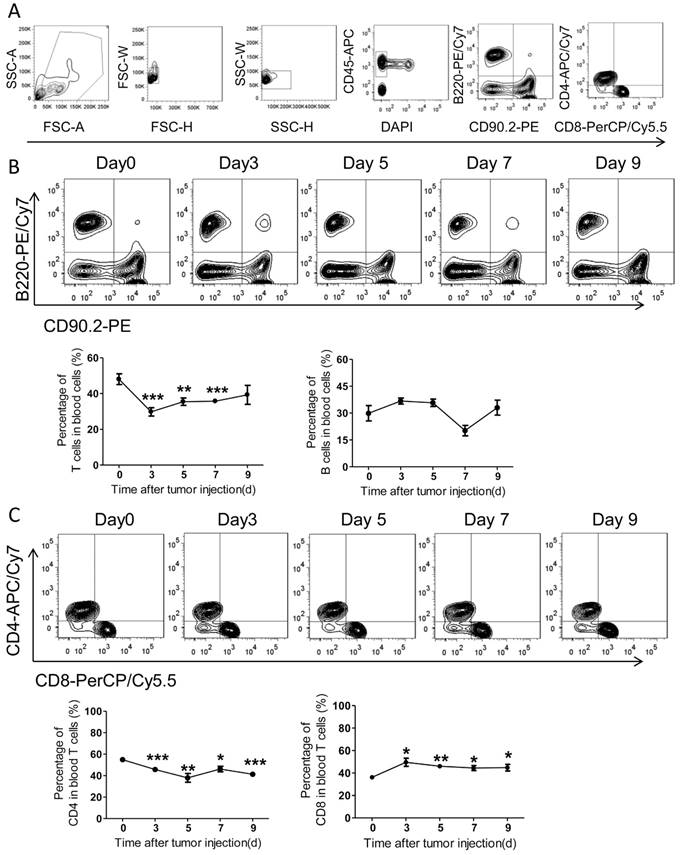
The adaptive immunity in blood of the colorectal PC model. (A) FACS gating strategy for blood T/B cells as described above in Figure 4A. (B-C) The levels of blood T/B cells at different time points. Blood of the mice described in Figure 4A was collected for flow cytometric analysis of T/B cells (B) and CD8+/CD4+ T cells (C). Representative images were displayed. Data are mean± s.e.m. (n=3, *P< 0.05, **P<0.01, ***P<0.005, student's t-test)
Figure 6
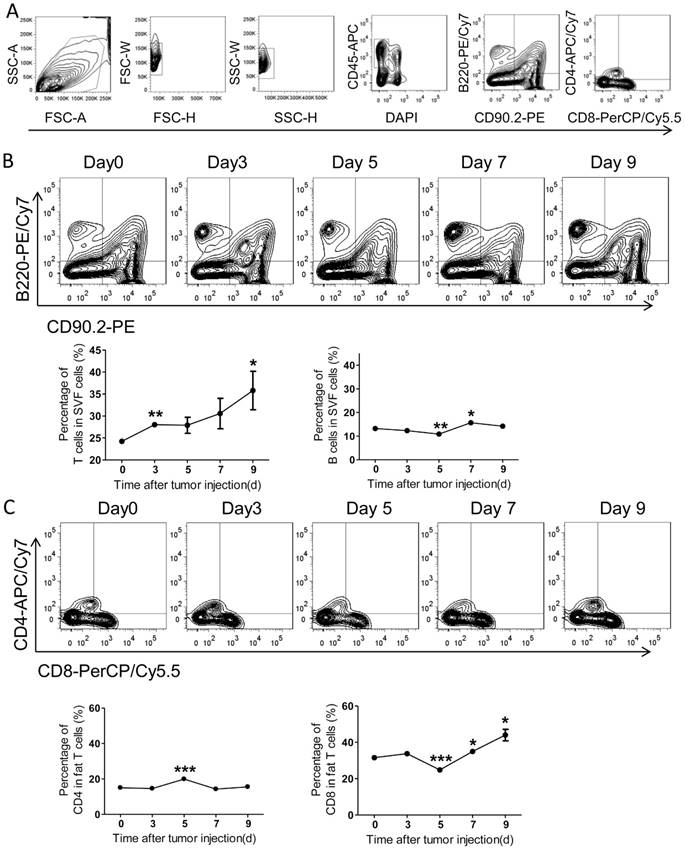
The adaptive immunity in epididymal fats of the colorectal PC model. (A) FACS gating strategy for epididymal fat T/B cells as described above in Figure 4A. (B-C) The levels of T/B cells in epididymal fats. The mice were treated as above, and then the adipose stromal cells were isolated for flow cytometric analysis of T/B cells (B) and CD8+/CD4+ T cells (C). Shown are representative images. Data are mean± s.e.m. (n=3, *P< 0.05, **P<0.01, ***P<0.005, student's t-test)
The previous studies suggested that adipocytes derived fatty acids might be the major resources of substances and energy for the development of cancer cells13, 28-29. However, those studies were mainly performed in PC models of immune-deficient mice. Actually, cancer progression is accompanied by immune alteration. Immunotherapy (such as PD1 and PDL-1 blockage30-31) has shown a great potential in cancer treatment. Our colorectal PC model was constructed in the immune complement mice, providing an ideal chance for immune studies. In the present study, we demonstrated that the quantity and quality of T cells in fat tissues altered dynamically with colorectal PC development. We speculated that modulation of T cell activity might be a good way to treat colorectal PC. The present model could be used for functional screening of immunotherapy-related drugs. The colorectal PC progressed quickly in our experimental model, making related studies rather convenient and reproducible.
Figure 7
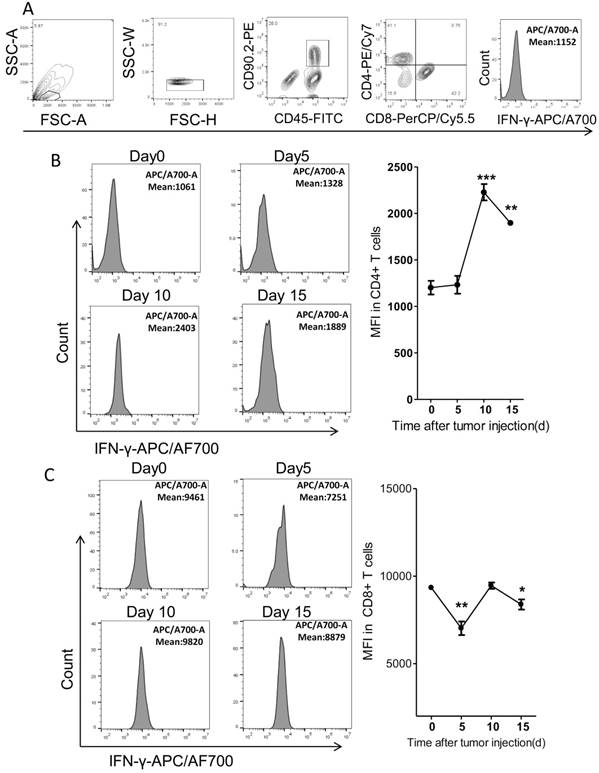
The expression of IFN-γ in Fat T cells of the colorectal PC model. (A) FACS gating strategy for the measurement of IFN-γ. Debris and doublets were removed, and then the CD4+ T cells were assessed as DAPI-CD45+CD90.2+CD4+ and the CD8+ T cells were assessed as DAPI-CD45+CD90.2+CD8+. The average fluorescence degree indicating the level of IFN-γ was measured. (B-C) The average fluorescence degree of IFN-γ in T cells. Six-week-old mice were intraperitoneally injected with MC-38 cells and then the adipose stromal cells were isolated for flow cytometric analysis of IFN-γ levels in CD4+ T cells (B) or CD8+ T cells (C). Shown are representative images. Data are mean± s.e.m. (n=3, *P<0.05, **P<0.01, ***P<0.005, student's t-test)
Figure 8
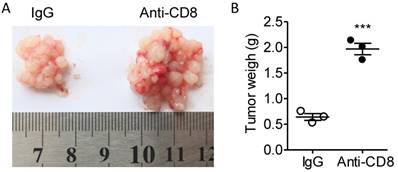
Anti-CD8 treatment potentiates the PC progression. (A) Six-week-old male C57BL/6 mice were intraperitoneally inoculated with MC-38 cells and then treated with anti-CD8 antibody or IgG as a control. The tumor nodes in visceral fats of each mouse were observed on day 15. Representative images were displayed. (B) The weight of tumor nodes in visceral fats as described above in (A). The data represent the means±s.e.ms. (n=3, ***P<0.005, Student's t-test).
In all, here we provided a convenient and reliable colorectal PC model in mice. In this model, we demonstrated that colorectal PC preferred to colonize in visceral fats and T cells were altered dynamically with the development of colorectal PC.
Abbreviations
CRC: Colorectal cancer; PC: peritoneal carcinomatosis; HIPEC: hyperthermic intraperitoneal chemotherapy; DMEM: Dulbecco's modified Eagle's medium; TMMU: Third Military Medical University; RPMI: Roswell Park Memorial Institute; FBS: fetal bovine serum.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81672693, to H.M.) and the Foundation and Frontier Research Project of Chongqing (cstc2017jcyjBX0071, to H.M.).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014;383:1490-1502
2. Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA: a cancer journal for clinicians. 2014;64:104-117
3. Baskaranathan S, Philips J, McCredden P, Solomon MJ. Free colorectal cancer cells on the peritoneal surface: correlation with pathologic variables and survival. Diseases of the colon and rectum. 2004;47:2076-2079
4. Koppe MJ, Boerman OC, Oyen WJ, Bleichrodt RP. Peritoneal carcinomatosis of colorectal origin: incidence and current treatment strategies. Annals of surgery. 2006;243:212-222
5. Solomon MJ, Egan M, Roberts RA, Philips J, Russell P. Incidence of free colorectal cancer cells on the peritoneal surface. Diseases of the colon and rectum. 1997;40:1294-1298
6. Sadeghi B, Arvieux C, Glehen O, Beaujard AC, Rivoire M, Baulieux J. et al. Peritoneal carcinomatosis from non-gynecologic malignancies: results of the EVOCAPE 1 multicentric prospective study. Cancer. 2000;88:358-363
7. Elias D, Lefevre JH, Chevalier J, Brouquet A, Marchal F, Classe JM. et al. Complete cytoreductive surgery plus intraperitoneal chemohyperthermia with oxaliplatin for peritoneal carcinomatosis of colorectal origin. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27:681-685
8. Sugarbaker PH. Peritonectomy procedures. Cancer treatment and research. 2007;134:247-264
9. Ceelen WP, Hesse U, de Hemptinne B, Pattyn P. Hyperthermic intraperitoneal chemoperfusion in the treatment of locally advanced intra-abdominal cancer. The British journal of surgery. 2000;87:1006-1015
10. Sugarbaker PH. Observations concerning cancer spread within the peritoneal cavity and concepts supporting an ordered pathophysiology. Cancer treatment and research. 1996;82:79-100
11. Arjona-Sanchez A, Medina-Fernandez FJ, Munoz-Casares FC, Casado-Adam A, Sanchez-Hidalgo JM, Rufian-Pena S. Peritoneal metastases of colorectal origin treated by cytoreduction and HIPEC: An overview. World journal of gastrointestinal oncology. 2014;6:407-412
12. Jayne DG, Fook S, Loi C, Seow-Choen F. Peritoneal carcinomatosis from colorectal cancer. The British journal of surgery. 2002;89:1545-1550
13. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR. et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nature medicine. 2011;17:1498-1503
14. Lengyel E. Ovarian cancer development and metastasis. The American journal of pathology. 2010;177:1053-1064
15. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350-1355
16. Xiang W, Shi R, Kang X, Zhang X, Chen P, Zhang L. et al. Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression. Nat Commun. 2018;9:2574
17. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361-1365
18. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nature immunology. 2013;14:1014-1022
19. Jenkins MK, Khoruts A, Ingulli E, Mueller DL, McSorley SJ, Reinhardt RL. et al. In vivo activation of antigen-specific CD4 T cells. Annual review of immunology. 2001;19:23-45
20. Hess Michelini R, Manzo T, Sturmheit T, Basso V, Rocchi M, Freschi M. et al. Vaccine-instructed intratumoral IFN-gamma enables regression of autochthonous mouse prostate cancer in allogeneic T-cell transplantation. Cancer research. 2013;73:4641-4652
21. Gremonprez F, Descamps B, Izmer A, Vanhove C, Vanhaecke F, De Wever O. et al. Pretreatment with VEGF(R)-inhibitors reduces interstitial fluid pressure, increases intraperitoneal chemotherapy drug penetration, and impedes tumor growth in a mouse colorectal carcinomatosis model. Oncotarget. 2015;6:29889-29900
22. Stoeltzing O, Ahmad SA, Liu W, McCarty MF, Parikh AA, Fan F. et al. Angiopoietin-1 inhibits tumour growth and ascites formation in a murine model of peritoneal carcinomatosis. Br J Cancer. 2002;87:1182-1187
23. Zhang W, Cui T, Liu L, Wu Q, Sun L, Li L. et al. Improving Anti-Tumor Activity of Curcumin by Polymeric Micelles in Thermosensitive Hydrogel System in Colorectal Peritoneal Carcinomatosis Model. J Biomed Nanotechnol. 2015;11:1173-1182
24. Wu QJ, Gong CY, Luo ST, Zhang DM, Zhang S, Shi HS. et al. AAV-mediated human PEDF inhibits tumor growth and metastasis in murine colorectal peritoneal carcinomatosis model. BMC Cancer. 2012;12:129
25. Lee IK, Vansaun MN, Shim JH, Matrisian LM, Gorden DL. Increased metastases are associated with inflammation and matrix metalloproteinase-9 activity at incision sites in a murine model of peritoneal dissemination of colorectal cancer. J Surg Res. 2013;180:252-259
26. Facy O, Radais F, Ladoire S, Delroeux D, Tixier H, Ghiringhelli F. et al. Comparison of hyperthermia and adrenaline to enhance the intratumoral accumulation of cisplatin in a murine model of peritoneal carcinomatosis. J Exp Clin Cancer Res. 2011;30:4
27. Mei LJ, Yang XJ, Tang L, Hassan AH, Yonemura Y, Li Y. Establishment and identification of a rabbit model of peritoneal carcinomatosis from gastric cancer. BMC Cancer. 2010;10:124
28. Tokuda Y, Satoh Y, Fujiyama C, Toda S, Sugihara H, Masaki Z. Prostate cancer cell growth is modulated by adipocyte-cancer cell interaction. BJU international. 2003;91:716-720
29. Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B. et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer research. 2011;71:2455-2465
30. Okada M, Chikuma S, Kondo T, Hibino S, Machiyama H, Yokosuka T. et al. Blockage of Core Fucosylation Reduces Cell-Surface Expression of PD-1 and Promotes Anti-tumor Immune Responses of T Cells. Cell reports. 2017;20:1017-1028
31. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56-61
Author contact
![]() Corresponding authors: hmmiaoedu.cn (H.M.M.), hefengtian06com (F.T.H.)
Corresponding authors: hmmiaoedu.cn (H.M.M.), hefengtian06com (F.T.H.)
Citation styles
APA
Shi, R., Xiang, W., Kang, X., Zhang, L., Wang, J., Miao, H., He, F. (2019). Alteration of Adaptive Immunity in a Colorectal Peritoneal Carcinomatosis Model. Journal of Cancer, 10(2), 367-377. https://doi.org/10.7150/jca.27947.
ACS
Shi, R.; Xiang, W.; Kang, X.; Zhang, L.; Wang, J.; Miao, H.; He, F. Alteration of Adaptive Immunity in a Colorectal Peritoneal Carcinomatosis Model. J. Cancer 2019, 10 (2), 367-377. DOI: 10.7150/jca.27947.
NLM
Shi R, Xiang W, Kang X, Zhang L, Wang J, Miao H, He F. Alteration of Adaptive Immunity in a Colorectal Peritoneal Carcinomatosis Model. J Cancer 2019; 10(2):367-377. doi:10.7150/jca.27947. https://www.jcancer.org/v10p0367.htm
CSE
Shi R, Xiang W, Kang X, Zhang L, Wang J, Miao H, He F. 2019. Alteration of Adaptive Immunity in a Colorectal Peritoneal Carcinomatosis Model. J Cancer. 10(2):367-377.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.