Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(4):918-926. doi:10.7150/jca.27226 This issue Cite
Research Paper
Downregulation of PDK4 Increases Lipogenesis and Associates with Poor Prognosis in Hepatocellular Carcinoma
Chen Yang1,2*, Siying Wang2*, Haoyu Ruan1, Botai Li3, Zhuoan Cheng3, Jia He2, Qiaozhu Zuo2, Chengtao Yu3, Hui Wang2, Yuanyuan Lv2, Dishui Gu4, Guangzhi Jin5, Ming Yao2, Wenxin Qin2, Haojie Jin2 ![]()
1. Shanghai Medical College of Fudan University, Shanghai, China
2. State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Renji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
3. School of Biomedical Engineering, Shanghai Jiao Tong University, Shanghai, China
4. Department of Pathophysiology, School of Basic Medical Sciences, Guangdong Medical University, Dongguan, Guangdong, 523808, China.
5. Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, China.
*Equal contributors.
Received 2018-5-13; Accepted 2018-11-9; Published 2019-1-29
Citation:
Yang C, Wang S, Ruan H, Li B, Cheng Z, He J, Zuo Q, Yu C, Wang H, Lv Y, Gu D, Jin G, Yao M, Qin W, Jin H. Downregulation of PDK4 Increases Lipogenesis and Associates with Poor Prognosis in Hepatocellular Carcinoma. J Cancer 2019; 10(4):918-926. doi:10.7150/jca.27226. https://www.jcancer.org/v10p0918.htm
Other stylesAbstract
Alterations in cellular metabolism are one of the characteristics in cancer. They are not only the result of tumor progression but also the cause of cancer initiation. Pyruvate dehydrogenase kinase 4 (PDK4) is a key metabolic enzyme, which regulates cell metabolism by inhibiting pyruvate dehydrogenase (PDH). However, the function and regulating mechanism of PDK4 in HCC remain unclear. Here, we found that the expression of PDK4 was significantly decreased in HCC tissues, and its downregulation could predict poor prognosis of HCC patients. Silencing PDK4 significantly facilitated proliferation and migration of HCC cells. Knockdown of PDK4 didn't influence the oxidative phosphorylation and glycolysis capacity of HCC cells in vitro. However, knockdown of PDK4 increased expression of key lipogenic enzymes, fatty acid synthase (FASN) and stearoyl-CoA desaturase (SCD), which finally induced lipogenesis. These data suggest that PDK4 inhibits proliferation and migration of HCC cells probably via suppressing lipogenesis.
Keywords: hepatocellular carcinoma, PDK4, proliferation, migration, lipogenesis
Introduction
Hepatocellular carcinoma (HCC) is one of the most malignant solid tumors and the fourth major causes of cancer related death worldwide [1]. There were 854,000 incident liver cancer cases and 810,000 deaths globally in 2015 [1]. Many factors lead to the development and progression of HCC such as virus infection, alcohol and metabolic diseases [2]. Although surgical resection, liver transplantation, and chemotherapy are the typical treatments for early-stage HCC, the prognosis of patients with advanced tumors still remains poor: the 5-year survival rate and recurrence rate is 25%-39% and 80%, respectively [3, 4]. Therefore, it is urgent to further clarify the mechanism underlying HCC progression and identify potential therapeutic targets for HCC treatment.
The study of cancer cellular metabolism has led to several key findings on the impact of energy availability for cell proliferation and metastasis [5, 6]. Pyruvate dehydrogenase kinase 4 (PDK4), inhibits pyruvate oxidation, and is shown to mediate the metabolic switch from glucose metabolism to fatty acid metabolism [7]. It belongs to PDK protein kinase family, which includes PDK1, PDK2, PDK3, and PDK4. This family can inhibit pyruvate dehydrogenase (PDH) by phosphorylating its subunit [8]. Previous studies reported that PDK4 was closely associated with diabetes, heart disease and cancer [9, 10]. Moreover, it is reported that PDK4 was downregulated in a number of different cancers such as prostate cancer, breast cancer, lung cancer and liver cancer [11-14]. PDK4 could inhibit the proliferation and induce the apoptosis in lung and breast cancer cells [14, 15]. In addition, loss of PDK4 could activate the EMT program and promote migration and invasion of ovarian cancer [16]. However, the role of PDK4 in HCC is still largely unclear.
In this study, we found that PDK4 was significantly downregulated in HCC tissues, and HCC patients with lower level of PDK4 had lower overall survival rate and higher recurrence rate. Knockdown of PDK4 significantly increased the proliferation and migration of HCC cells in vitro. Moreover, knockdown of PDK4 increased lipogenesis by elevating the expression of fatty acid synthase (FASN) and stearoyl-CoA desaturase (SCD). Taken together, PDK4 may play a tumor suppressive role in proliferation and migration of HCC cells via suppressing lipogenesis.
Materials and methods
Cell culture
HepG2, Hep3B and SK-Hep1 were purchased from the American Type Culture Collection (ATCC, USA). SMMC7721 and L02 were obtained from Chinese Academy of Sciences (Shanghai, China). MHCC97H and HCCLM3 were provided by Liver Cancer Institute of Fudan University (Shanghai, China). All the cell lines were cultured in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% antibiotics (100 U/ml penicillin, 100 mg/ml streptomycin) at 37℃ in a humidified atmosphere of 5% CO2.
Clinical samples and tissue microarray
A total 188 cases of HCC samples was enrolled in our study: 86 cases for quantitative real-time PCR, 8 cases for western blotting, 80 cases for IHC analysis, and 14 cases for mRNA microarray analysis. The samples were collected from HCC patients at Eastern Hepatobiliary Surgery Hospital (EHBH), Second Military Medical University, Shanghai, China. The clinical parameters of HCC samples used for quantitative real-time PCR (n=86) and western blotting (n=8) analyses were listed in Table S1. The clinical parameters of HCC samples used for IHC (n=80) analyses were listed in Table S2. The detailed clinicopathological characteristics of the 14 HCC patients used for mRNA microarray analysis has been described in our previous published paper [17], and summarized in Table S1. No patients in this study received any therapies before the surgery and written informed consent was obtained from each patient. Clinical data of 180 patients with HCC from The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov) were analyzed using a Kaplan-Meier survival plot. Overall survival (OS) was the interval between the date of surgery and death. The time to recurrence (TTR) was calculated from the date of tumor resection to the date of the detection of tumor recurrence.
Immunohistochemistry staining and scoring
Detailed protocol was described in our previous study [18]. The GTVisionTM Ⅲ Detection System/Mo&Rb kit (Dako, Danmark) was used in this study. Briefly, tissue sections were incubated overnight at 4°C with primary antibody. Primary antibodies were mouse anti-PDK4 monoclonal antibody (1:500, Abcam, USA), rabbit anti-FASN polyclonal antibody (1:200, Proteintech, Chinese), rabbit anti-SCD polyclonal antibody (1:200, Proteintech, Chinese). Average sum of integrated optical density (IOD) of each sample was measured by Image-Pro Plus software.
Semiquantitative scores were used to analyze PDK4 immunostaining of each HCC case in tissue microarray. Intensity of staining was categorized into -, +, ++, or +++, denoting negative, weak, moderate, or strong PDK4 staining, respectively. Extent of immunostaining was categorized into 0 (0-5%), 1 (6-25%), 2 (26-50%), or 3 (>51%) on the basis of the percentage of positive cells. Three random microscope fields per tissue were calculated. The sum of intensity and extent of staining was used as final score of expression level, and determined by the formula: final score = intensity score × percentage score. The final score of ≤ 3 was defined as PDK4 low expression, and > 3 as PDK4 high expression.
Quantitative real-time PCR
Total RNA of cultured cells and human HCC samples were extracted using Trizol Reagent (Invitrogen, USA). PrimeScript™ RT Reagent kit (TaKaRa, Japan) was used to perform reverse transcription. Quantitative real-time PCR was performed using an ABI 7500 instrument (Applied Biosystems, USA) and SYBR Premix Ex Taq Ⅱ (TaKaRa, Japan). Primers used are listed in Table S3 along with their sequences. Expression levels were normalized against the internal reference gene β-actin, and relative expression levels were displayed using the 2-ΔΔCt method.
Western blotting
Equal amounts of protein from different sample were separated by SDS-PAGE and transferred onto PVDF membrane. Membranes were blocked with 5% skim milk and incubated with the specific primary antibody at 4°C overnight. Membranes were then washed three times with PBS-Tween, and incubated with HRP-conjugated secondary antibodies, followed by three times' wash with PBS-Tween. Protein bands were visualized using Bio-rad ChemiDoc XRS system (USA).
Establishment of stable cell lines with PDK4 knockdown
Stable SMMC7721 and HepG2 cells silenced for PDK4 expression were established using lentiviral constructs expressing shPDK4 and negative control. The specific shRNA sequences of PDK4 (Table S3) were cloned into the vector PuroGreen (Addgene) and vectors were packaged into 293T cells to produce viruses. Viruses were harvested 48h after transfection, and viral titers were determined. Target cells (1×105 cells) were infected with 1×106 recombinant lentivirus-transducing units in the presence of 6 μg/ml polybrene (Sigma, USA).
CCK8 and colony formation assay
For CCK8 assay, 1×103 cells were seeded into 96-well plates. We performed Cell Counting Kit-8 assay (Dojindo Laboratories, Kumamoto, Japan) to measure cell viability at different time points according to manufacturer's instructions.
For the colony formation assay, 0.5×103 cells were added in six-well plates in the culture medium and the culture medium was replaced every 3 days. After 10 days, the cells were fixed with 4% formaldehyde and visualized by crystal violet staining, and then the numbers of the colony were counted.
Cell migration and invasion assays
Cell migration and invasion were performed as previously described [19].
Xenograft experiment
2×106 cells with PDK4 knockdown or control were injected subcutaneously into 6-week-old female nude mice. The mice were killed 45 days later, and then tumors were surgically removed at the end of the experiments. The body weight and the size of the tumor were measured. The tumor growth was monitored based on the tumor weight and volume, which was calculated as follows: V (volume) = (width2×length) /2. All the use of animals in this study was approved by Shanghai Medical Experimental Animal Care Commission.
Oil Red O staining
Frozen sections were dried at 60℃ for 30 min, fixed with 4% paraformaldehyde and washed with H2O. After washed by 60% isopropanol, the tissue sections were stained for 15 min at room temperature with freshly prepared Oil Red O solution (5 mg/ml in 60% isopropanol). Then sections were washed with 60% isopropanol and were stained with hematoxylin (Sigma-Aldrich, USA). Finally, Oil Red O stained sections were observed under a microscope.
Metabolic assays
Extracellular acidification rate (ECAR) and Oxygen consumption rate (OCR) were evaluated using the Seahorse XF96 extracellular flux analyzer as previously described [20]. Briefly, 1×104 cells were seeded and incubated overnight in XF96 Cell Culture Microplate (Seahorse Bioscience, USA) in growth medium. Next day, culture medium was changed to non-buffered assay medium. For glycolysis stress test, ECARs in the indicated HCC cells were detected under basal conditions and in response to indicated inhibitors including glucose, oligomycin (OLI) and 2-Deoxy-D-glucose (2-DG). For cell mitochondrial stress test, OCRs were performed at basal conditions and after injection of OLI, Carbonyl cyanide 4- (trifluoromethoxy) phenylhydrazone (FCCP), and rotenone supplemented with antimycin A (Rtn/AA) at indicated time points.
Statistical analysis
Statistical analysis was carried out by GraphPad Prism 7 (GraphPad Software Inc., La Jolla, CA, USA) and SPSS version 20.0 (SPSS Inc., Chicago, IL, USA). Quantitative variables were analyzed by Student t tests. Patient survival was calculated using Kaplan-Meier's method and log-rank analysis. All values were presented as mean ± standard deviation (SD). P < 0.05 was considered to be statistically significant.
Results
Downregulation of PDK4 in HCC
By analyzing our global gene expression profile in 14 pairs of tumorous tissues (T) and corresponding adjacent nontumorous liver tissues (N) from HCC patients (database GSE84402) [17], we found that PDK4 was significantly downregulated in HCC tissues (Fig 1A, B). We further analyzed mRNA levels of PDK4 in 86 pairs clinical samples from Eastern Hepatobiliary Hospital of the Second Military Medical University. As shown in Figure 1C-E, PDK4 expression was significantly decreased in tumorous tissues compared with corresponding adjacent noncancerous liver tissues (66/86, 76.74%). This result was validated by western blotting analysis (Figure 1F). We also evaluated the expression of PDK4 in 80 paired samples of patients with HCC by immunohistochemical staining. The results showed that staining density of PDK4 in nontumorous tissue group was obviously stronger than that in tumorous tissue group (Figure S1A, B). Oncomine Dataset also showed that PDK4 was downregulated in HCC samples (Figure S1C).
Clinical significance of PDK4 in HCC patients
In order to explore the clinical significance of PDK4 in HCC patients, IHC staining was performed in a tissue microarray which contained 80 HCC samples. The samples were classified into two groups: low PDK4 group and high PDK4 group. Kaplan-Meier analysis showed that HCC patients with high PDK4 expression exhibited better overall survival (OS, P=0.0360) and shorter time to recurrence (TTR, P=0.0010) than patients with low PDK4 expression (Figure 2B, C). The same result was also observed in the TCGA database (Figure 2D).
Correlations between PDK4 expression and clinicopathological variables were shown in Table S2. The expression of PDK4 was not related to clinicopathological characteristics, such as sex, age and tumor size. However, Cox's multivariate analysis showed that PDK4 was an independent prognostic factor for OS (P=0.049) and TTR time (P=0.007) in HCC (Table 1).
Figure 1
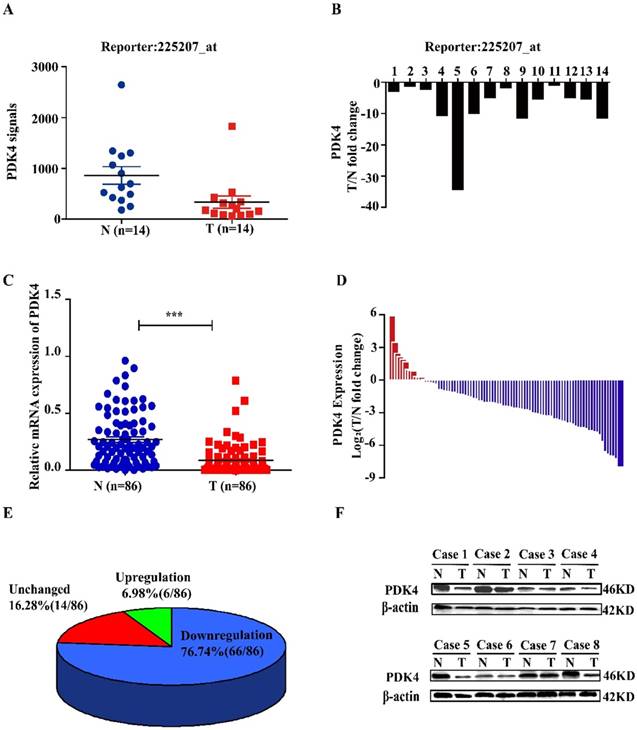
PDK4 is frequently downregulated in HCC. (A, B) Downregulation of PDK4 was found in 14 paired samples of HCC by global gene expression analysis. (C-E) mRNA levels of PDK4 in 86 paired samples of tumorous tissues (T) and corresponding adjacent nontumorous tissues (N) from patients with HCC were detected by quantitative real-time PCR. (F) Representative images of western blotting analysis for the expression of PDK4 in eight paired samples of HCC. ***P<0.001
Figure 2
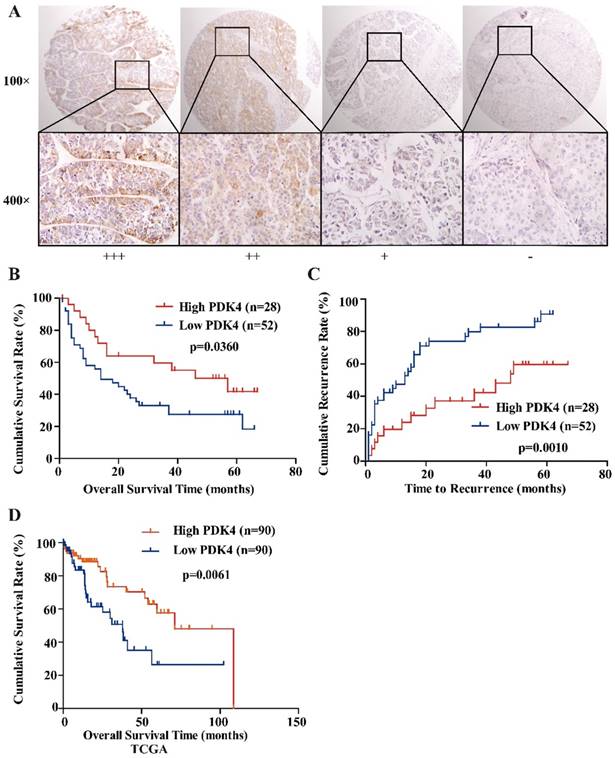
Downregulation of PDK4 is associated with poor prognosis in HCC patients. (A) Typical images show strong (+++), moderate (++), weak (+), or negative (-) staining of PDK4 in HCC specimens (magnification: 100×, 400×). (B, C) Kaplan-Meier analysis of overall survival time and time to recurrence in HCC cases based on PDK4 expression. (D) Kaplan-Meier analysis of overall survival time in HCC cases based on PDK4 expression from TCGA database.
Table 1
Univariate and multivariate analyses of factors associated with OS and TTR of HCC (n=80)
| Variable | OS | TTR | ||||||
|---|---|---|---|---|---|---|---|---|
| Multivariate analysis | Multivariate analysis | |||||||
| Univariate P | HR | 95% CI | P value | Univariate P | HR | 95% CI | P value | |
| PDK4 (Low vs. High) | 0.036 | 0.525 | 0.276-0.999 | 0.049 | 0.001 | 0.384 | 0.203-0.726 | 0.007 |
| Sex (male vs. female) | 0.757 | NA | 0.578 | NA | ||||
| Age (≤ 50 vs. >50) | 0.125 | NA | 0.272 | NA | ||||
| TNM stage (I vs. II vs. III-IV) | 0.019 | 1.618 | 1.074-2.437 | 0.021 | 0.018 | NS | ||
| Tumor size ( ≤ 3cm vs. >3cm ) | 0.617 | NA | 0.326 | NA | ||||
| Tumor number (1vs. >1) | 0.1 | NA | 0.480 | NA | ||||
| AFP ( ≤20 ng/mL vs. > 20 ng/mL) | 0.361 | NA | 0.253 | NA | ||||
| HBsAg (negative vs. positive) | 0.634 | NA | 0.897 | NA | ||||
| Tumor differentiation (I-II vs. III-IV) | 0.914 | NA | 0.641 | NA | ||||
| Vascular invasion (no vs. yes) | 0.042 | NS | 0.774 | NA | ||||
| Child-Pugh stage (A vs. B) | 0.603 | NA | 0.489 | NA | ||||
| Liver cirrhosis (no vs. yes) | 0.006 | 0.346 | 0.135-0.886 | 0.027 | 0.017 | 0.378 | 0.148-0.967 | 0.042 |
NOTE. Bold values indicate statistical significance.
Abbreviations: HBsAg: hepatitis B surface antigen; AFP: alpha-fetoprotein; TNM: tumor-node-metastasis staging system; CI: confidence interval; HR: hazard ratio; OS: overall survival; TTR: time to recurrence; NA: not applicable; NS: not significant.
Figure 3
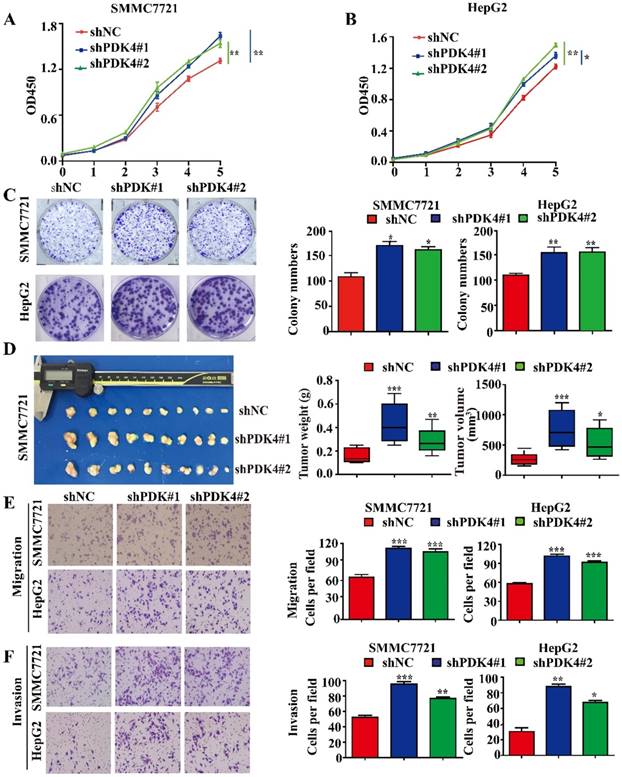
Downregulation of PDK4 promotes proliferation and migration in HCC cells. (A, B) CCK-8 assays for the effects of PDK4 silence on proliferation of SMMC7721 and HepG2 cells. (C) Colony formation assays for the effects of PDK4 silence in SMMC7721 and HepG2 cells. (D) In vivo assays for the effects of PDK4 silence on proliferation of SMMC7721 cells. (E, F) Transwell assays for the effects of PDK4 silence on migration and invasion of SMMC7721 and HepG2 cells (magnification: 400×). ***P<0.001, **P<0.01, *P<0.05
PDK4 inhibited the aggressive behavior of HCC cells
To investigate the functional roles of PDK4 in HCC progression, we first detected the expression levels of PDK4 in an immortal liver cell line L02 and a panel of HCC cell lines (Figure S2A). The expression of PDK4 was knocked down in two HCC cell lines (SMMC7721 and HepG2) using PDK4 shRNAs. The knockdown efficiency of PDK4 was confirmed by western blotting (Figure S2B, C). The CCK-8 assays showed that PDK4 knockdown increased proliferation of SMMC7721 and HepG2 cells in vitro (Figure 3A, B). Our colony formation assays showed that PDK4 knockdown also increased colony formation of SMMC7721 and HepG2 cells in vitro (Figure 3C). In addition, knockdown of PDK4 significantly facilitated the migration and invasion of HCC cells in vitro (Figure 3E, F).
To test the role of PDK4 in tumor growth in vivo, SMMC7721 cells with PDK4 knockdown were injected into nude mice subcutaneously. The tumors with PDK4 knockdown displayed an advantage in growth (Figure 3D).
PDK4 regulates lipogenesis of liver cancer cells
As a member of PDK protein kinase family, PDK4 can inhibit PDH by phosphorylating its E1α subunit [21]. PDH catalyzes conversion of pyruvate to acetyl-CoA and thus controls the carbon flow switch from lactate production to TCA cycle and lipogenesis. Therefore, we speculated that downregulation of PDK4 might influence the glucose metabolism and lipid metabolism. First of all, we examined the effects of PDK4 silence on HCC glucose metabolism. Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were analyzed by using the Seahorse XF96 extracellular flux analyzer. We found that knockdown of PDK4 didn't influence the OCR or ECAR (Figure S3A, B). Then, we analyzed lipogenesis in tumor tissues of HCC xenograft by Oil Red O staining. As shown in Figure 4A, we found that the staining of the tumor tissues with PDK4 knockdown was stronger than control group. This result indicates that downregulation of PDK4 may induce lipogenesis of HCC cells. Therefore, we further analyzed the expression of key lipogenic enzymes including FASN and SCD in tumor tissues of xenograft and in HCC cell lines. As the results shown, knockdown of PDK4 increased the expression of FASN and SCD (Figure 4B, C). Our data suggest that PDK4 may affect proliferation and migration of HCC cells by regulating lipogenesis.
Figure 4
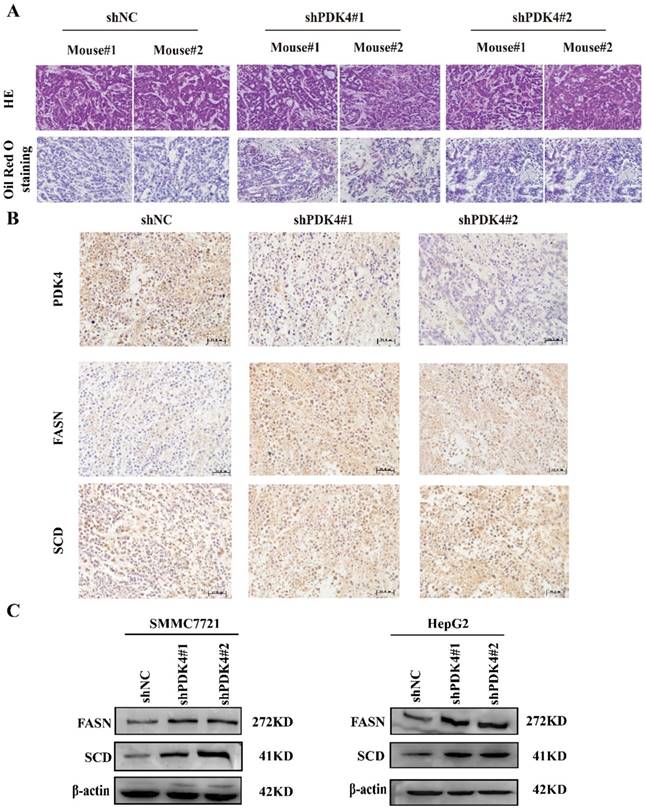
Knockdown of PDK4 influences the lipid metabolism. (A) Oil Red O staining for the effects of PDK4 silence on the lipogenesis of xenograft tumor. (B) Representative images of IHC staining for PDK4, FASN and SCD in tumor tissues of subcutaneous HCC implantation models (magnification: 400×). (C) Western blotting analyses of FASN, SCD in HCC cells with PDK4 knockdown.
Discussion
Due to the fast progression and high rate of recurrence, the prognosis of HCC is very poor. It is necessary to identify molecular mechanism responsible for HCC progression. In this study, we found PDK4 was downregulated in HCC tissues and its downregulation could predict poor prognosis of HCC patients. Moreover, knockdown of PDK4 could promote proliferation and metastasis of HCC, which were probably mediated by promoting lipogenesis. However, more detailed mechanism should be explored in the future.
The function of PDK4 in solid tumors is still controversial. PDK4 displays both oncogenic and tumor suppressive effect in different cancer types. The tumor suppressive effect of PDK4 was widely reported in various types of cancers. For instance, PDK4 was significantly down-regulated in prostate cancer and was suggested as a potential marker for the diagnosis of prostate cancer [11]. The downregulation of PDK4 in lung cancer promoted cell proliferation and tumor growth by modulating the activity of PDH, and subsequently de novo lipogenesis of cancer cells [15]. In another study, it was reported that the anti-proliferative effect of PPAR activation could be inhibited by suppressing PDK4 expression, and overexpression of PDK4 could increase the ROS levels and lead to RB hypophosphorylation and tumor growth arrest [22]. However, there are few research studies focused on the role of PDK4 in HCC. One study showed that mRNA level of PDK4 was decreased in 21 HCC samples, and knockdown of PDK4 in HCC cells showed rapid G2/M cell cycle progression and induced cell cycle proteins such as cyclins E1 and A2 [13]. In our current study, we further provided the evidence that PDK4 acted as a tumor suppressor in HCC. We showed that both mRNA and protein levels of PDK4 were significantly decreased in large HCC samples. Moreover, we firstly found that downregulation of PDK4 could predict poor prognosis of HCC patients. In line with the above studies, our results also showed that knockdown of PDK4 could enhance the proliferation and tumor growth in HCC. Interestingly, we found that knockdown of PDK4 could strongly promote the migration and invasion of HCC cells in vitro. The underling mechanisms need further investigation. On the contrary, several studies also reported the oncogenic effect of PDK4 in tumors. For example, PDK4 was increased in normal mucosa of colorectal cancer (CRC) due to the decreased methylation of its CpGs, and played oncogenic role in human colon cancer cells [23, 24]. In another study, PDK4 was showed to promote tumorigenesis through activation of mTOR signaling and increase of aerobic glycolysis in various cell lines such as A549 cells ( lung cancer cell line), PC3 cells (prostate cancer cell line), and MCF7 (breast cancer cell line) [25]. Taken together, these studies suggested that, on the one hand, tissue- or cell-specificity may account for the distinct function of PDK4 in different cancer types; on the other hand, the metabolic microenvironment and thus the metabolic requirements of cancer cells in various organs may not be the same, which could lead to multifarious function of PDK4.
PDK4, a member of PDK protein family, inhibits PDH activity, and its downregulation can elevate the level of acetyl CoA. Acetyl-CoA is the substrate of oxidative phosphorylation and de novo biogenesis of fatty acids. Therefore, we suppose that PDK4 may exert its roles by involving in glucose and lipid metabolism. Warburg effect is the most understood metabolic reprogramming in cancer cells [26]. Most cancer cells prefer glycolysis to produce their energy even in the presence of abundant oxygen. Although glycolysis is an energetically wasteful alteration, it can promote drug resistance and metastasis of cancer cells [27]. We detected the effect of PDK4 on glucose metabolism of HCC cells, and found that knockdown of PDK4 did not influence the capacity of glycolysis or mitochondrial oxidative phosphorylation in HCC cells. It might be related to replenishment of the TCA cycle, which needs further investigation.
Metabolism reprogramming, including increased de novo lipogenesis, is one of the hallmarks of cancer [26]. We observed that knockdown of PDK4 could promote de novo biogenesis of fatty acids and increase the expression levels of key lipogenic enzymes FASN and SCD of HCC cells. FASN catalyzes the de novo synthesis of long-chain fatty acids from acetyl-CoA and malonyl-CoA. It has been described that FASN is elevated in multiple tumor types, and malignant behaviors of cancer are inhibited in suppression of FASN [28]. SCD belongs to the fatty acid desaturase family and is involved in monounsaturated fatty acids synthesis. SCD expression correlates with malignant transformation, proliferation and survival. SCD can inhibit autophagy-induced cell death in human hepatocellular carcinoma through inactivation of the AMPK signaling pathway [29]. High levels of E2F transcription factor 1 (E2F1) is observed in HCC cells with knockdown of PDK4 [13]. E2F1, a transcription factor, is known to regulate the cell cycle, and proliferation and can directly bind to the promoters of FASN and SCD1 and induce their expression to sustain lipogenesis [30]. As evidence above, we suppose that knockdown of PDK4 may induce the expression of FASN and SCD by upregulating the levels of E2F1.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
Financial support: This work was supported by grants from Shanghai Natural Science Foundation of China (16ZR1434700), National Key Basic Research Program of China (973 Program: 2015CB553905), National Natural Science Foundation of China (81702838), Shanghai Jiao Tong University School of Medicine (YG2015QN34), Shanghai Municipal Commission of Health and Family Planning (201640055, 2018YQ20), Shanghai Jiao Tong University School of Medicine(PYII-17-013).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Global Burden of Disease Liver Cancer Collaboration. et al. The Burden of Primary Liver Cancer and Underlying Etiologies From 1990 to 2015 at the Global, Regional, and National Level: Results From the Global Burden of Disease Study 2015. JAMA oncology. 2017;3:1683-91
2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA: a cancer journal for clinicians. 2015;65:87-108
3. Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut. 2014;63:844-55
4. Bruix J, Reig M, Sherman M. Evidence-Based Diagnosis, Staging, and Treatment of Patients With Hepatocellular Carcinoma. Gastroenterology. 2016;150:835-53
5. Bu P, Chen KY, Xiang K, Johnson C, Crown SB, Rakhilin N. et al. Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell metabolism. 2018;27:1249-62 e4
6. Jiang SH, Li J, Dong FY, Yang JY, Liu DJ, Yang XM. et al. Increased Serotonin Signaling Contributes to the Warburg Effect in Pancreatic Tumor Cells Under Metabolic Stress and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology. 2017;153:277-91 e19
7. Rowles J, Scherer SW, Xi T, Majer M, Nickle DC, Rommens JM. et al. Cloning and characterization of PDK4 on 7q21.3 encoding a fourth pyruvate dehydrogenase kinase isoenzyme in human. The Journal of biological chemistry. 1996;271:22376-82
8. Buck MJ, Squire TL, Andrews MT. Coordinate expression of the PDK4 gene: a means of regulating fuel selection in a hibernating mammal. Physiological genomics. 2002;8:5-13
9. Holness MJ, Bulmer K, Gibbons GF, Sugden MC. Up-regulation of pyruvate dehydrogenase kinase isoform 4 (PDK4) protein expression in oxidative skeletal muscle does not require the obligatory participation of peroxisome-proliferator-activated receptor alpha (PPARalpha). The Biochemical journal. 2002;366:839-46
10. Furuyama T, Kitayama K, Yamashita H, Mori N. Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. The Biochemical journal. 2003;375:365-71
11. Mengual L, Ars E, Lozano JJ, Burset M, Izquierdo L, Ingelmo-Torres M. et al. Gene expression profiles in prostate cancer: identification of candidate non-invasive diagnostic markers. Actas urologicas espanolas. 2014;38:143-9
12. Sun Y, Daemen A, Hatzivassiliou G, Arnott D, Wilson C, Zhuang G. et al. Metabolic and transcriptional profiling reveals pyruvate dehydrogenase kinase 4 as a mediator of epithelial-mesenchymal transition and drug resistance in tumor cells. Cancer & metabolism. 2014;2:20
13. Choiniere J, Wu J, Wang L. Pyruvate Dehydrogenase Kinase 4 Deficiency Results in Expedited Cellular Proliferation through E2F1-Mediated Increase of Cyclins. Molecular pharmacology. 2017;91:189-96
14. Grassian AR, Metallo CM, Coloff JL, Stephanopoulos G, Brugge JS. Erk regulation of pyruvate dehydrogenase flux through PDK4 modulates cell proliferation. Genes & development. 2011;25:1716-33
15. Li G, Li M, Hu J, Lei R, Xiong H, Ji H. et al. The microRNA-182-PDK4 axis regulates lung tumorigenesis by modulating pyruvate dehydrogenase and lipogenesis. Oncogene. 2017;36:989-98
16. Sun S, Liu J, Zhao M, Han Y, Chen P, Mo Q. et al. Loss of the novel mitochondrial protein FAM210B promotes metastasis via PDK4-dependent metabolic reprogramming. Cell death & disease. 2017;8:e2870
17. Jin H, Wang C, Jin G, Ruan H, Gu D, Wei L. et al. Regulator of Calcineurin 1 Gene Isoform 4, Down-regulated in Hepatocellular Carcinoma, Prevents Proliferation, Migration, and Invasive Activity of Cancer Cells and Metastasis of Orthotopic Tumors by Inhibiting Nuclear Translocation of NFAT1. Gastroenterology. 2017;153:799-811.e33
18. Jin H, Zhang Y, You H, Tao X, Wang C, Jin G. et al. Prognostic significance of kynurenine 3-monooxygenase and effects on proliferation, migration, and invasion of human hepatocellular carcinoma. Scientific reports. 2015;5:10466
19. Ruan HY, Yang C, Tao XM, He J, Wang T, Wang H. et al. Downregulation of ACSM3 promotes metastasis and predicts poor prognosis in hepatocellular carcinoma. American journal of cancer research. 2017;7:543-53
20. Ferrick DA, Neilson A, Beeson C. Advances in measuring cellular bioenergetics using extracellular flux. Drug discovery today. 2008;13:268-74
21. Roche TE, Hiromasa Y. Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cellular and molecular life sciences: CMLS. 2007;64:830-49
22. Srivastava N, Kollipara RK, Singh DK, Sudderth J, Hu Z, Nguyen H. et al. Inhibition of cancer cell proliferation by PPARgamma is mediated by a metabolic switch that increases reactive oxygen species levels. Cell metabolism. 2014;20:650-61
23. Leclerc D, Levesque N, Cao Y, Deng L, Wu Q, Powell J. et al. Genes with aberrant expression in murine preneoplastic intestine show epigenetic and expression changes in normal mucosa of colon cancer patients. Cancer prevention research (Philadelphia, Pa). 2013;6:1171-81
24. Leclerc D, Pham DN, Levesque N, Truongcao M, Foulkes WD, Sapienza C. et al. Oncogenic role of PDK4 in human colon cancer cells. British journal of cancer. 2017;116:930-6
25. Liu Z, Chen X, Wang Y, Peng H, Wang Y, Jing Y. et al. PDK4 protein promotes tumorigenesis through activation of cAMP-response element-binding protein (CREB)-Ras homolog enriched in brain (RHEB)-mTORC1 signaling cascade. The Journal of biological chemistry. 2014;289:29739-49
26. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
27. Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134:703-7
28. Gong J, Shen S, Yang Y, Qin S, Huang L, Zhang H. et al. Inhibition of FASN suppresses migration, invasion and growth in hepatoma carcinoma cells by deregulating the HIF-1alpha/IGFBP1 pathway. International journal of oncology. 2017;50:883-92
29. Lai KKY, Kweon SM, Chi F, Hwang E, Kabe Y, Higashiyama R. et al. Stearoyl-CoA Desaturase Promotes Liver Fibrosis and Tumor Development in Mice via a Wnt Positive-Signaling Loop by Stabilization of Low-Density Lipoprotein-Receptor-Related Proteins 5 and 6. Gastroenterology. 2017;152:1477-91
30. Bhatia B, Hsieh M, Kenney AM, Nahle Z. Mitogenic Sonic hedgehog signaling drives E2F1-dependent lipogenesis in progenitor cells and medulloblastoma. Oncogene. 2011;30:410-22
Author contact
![]() Corresponding author: Haojie Jin; Mailing Address: State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Renji Hospital, Shanghai Jiao Tong University School of Medicine, No. 25/Ln 2200 Xie-Tu Road, Shanghai 200032, China; Email: hjjin1986org
Corresponding author: Haojie Jin; Mailing Address: State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Renji Hospital, Shanghai Jiao Tong University School of Medicine, No. 25/Ln 2200 Xie-Tu Road, Shanghai 200032, China; Email: hjjin1986org
Citation styles
APA
Yang, C., Wang, S., Ruan, H., Li, B., Cheng, Z., He, J., Zuo, Q., Yu, C., Wang, H., Lv, Y., Gu, D., Jin, G., Yao, M., Qin, W., Jin, H. (2019). Downregulation of PDK4 Increases Lipogenesis and Associates with Poor Prognosis in Hepatocellular Carcinoma. Journal of Cancer, 10(4), 918-926. https://doi.org/10.7150/jca.27226.
ACS
Yang, C.; Wang, S.; Ruan, H.; Li, B.; Cheng, Z.; He, J.; Zuo, Q.; Yu, C.; Wang, H.; Lv, Y.; Gu, D.; Jin, G.; Yao, M.; Qin, W.; Jin, H. Downregulation of PDK4 Increases Lipogenesis and Associates with Poor Prognosis in Hepatocellular Carcinoma. J. Cancer 2019, 10 (4), 918-926. DOI: 10.7150/jca.27226.
NLM
Yang C, Wang S, Ruan H, Li B, Cheng Z, He J, Zuo Q, Yu C, Wang H, Lv Y, Gu D, Jin G, Yao M, Qin W, Jin H. Downregulation of PDK4 Increases Lipogenesis and Associates with Poor Prognosis in Hepatocellular Carcinoma. J Cancer 2019; 10(4):918-926. doi:10.7150/jca.27226. https://www.jcancer.org/v10p0918.htm
CSE
Yang C, Wang S, Ruan H, Li B, Cheng Z, He J, Zuo Q, Yu C, Wang H, Lv Y, Gu D, Jin G, Yao M, Qin W, Jin H. 2019. Downregulation of PDK4 Increases Lipogenesis and Associates with Poor Prognosis in Hepatocellular Carcinoma. J Cancer. 10(4):918-926.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.