Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(3):757-764. doi:10.7150/jca.28087 This issue Cite
Research Paper
Oxidized Vitamin C (DHA) Overcomes Resistance to EGFR-targeted Therapy of Lung Cancer through Disturbing Energy Homeostasis
Mingtong Ye3#, Nengzhi Pang1#, Ting Wan4, Yuanling Huang1, Tianyi Wei3, Xuye Jiang1, Yujia Zhou1, Yufeng Huang2, Hainan Yang2, Zhenfeng Zhang2 ![]() , Lili Yang1
, Lili Yang1 ![]()
1. Department of Nutrition, Guangdong Provincial Key Laboratory of Food, Nutrition and Health, School of Public Health, Sun Yat-Sen University, Guangzhou, Guangdong, PR China.
2. Department of Radiology, The Second Affiliated Hospital of Guangzhou Medical University, Guangzhou, Guangdong, PR China.
3. The First Women and Children's Hospital of Huizhou, Huizhou, Guangdong, PR China.
4. Huizhou First People's Hospital, Huizhou, Guangdong, PR China.
#These authors contribute equally to this work.
Received 2018-6-24; Accepted 2018-10-28; Published 2019-1-1
Citation:
Ye M, Pang N, Wan T, Huang Y, Wei T, Jiang X, Zhou Y, Huang Y, Yang H, Zhang Z, Yang L. Oxidized Vitamin C (DHA) Overcomes Resistance to EGFR-targeted Therapy of Lung Cancer through Disturbing Energy Homeostasis. J Cancer 2019; 10(3):757-764. doi:10.7150/jca.28087. https://www.jcancer.org/v10p0757.htm
Other stylesAbstract
Switching aerobic respiration to anaerobic glycolysis of cancer cells plays an important role in development and progression of acquired resistance. Since vitamin C enabled the inhibition of glycolysis of cancer cells, and erlotinib-resistant sub-line of HCC827 (ER6 cells) switched its metabolic features to higher glycolysis for survival, we hypothesize that vitamin C is able to inhibit glycolysis of ER6 cells. In this study, we found that both reduced vitamin C and oxidized vitamin C (DHA) could selectively suppress the viability of ER6 cells. DHA was efficient in inhibiting glycolysis and leading to energy crisis, which could be one mechanism for overcoming drug resistance to erlotinib of ER6 cells. Our data suggest that applying DHA could be a novel treatment strategy for NSCLC with acquired resistance to targeted therapy.
Keywords: Drug resistance, oxidized vitamin C (DHA), energy homeostasis, glycolysis.
1. Introduction
It has been well established that the first-line tyrosine kinase inhibitors (TKIs) which target epidermal growth factor receptor (EGFR) mutations in advanced non-small cell lung cancer (NSCLC) patients show better response than those patients treated with general chemotherapy [1-3]. Nevertheless, acquired resistance often occurs after 12 months TKIs treatment on average [4, 5]. A major acquired resistant mechanism of NSCLC is molecular abnormalities, including EGFR-T790M mutation, AXL or MET over-expressions [6-10].
Metabolism adaption of cancer, for example, the Warburg effect, has appealed great attention in drug resistance during tumor therapy [11]. Reprogramming of glycolytic activity and its corresponding changes of metabolites had been found in drug resistant cells and tissues of breast cancer, breast cancer-associated fibroblasts, human glioblastoma, pancreatic adenocarcinoma, gastric cancer, myeloid leukemia and non-small cell lung cancer [12-18].
Targeting metabolite changes and its corresponding metabolic pathways has been novel ways to overcome drug resistance [19]. Data had proven that inhibition of GLUT1 activity and expression can sensitize head and neck cancer cells (Cal27 cells) to cisplatin treatment in both normoxic and hypoxic conditions [20]. Another data had shown that development of tamoxifen resistance may be driven by HIF-1α hyper-activation via modulation of Akt/mTOR and/or AMPK signaling pathways, and that inhibition of aerobic glycolysis and repression of this signaling pathway enable the restoration of tamoxifen sensitivity in antiestrogen-resistant breast cancer cells [21]. Similar outcomes that inhibiting glycolysis with 2-deoxyglucose and 3-bromopyruvate propylester is able to reverse the drug resistance were also reported in oxaliplatin-resistant colon cancer cells, sorafenib-resistant HCC cells and leukemia cells [22-24].
Vitamin C or ascorbic acid had been proposed as an anticancer agent that enables to reduce cancer incidence and cancer treatment-related side effects [25]. Data showed that vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH due to increased uptake of oxidized vitamin C, dehydroascorbate (DHA), via the glucose transporter-GLUT1, which induces oxidative stress and suppresses GAPDH [26]. Other data also showed that vitamin C treatment generates reactive oxygen species (ROS) resulting in cell death in multiple myeloma tumor cells, cholangiocarcinoma cells, breast cancer cells and malignant melanoma cells [27-30]. Moreover, vitamin C treatment was dose-independent and transporter-independent, such as sodium-dependent vitamin C transporter 2 (SVCT-2) [28, 30, 31]. In vivo xenograft experiment and case-control study had proven that vitamin C treatment is able to inhibit tumor growth and dietary intake of vitamin C protects against cancer [28, 32, 33]. A recent report found that vitamin C treatment mimics Tet2 restoration to block leukemia progression and vitamin C treatment in leukemia cells enhances their sensitivity to PARP inhibition [34].
We have successfully established a series of erlotinib-resistant subclonal cells (ER1-6) originated from HCC827 cells through de-sensitizing the HCC827 cells in gradually increasing erlotinib concentrations until 10 μM in the culture media and reported AXL kinase as a novel resistance molecule in ER1-5 cells [8, 18]. We also found that ER6 cells switch their metabolic features to higher glycolysis for survival and combining inhibitions of glycolysis and AKT/autophagy could overcome drug resistance in ER6 cells [18]. In this study, we explored the effects of reduced vitamin C and DHA on ER6 cells comparing to HCC827 cells, investigated the underlying mechanism of vitamin C to ER6 cells and HCC827 cells, and evaluated the potential application of reduced or oxidized vitamin C for a potential adjuvant treatment of NSCLC with EGFR mutations.
2. Materials and Methods
2.1 Reagents & materials
Reduced vitamin C (A103539-25g), nevirapine (N129779), glucose, NAD+, ATP, ADP and AMP were purchased from Aladdin chemicals (Shanghai, China). Oxidized vitamin C (261556-250mg), PMS (P9625) were purchased from Sigma-Aldrich (Shanghai, China). MTS Reagent Powder (G1112) was purchased from Promega Corportion (WI, USA). The antibody of GAPDH was purchased from abcam (Shanghai, China). The antibody of goat anti-rabbit was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The antibodies of phosphorlation-AMPK and AMPK were purchased from Cell Signaling Technologies (Beverly, MA, USA). The antibodies of HK-2, PK-M2 and PFK3 were purchased from Proteintech (Wuhan, China). RPMI-1640 (8116322) was purchased from Gibco Thermo Fisher Scientific (Guangzhou, China). GSH/GSSG test kit was purchased from Beyotime Biotechnology (Shanghai, China). HPLC acetonitrile and HPLC methanol were purchased from Oceanpak (Goteborg, Sweden). Deionized water was purchased from Watsons (Guangzhou, China). Seahorse XF24 Cell culture microplate, XF24 extracellular flux assay kit, base medium, calibrate medium were purchased from Seahorse Agilent technologies (Beijing, China).
2.2 Cell culture
All cell lines were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum and 1% Penicillin & Streptomycin in incubator under 37℃, 5% carbon dioxide condition.
2.3 Cell viability assay
Cells were transformed to a 96-well plate in 100 microliter full medium. When cells reached 50% confluence, flash medium or medium supplemented with reduced vitamin C or DHA replaced original medium. MTS-PMS mixed solution was added and incubated in incubator under 37℃, 5% carbon dioxide condition. Absorbance was measured following the instructions of the assay kit (Ye, M., et al. 2017).
2.4 Protein expression assays
Protein was isolated from cells using RIPA with 10 μM PMSF. Lysates were standardized for protein content. The following procedure followed by the guideline of manufacturer. The immune-reactive proteins were visualized using ECL (Thermo Fisher Scientific).
2.5 Extracellular flux analysis (OCR and ECAR)
Cells were transferred into XF24 assay well then pretreated with or without DHA in normal medium. Finally, replacing and washing the normal medium with Seahorse assay medium without glucose. OCR and ECAR analysis followed the instructions of Seahorse Analyzer as reported previously (Ye, M., et al. 2017).
2.6 GSH and GSSG measurement
Cells were cultured in 6-well plate. When 80% confluence was reached, cells were treated with DHA. Then cells were washed for three times with PBS and collected cells after trypsinization. The following procedure followed by the guideline of manufacturer.
2.7 Metabolites measurement applied by UPLC-TOF
Cells were cultured in 6-well plate. When cells had reached 80% confluence, adding DHA into medium and collecting metabolites after 2 hours. The whole metabolites test procedure was reported previously (Ye, M., et al. 2017).
2.8 Statistical analysis
Mean values and SD were calculated by GraphPad Prism 5 software. The analysis of variance (ANOVA) was used to identify statistical differences between experimental factors using SPSS17.0. P<0.05 was considered as statistically significant.
3. Results
3.1 Reduced vitamin C decreases viability of ER6 cells in both normal and glucose free condition
ER6 cells have higher glycolysis than their parental cells HCC827 [18]. We wondered whether some key kinases of glycolysis such as HK-2, PK-M2 are overexpressed. Unfortunately, the expression of both HK-2 and PK-M2 did not increase in ER6 cells compared to HCC827 cells (Figure 1A). The expression of PFK3, another key kinase of glycolysis was not detected in both ER6 cells and HCC827 cells (data not shown). When treated with reduced vitamin C for 24 hours, cell viability of ER6 cells and HCC827 cells was not impacted in the normal glucose medium (Figure 1B). When treated with reduced vitamin C for 48 hours, cell viability of ER6 cells was depressed in 2 mM dosage, while this same treatment was not able to inhibit viability of HCC827 cells (Figure 1C).
We had reported that ER6 cells are more addicted to glucose previously [18]. When treated with reduced vitamin C after glucose deprivation for 24 and 48 hours, the viability of ER6 cells was suppressed in 1 mM dosage treatment, while this same dosage treatment was not able to affect the viability of HCC827 cells (Figure 1D, 1E). This indicates that reduced vitamin C enables to suppress ER6 cells selectively in both normal and glucose free medium.
3.2 DHA decreases viability of ER6 cells more effectively than reduced vitamin C in normal glucose medium, and DHA decreases viability of both ER6 cells and HCC827 cells in glucose free condition
When treated with 1 mM DHA for 24 and 48 hours, the viability of ER6 cells was depressed, while the viability of HCC827 cells was not inhibited in the normal glucose medium (Figure 2A, 2B).
Figure 1
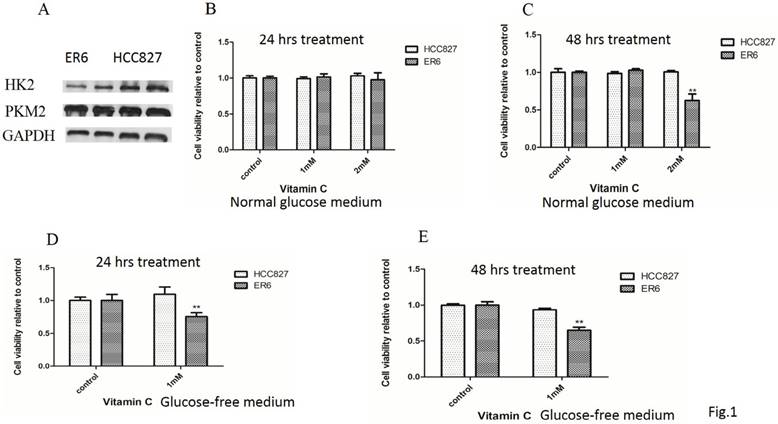
Reduced vitamin C decreases viability of ER6 cells in both normal and glucose free condition. A. Protein expressions of HK-2 and PK-M2 of ER6 and HCC827 cells analyzed by western blot; B&C. Viability analysis of ER6 and HCC827 cells with supplement of reduced vitamin C for 24 hrs (B) and 48 hrs (C) in normal glucose medium. ** represents p<0.01 comparing to ER6 control group; D&E. Viability analysis of ER6 and HCC827 cells with reduced vitamin C and glucose starvation for 24 hrs (D) and 48 hrs (E). ** represents p<0.01 comparing to ER6 control group.
Figure 2
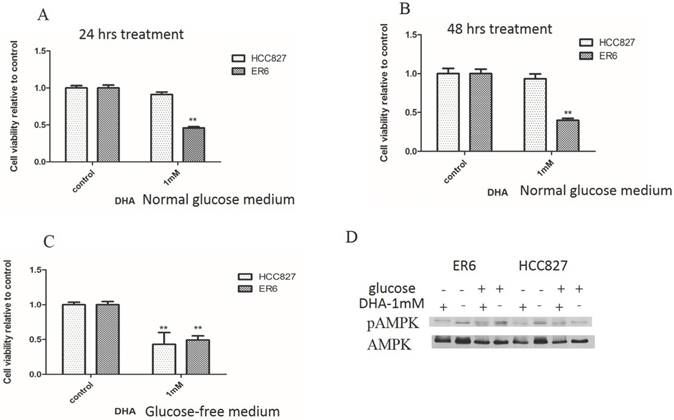
DHA decreases viability of ER6 cells more effectively than reduced vitamin C in normal glucose medium, and DHA decreases viability of both ER6 cells and HCC827 cells in glucose free condition. A&B. Viability analysis of ER6 and HCC827 cells with supplement of DHA for 24 hrs (A) and 48 hrs (B) in normal glucose medium. ** represents p<0.01 comparing to ER6 control group; C. Viability analysis of ER6 and HCC827 cells with DHA and glucose starvation for 24 hrs. ** represents p<0.01 comparing to control group; D. Western blot analysis of protein levels of p-AMPK and t-AMPK after 6 hrs treatment with glucose deprivation and/or DHA in both ER6 and HCC827 cells.
When cells were treated with 1 mM DHA in glucose free medium for 24 hours, the viabilities of both ER6 cells and HCC827 cells were suppressed (Figure 2C). Previously, we had reported that glucose deprivation is able to increase the phosphorylation of AMPK [18]. We wondered whether DHA treatment could affect the activation of AMPK. We found that DHA treatment was able to depress the activation of AMPK, especially after glucose deprivation in ER6 cells, while the depression of AMPK phosphorylation was only significant in glucose deprivation groups in HCC827 cells (Figure 2D). These data revealed that at the dosage of 1 mM DHA decreased the viability of ER6 cells selectively in normal glucose condition, while decreased the viability of both ER6 and HCC827 cells in glucose free medium. This phenomenon is closely related to the decrease of AMPK phosphorylation under both conditions, which might imply inhibition of further self-protection mechanisms by DHA, such as energy balance or autophagy etc. Clinically, it is difficult to deprive cells of glucose, so the selective depression of cell viability of ER6 cells by DHA with the presence of glucose is more valuable, which is a key focus of our following mechanism study.
3.3 DHA treatment interrupts the intracellular energy balance of ER6 cells and HCC827 cells
When treated with 1 mM DHA for 2 hours, the abundance of NAD+ was decreased both in ER6 cells and HCC827 cells comparing with control group (Figure 3A). Importantly, we found the dramatically decreased ATP abundance in ER6 cells in 1 mM DHA treatment group, while relatively higher abundance of AMP and ADP levels compared to HCC827 cells under the same treatment (Figure 3B-D). When the ratio of AMP/ATP was calculated, we found significant increase of the ratio of AMP/ATP in ER6 cells compared to HCC827 cells in 1 mM DHA treatment group (Figure 3E). In addition, the ratio of AMP/ATP was also much higher in 1 mM DHA treatment group compared to control group in ER6 cells. This data showed the significant energy disturbance in ER6 cells when 1 mM DHA was applied.
3.4 DHA treatment does not affect oxidative stress status in ER6 cells and HCC827 cells
Intracellular DHA was not able to be detected in control group without DHA addition into the culture medium, while 1 mM DHA supplement indeed enabled to increase intracellular DHA abundance. The concentration of intracellular DHA was higher in ER6 cells than that in HCC827 cells (figure 4A). Oxidative stress status in ER6 cells and HCC827 cells was not capable to be disturbed when they were treated with DHA in 1 mM or 2 mM dosage for 2 hours (figure 4B, 4C). This indicates that DHA treatment might not affect intracellular redox status in ER6 and HCC827 cells.
Figure 3
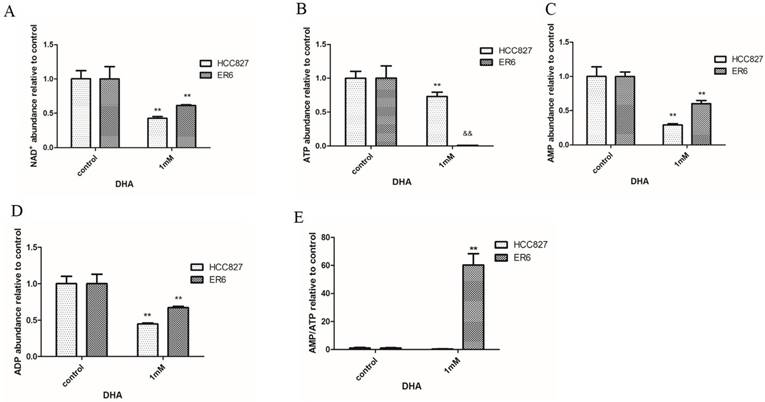
DHA treatment interrupts the intracellular energy balance of ER6 cells and HCC827 cells. Intracellular abundance of NAD+ (A), ATP (B), AMP (C), AMP (D) and AMP/ATP (E) in ER6 and HCC827 cells supplemented with 1 mM DHA for 2 hrs. ** represents p<0.01 comparing to corresponding control group; && represents p<0.01 comparing to corresponding ER6 group.
Figure 4
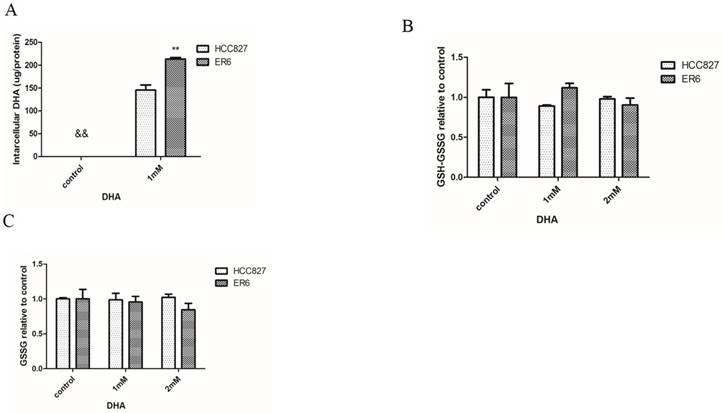
DHA treatment does not affect oxidative stress status in ER6 cells and HCC827 cells. A. Intracellular DHA abundance supplemented with DHA for 2 hrs, ** represents p<0.01 comparing to corresponding HCC827 group; && represents DHA was non-detected in control group; B. Total of intracellular GSH and GSSG, control group was set as 100% in both ER6 cells and HCC827 cells; C. Intracellular GSSG abundance, control group was set as 100% in both ER6 cells and HCC827 cells.
3.5 DHA treatment depresses glycolysis of ER6 cells and HCC827 cells
When pretreated with 2 mM DHA for 2 hours then replaced it with medium without glucose and DHA, glycolysis of ER6 cells and HCC827 cells was depressed after adding 10 mM glucose (Figure 5A, 5B). Then, we expected to see whether supplement of DHA can affect glycolysis of ER6 cells and HCC827 cells with 10 mM glucose. Fortunately, glycolysis of ER6 cells and HCC827 cells was also depressed immediately by 2 mM DHA in 10 mM glucose medium (Figure 5C). After that, we further explored whether pretreatment of cells with DHA for 2 hours and then supplement of DHA after adding 10 mM glucose could depress glycolysis of ER6 cells and HCC827 cells. Unfortunately, reinforcement effect did not exist (Figure 5D). It indicates that DHA depresses glycolysis of both ER6 cells and HCC827 cells.
Figure 5
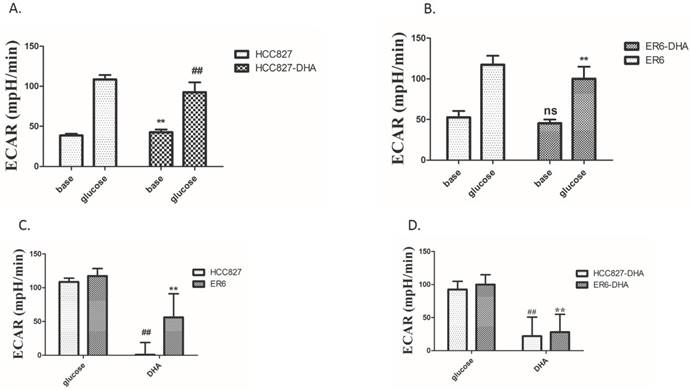
DHA treatment depresses glycolysis of ER6 cells and HCC827 cells. A-D. ER6 and HCC827 cells were plated on Seahorse plate, and cultured for 24 hrs. Extracellular acidification rate analysis (ECAR) was performed according to the instructions of the manufacturer; A. Extracellular acidification rate analysis (ECAR) of HCC827 cells pretreated with 2 mM DHA for 2 hrs, ** represents p<0.05 comparing to corresponding base group; ## represents p<0.05 comparing to corresponding glucose group; base group (glucose-free); glucose group (10 mM glucose); B. ECAR of ER6 cells pretreated with 2 mM DHA for 2 hrs, ** represents p<0.05 comparing to corresponding glucose group, ns represents p>0.05 comparing to corresponding base group; C. ECAR of ER6 and HCC827 cells supplemented with 2 mM DHA in 10 mM glucose; ** represents p<0.05 comparing to corresponding ER6 glucose group; ## represents p<0.05 comparing to corresponding HCC827 glucose group; D. ECAR of ER6 and HCC827 cells pretreated with 2 mM DHA for 2 hrs, then extra 1 mM DHA was added in 10 mM glucose, ** represents p<0.05 comparing to corresponding ER6 DHA glucose group; ## represents p<0.05 comparing to corresponding HCC827 DHA glucose group.
4. Discussion
The fact that cancer cells increase glycolysis rather than oxidative phosphorylation to meet energy requirements under physiological oxygen conditions had been reported to have strong correlation with drug resistance [11]. Recently, we reported that ER6 cells have higher glycolysis potential and overexpress GLUT1 and MCT4, which are more sensitive to glucose deprivation. Deficiency of energy under glucose deprivation is one cause of decreased viability of ER6 cells [18]. However, clinical deprivation of glucose only to cancer cells is not practical, other intervention methods have to be considered. One study indicated that reduced vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH, which is due to increased uptake of the oxidized vitamin C via GLUT1 [26]. A recent article showed that pharmacologically-dosed vitamin C enables to selectively kill multiple myeloma tumor cells [27]. High level vitamin C was reported to be able to be autoxidized, which provides an electron to oxygen to generate H2O2, thus excessive intracellular H2O2 leads to death of breast cancer cells [29]. To have better understanding of the effects of vitamin C on ER6 cells and HCC827 cells, we applied reduced vitamin C on ER6 cells and HCC827 cells and found that treating cells with 2 mM reduced vitamin C for 48 hours was able to decrease the viability of ER6 cells in the presence of 2 g/L glucose. Since ER6 cells are more sensitive to glucose deprivation, as mentioned above, lower concentration (1 mM) and shorter incubation time (24 hrs) of reduced vitamin C could selectively decrease viability of ER6 cells under glucose starvation condition. It is consistent to the report that high-dose vitamin C treatment selectively induces death of glycolysis-addicted KRAS and BRAF driven cancer cells [26]. Other article showed that doxycycline combined with vitamin C is a way to eradicate doxycycline-resistant cancer stem cells (CSCs) population [35].
Vitamin C uptake is mediated by two families of transport proteins, including sodium-dependent vitamin C transporters (SVCTs) 1 and 2, and the glucose transporters (GLUTs). GLUTs, mainly GLUT1 and GLUT3, transport the DHA, into cells and SVCTs transport reduced vitamin C directly into the cell [26, 31]. Considering that ER6 cells overexpress GLUT1, we wondered whether DHA treatment would be more efficient to suppress ER6 cells than HCC827 cells. We found that treatment with 1 mM DHA for 24 and 48 hours selectively decreased the viability of ER6 cells in normal glucose level, and 1 mM DHA combined with glucose deprivation treatment for 24 hours enabled to suppress both ER6 cells and HCC827 cells. Consistent to the reports that overexpression of GLUT1 enables to transport DHA more efficiently into cells [26, 36], we did find that ER6 cells have higher intracellular DHA concentration after DHA treatment than HCC827 cells. It indicates that overexpression of GLUT1 in ER6 cells facilitates ER6 cells to transport more DHA than HCC827 cells, which is the underlying mechanism of the selective suppression of reduced vitamin C and DHA on ER6 cells.
Studies had shown that vitamin C treatment kills cancer cells is ascribed to the cause of energy crisis due to glycolysis inhibition [26, 28, 30, 37]. We have recently proven that glucose deprivation enables to cause energy crisis and selectively depress the viability of ER6 cells [18]. Hence we hypothesized that DHA treatment is able to reduce energy production. We found that treatment with 1 mM DHA for 2 hours enabled to decrease intracellular abundance of NAD+ in both ER6 cells and HCC827 cells. And we also found the dramatically decreased ATP abundance in ER6 cells in 1 mM DHA treatment, while relatively higher abundance of AMP and ADP levels compared to HCC827 cells under the same treatment. It is consistent to the results of these metabolite levels under glucose deprivation treatment as we reported before [18]. We also found that DHA treatment suppressed AMPK both in normal glucose and glucose deprivation, which is in accordance with the previous finding [18, 26]. We have found that glucose deprivation stimulates AMPK and increases autophagy [18]. AMPK protects tumor cells from metabolic crisis through different mechanisms: autophagy induction, maintaining proper ATP levels [38, 39]. Hence, it suggests that DHA might depress the viability of ER6 cells through disturbing intracellular energy equilibrium and inhibiting AMPK phosphorylation. Studies had shown that metformin targets cancer stem cells in breast cancer, pancreatic cancer, glioblastoma and colon cancer by inhibiting oxidative phosphorylation and by reducing mitochondrial ATP production resulting in energy crisis [40]. Another research showed that neoalbaconol, a novel small-molecular compound isolated from the fungus, induces energy depletion by reducing the consumption of glucose and ATP generation [41]. It indicates that disturbance of energy balance by suppressing ATP production of ER6 cells is the underlying mechanism of viability depression of ER6 cells by DHA.
To better understand the underlying suppression mechanism of DHA to ER6 cells, we applied XF24 extracellular flux analyzer (Seahorse Biosciences Agilent technologies) to measure real time glycolysis ability, and found that DHA pretreatment, DHA supplement and combined DHA pretreatment with DHA supplement were able to suppress glycolysis in ER6 cells and HCC827 cells. What we found is consistent with previous reports [26, 28, 30, 37]. Studies had shown that increased DHA uptake causes oxidative stress as intracellular DHA is reduced to vitamin C, which depletes glutathione [26-29]. Unfortunately, we did not find DHA treatment impacted redox state of ER6 cells and HCC827 cells. We had previously reported that the antioxidation ability of ER6 cells is destroyed and produces more ROS than HCC827 cells without any treatments ascribed to mitochondria dysfunction [18]. A paper showed that treatment of cells with vitamin C and quercetin did not modulate cellular reduced glutamine levels in breast cancer cells [42]. It suggests that DHA treatment disrupts energy production of ER6 cells by inhibiting glycolysis directly rather than promoting oxidative stress in the way of consuming reduced glutathione.
In conclusion, DHA is more efficient than reduced vitamin C in depressing the viability of ER6 cells in the presence of glucose. Inhibiting glycolysis which results in energy crisis might be a mechanism of the suppression of the viability of ER6 cells by DHA. DHA supplement could be a potential strategy against acquired resistance to TKIs in NSCLCs' targeted therapies.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81872613 and 81573142 to Lili Yang; No. 81672276 and 81461168028 to Zhenfeng Zhang, 81872069 to Hainan Yang); Guangzhou Science and Technology program (201803010038) to Zhenfeng Zhang.
Abbreviations
TKIs: tyrosine kinase inhibitors; NSCLCs: non-small cell lung cancers; DHA: oxidized vitamin C or dehydroascorbate; GLUT: glucose transporter; ECAR: extracellular acidification rate; OCR: oxygen consumption rate; GSH: glutathione; GSSG: oxidized glutathione; AMPK: adenosine 5'-monophosphate (AMP)-activated protein kinase; p-AMPK: phosphorylated AMPK; HK-2: hexokinase 2; PK-M2: pyruvate kinase M2; PFK3: pyrophosphate-dependent phosphofructokinase 3; NAD+: nicotinamide adenine dinucleotide.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mok TS, Wu Y-L, Thongprasert S, Yang C-H, Chu D-T, Saijo N. et al. Gefitinib or Carboplatin-Paclitaxel in Pulmonary Adenocarcinoma. N Engl J Med. 2009;361:947-57
2. Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H. et al. Gefitinib or Chemotherapy for Non-Small-Cell Lung Cancer with Mutated EGFR. N Engl J Med. 2010;362:2380-8
3. Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J. et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121-8
4. Oxnard GR, Arcila ME, Chmielecki J, Ladanyi M, Miller VA, Pao W. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin Cancer Res. 2011;17:5530-7
5. Lee DH. Treatments for EGFR-mutant non-small cell lung cancer (NSCLC): The road to a success, paved with failures. Pharmacol Ther. 2017;174:1-21
6. Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M. et al. EGFR Mutation and Resistance of Non-Small-Cell Lung Cancer to Gefitinib. N Engl J Med. 2005;352:786-92
7. Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J Clin Oncol. 2013;31:1070-80
8. Zhang Z, Lee JC, Lin L, Olivas V, Au V, Laframboise T. et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852-60
9. Wu X, Liu X, Koul S, Lee CY, Zhang Z, Halmos B. AXL kinase as a novel target for cancer therapy. Oncotarget. 2014;5:9546-63
10. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Joon OP. et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039-43
11. Bhattacharya B, Mohd Omar MF, Soong R. The Warburg effect and drug resistance. Br J Pharmacol. 2016;173:970-9
12. Ruprecht B, Zaal EA, Zecha J, Wu W, Berkers CR, Kuster B. et al. Lapatinib resistance in breast cancer cells is accompanied by phosphorylation-mediated reprogramming of glycolysis. Cancer Res. 2017;77:1842-53
13. Hudson CD, Hagemann T, Mather SJ, Avril N. Resistance to the tyrosine kinase inhibitor axitinib is associated with increased glucose metabolism in pancreatic adenocarcinoma. Cell Death Dis. 2014;5(4):e1160
14. Yu T, Yang G, Hou Y, Tang X, Wu C, Wu XA. et al. Cytoplasmic GPER translocation in cancer-associated fibroblasts mediates cAMP/PKA/CREB/glycolytic axis to confer tumor cells with multidrug resistance. Oncogene. 2017;36:2131-45
15. Qian X, Xu W, Xu J, Shi Q, Li J, Weng Y. et al. Enolase 1 stimulates glycolysis to promote chemoresistance in gastric cancer. Oncotarget. 2017;8:47691-708
16. Le Calvé B, Rynkowski M, Le Mercier M, Bruyère C, Lonez C, Gras T. et al. Long-term in vitro treatment of human glioblastoma cells with temozolomide increases resistance in vivo through up-regulation of GLUT transporter and aldo-keto reductase enzyme AKR1C expression. Neoplasia. 2010;12:727-39
17. Song K, Li M, Xu X, Xuan L, Huang G, Liu Q. Resistance to chemotherapy is associated with altered glucose metabolism in acute myeloid leukemia. Oncol Lett. 2016;12:334-42
18. Ye M, Wang S, Wan T, Jiang R, Qiu Y, Pei L. et al. Combined inhibitions of glycolysis and AKT/autophagy can overcome resistance to EGFR-targeted therapy of lung cancer. J Cancer. 2017;8:3774-84
19. Butler EB, Zhao Y, Muñoz-Pinedo C, Lu J, Tan M. Stalling the engine of resistance: Targeting cancer metabolism to overcome therapeutic resistance. Cancer Res. 2013;73:2709-17
20. Wang YD, Li SJ, Liao JX. Inhibition of Glucose Transporter 1 (GLUT1) Chemosensitized Head and Neck Cancer Cells to Cisplatin. Technol Cancer Res Treat. 2013;12:525-35
21. Woo YM, Shin Y, Lee EJ, Lee S, Jeong SH, Kong HK. et al. Inhibition of aerobic glycolysis represses Akt/mTOR/HIF-1α axis and restores tamoxifen sensitivity in antiestrogen-resistant breast cancer cells. PLoS One. 2015:10 (7), e0132285
22. Park GB, Chung YH, Kim D. 2-Deoxy- d -glucose suppresses the migration and reverses the drug resistance of colon cancer cells through ADAM expression regulation. Anticancer Drugs. 2017;28:410-20
23. Reyes R, Wani NA, Ghoshal K, Jacob ST, Motiwala T. Sorafenib and 2-deoxyglucose synergistically inhibit proliferation of both sorafenib-sensitive and -resistant HCC cells by inhibiting ATP production. Gene Expr. 2017;17:129-40
24. Huang A, Ju HQ, Liu K, Zhan G, Liu D, Wen S. et al. Metabolic alterations and drug sensitivity of tyrosine kinase inhibitor resistant leukemia cells with a FLT3/ITD mutation. Cancer Lett. 2016;377:149-57
25. Gonzalez MJ, Miranda-Massari JR. New insights on Vitamin C and cancer. New York, USA: Springer New York. 2014
26. Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K. et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015;350:1391-6
27. Xia J, Xu H, Zhang X, Allamargot C, Coleman KL, Nessler R. et al. Multiple Myeloma Tumor Cells are Selectively Killed by Pharmacologically-dosed Ascorbic Acid. EBioMedicine. 2017;18:41-9
28. Wang C, Lv H, Yang W, Li T, Fang T, Lv G. et al. SVCT-2 determines the sensitivity to ascorbate-induced cell death in cholangiocarcinoma cell lines and patient derived xenografts. Cancer Lett. 2017;398:1-11
29. Wang L, Luo X, Li C, Huang Y, Xu P, Lloyd-Davies LH. et al. Triethylenetetramine Synergizes with Pharmacologic Ascorbic Acid in Hydrogen Peroxide Mediated Selective Toxicity to Breast Cancer Cell. Oxid Med Cell Longev. 2017:2017
30. Yang G, Yan Y, Ma Y, Yang Y. Vitamin C at high concentrations induces cytotoxicity in malignant melanoma but promotes tumor growth at low concentrations. Mol Carcinog. 2017;56:1965-76
31. Wohlrab C, Phillips E, Dachs GU. Vitamin C Transporters in Cancer: Current Understanding and Gaps in Knowledge. Front Oncol. 2017;7:74
32. Roomi MW, Kalinovsky T, Rath M, Niedzwiecki A. A specific mixture of nutrients suppresses ovarian cancer A-2780 tumor incidence, growth, and metastasis to lungs. Nutrients. 2017;9(3):303
33. Shareck M, Rousseau M-C, Koushik A, Siemiatycki J, Parent M-E. Inverse Association between Dietary Intake of Selected Carotenoids and Vitamin C and Risk of Lung Cancer. Front Oncol. 2017;7:23
34. Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J. et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell. 2017;170:1079-1095.e20
35. De Francesco EM, Bonuccelli G, Maggiolini M, Sotgia F, Lisanti MP, De Francesco EM. et al. Vitamin C and Doxycycline: A synthetic lethal combination therapy targeting metabolic flexibility in cancer stem cells (CSCs). Oncotarget. 2015;5:14-9
36. Guo Y, Zhang Y, Li J, Zheng Y, Lu Y, Jiang X. et al. Cell microenvironment-controlled antitumor drug releasing-nanomicelles for GLUT1-targeting hepatocellular carcinoma therapy. ACS Appl Mater Interfaces. 2015;7:5444-53
37. Ma E, Chen P, Wilkins HM, Wang T, Swerdlow RH, Chen Q. Pharmacologic ascorbate induces neuroblastoma cell death by hydrogen peroxide mediated DNA damage and reduction in cancer cell glycolysis. Free Radic Biol Med. 2017;113:36-47
38. Draz H, Goldberg AA, Titorenko VI, Tomlinson Guns ES, Safe SH, Sanderson JT. Diindolylmethane and its halogenated derivatives induce protective autophagy in human prostate cancer cells via induction of the oncogenic protein AEG-1 and activation of AMP-activated protein kinase (AMPK). Cell Signal. 2017;40:172-82
39. Hardie DG, Ross FA, Hawley SA. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251-62
40. Mayer MJ, Klotz LH, Venkateswaran V. Metformin and prostate cancer stem cells: a novel therapeutic target. Prostate Cancer Prostatic Dis. 2015;18:303-9
41. Deng Q, Yu X, Xiao L, Hu Z, Luo X, Tao Y. et al. Neoalbaconol induces energy depletion and multiple cell death in cancer cells by targeting PDK1-PI3-K/Akt signaling pathway. Cell Death Dis. 2013;4(9):e804
42. Mostafavi-Pour Z, Ramezani F, Keshavarzi F, Samadi N. The role of quercetin and vitamin c in NRF2-dependent oxidative stress production in breast cancer cells. Oncol Lett. 2017;13:1965-73
Author contact
![]() Corresponding authors: Zhenfeng Zhang, 250 Changgang Rd East, Guangzhou, 510260, China; E-mail: zhangzhfedu.cn; and Lili Yang, 74 zhongshan road 2, Guangzhou, Guangdong, PR China; E-mail: yangll7sysu.edu.cn.
Corresponding authors: Zhenfeng Zhang, 250 Changgang Rd East, Guangzhou, 510260, China; E-mail: zhangzhfedu.cn; and Lili Yang, 74 zhongshan road 2, Guangzhou, Guangdong, PR China; E-mail: yangll7sysu.edu.cn.
Citation styles
APA
Ye, M., Pang, N., Wan, T., Huang, Y., Wei, T., Jiang, X., Zhou, Y., Huang, Y., Yang, H., Zhang, Z., Yang, L. (2019). Oxidized Vitamin C (DHA) Overcomes Resistance to EGFR-targeted Therapy of Lung Cancer through Disturbing Energy Homeostasis. Journal of Cancer, 10(3), 757-764. https://doi.org/10.7150/jca.28087.
ACS
Ye, M.; Pang, N.; Wan, T.; Huang, Y.; Wei, T.; Jiang, X.; Zhou, Y.; Huang, Y.; Yang, H.; Zhang, Z.; Yang, L. Oxidized Vitamin C (DHA) Overcomes Resistance to EGFR-targeted Therapy of Lung Cancer through Disturbing Energy Homeostasis. J. Cancer 2019, 10 (3), 757-764. DOI: 10.7150/jca.28087.
NLM
Ye M, Pang N, Wan T, Huang Y, Wei T, Jiang X, Zhou Y, Huang Y, Yang H, Zhang Z, Yang L. Oxidized Vitamin C (DHA) Overcomes Resistance to EGFR-targeted Therapy of Lung Cancer through Disturbing Energy Homeostasis. J Cancer 2019; 10(3):757-764. doi:10.7150/jca.28087. https://www.jcancer.org/v10p0757.htm
CSE
Ye M, Pang N, Wan T, Huang Y, Wei T, Jiang X, Zhou Y, Huang Y, Yang H, Zhang Z, Yang L. 2019. Oxidized Vitamin C (DHA) Overcomes Resistance to EGFR-targeted Therapy of Lung Cancer through Disturbing Energy Homeostasis. J Cancer. 10(3):757-764.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.