Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(5):1275-1287. doi:10.7150/jca.28570 This issue Cite
Research Paper
Antitumor activity of histone deacetylase inhibitor chidamide alone or in combination with epidermal growth factor receptor tyrosine kinase inhibitor icotinib in NSCLC
Ningning Zhang1, Caixia Liang1, Wenya Song1, Dan Tao1, Jiarui Yao1, Shuai Wang1, Li Ma1, Yuankai Shi1 ![]() , Xiaohong Han1,2
, Xiaohong Han1,2 ![]()
1. Department of Medical Oncology, Beijing Key Laboratory of Clinical Study on Anticancer Molecular Targeted Drugs, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, 100021, China.
2. Department of Clinical Laboratory, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, 100021, China.
Received 2018-7-16; Accepted 2019-1-2; Published 2019-1-29
Citation:
Zhang N, Liang C, Song W, Tao D, Yao J, Wang S, Ma L, Shi Y, Han X. Antitumor activity of histone deacetylase inhibitor chidamide alone or in combination with epidermal growth factor receptor tyrosine kinase inhibitor icotinib in NSCLC. J Cancer 2019; 10(5):1275-1287. doi:10.7150/jca.28570. https://www.jcancer.org/v10p1275.htm
Other stylesAbstract
The study was performed to investigate the antitumor efficacy of histone deacetylase inhibitor (HDACi) chidamide alone or with epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI) icotinib in non-small cell lung cancer (NSCLC). The cell viability, cell cycle, apoptosis, protein expression, and the molecular mechanisms were explored among ten NSCLC cell lines with chidamide and icotinib alone or in combination, and further validated in xenograft models of nude mice. Chidamide significantly reduced the viability of A549, HCC827, HCC827IR (icotinib resistant) cells, increased the sensitivity of icotinib synergistically in EGFR-TKI resistant cell line, especially in H1975 cells. Chidamide alone or combined with icotinib induced cell cycle arrest by inhibiting the activation of RAS/MAPK, PI3K/AKT and/or JAK/STAT pathways, and caused apoptosis by activating caspase 3 and PARP. Chidamide alone or with icotinib suppressed β-catenin expression in HCC827, HCC827IR, and H1975 cells. The sensitivity of H1975 cells to icotinib was increased by chidamide through restoring E-cadherin expression. Furthermore, chidamide alone or in combination with icotinib inhibited HCC827IR and H1975 xenograft growth in athymic nude mice, respectively, with no appreciable side effects. Chidamide or combinating with icotinib exhibits antitumor activity in NSCLC cells, and has potential clinical implication for the treatment of NSCLC.
Introduction
In the past decade, molecularly targeted therapies for distinct patient molecular subgroups have led to a complete revolution in the treatment of non-small cell lung cancer (NSCLC). Epidermal growth factor receptor (EGFR) mutations which respond to tyrosine kinase inhibitor (TKI) were the first clinically relevant molecular alterations being well characterized in NSCLC. However, the overwhelming majority of these patients inevitably develop drug resistance. The resistance mechanism of EGFR-TKI is anfractuous, including secondary mutation (T790M), activation of alternative pathways (MET amplification), aberrance of the downstream pathways (KRAS mutations, loss of PTEN), epithelial-mesenchymal transition (EMT), etc [1, 2]. Among these mechanisms, T790M mutation and MET amplification are most common, accounting for 50% and 20%, respectively [3-5]. AZD9291 (osimertinib), a third-generation EGFR TKI, has shown promising clinical efficacy in patients who had acquired resistance to first- or second-generation EGFR-TKIs and was recently approved by Food and Drug Administration (FDA) for metastatic EGFR T790M mutation-positive NSCLC [6, 7]. However, acquired resistance to this drug eventually occurs after a median duration of response of ∼10 months on average [8-10]. Thus, drug resistance is the biggest barrier to hinder NSCLC patients to benefit from EGFR-TKI treatment. Therefore, exploring new therapeutic strategies is critical to prolong survival of NSCLC patients.
Histone deacetylase (HDAC) plays an important role in regulating chromatin conformation, protein-DNA interaction and gene expression [11]. Elevated expression or activity of HADC is involved in the mechanisms of development and progression of cancer [12], such as tumor suppressor silencing, cell migration, cell cycle abnormalities, signal transduction, cell adhesion, and so on. HDAC inhibitor (HDACi) can modulate cell responses through alterations in gene expression, inhibition of cell growth, induction of cell cycle arrest and cell apoptosis. It has been highlighted as a novel category of anti-cancer drugs in recent years. To date, several HDAC inhibitors, such as vorinostat, romidepsin, panobinostat, and belinostat, have been approved by FDA to treat hematologic cancers [11]. In addition, HDACi has also been investigated in the solid tumor as combination therapies. Previous studies have shown that the HDACi romidepsin and entinostat could enhance antitumor effect of EGFR-TKI in NSCLC cell lines [13, 14], and vorinostat combined with irreversible EGFR TKIs could overcome acquired resistance in EGFR T790M-mutated lung cancer [15], suggesting the potential clinical application value of HDACi in the treatment of NSCLC.
Chidamide (CS055), a benzamide HDAC inhibitor, is highly selective in class I HDAC1, 2, 3 and class IIb HDAC10 subtypes, which closely related to tumorigenesis development. Recent investigations have displayed antitumor effects mediated by chidamide in both hematologic and solid tumor malignancies [16-22]. And further, it could improve antitumor effect of gemcitabine in pancreatic cancer cells [23], and of platinum in NSCLC [24]. However, the antitumor effect of chidamide alone or in combination with EGFR-TKI in NSCLC has not yet been revealed. In this study, we exploit the therapeutic effect of chidamide alone or in combination with icotinib in NSCLC with varying mutation status in vitro and in vivo, aiming to provide more theoretical basis and experimental data for the clinical application of HDACi in NSCLC.
Materials and Methods
Cell lines and Drugs
Ten NSCLC cell lines were used in this study (Table 1). PC-9 (EGFR Exon19del E746-A750), HCC827 (EGFR Exon19del E746-A750), H1650 (EGFR Exon19del E746-A750 and PTEN del), H1975 (EGFR Exon 21 L858R and Exon 20 T790M), A549 (KRAS G12S), H460 (KRAS Q61H and PIK3CA E545K), H292 (EGFR and KRAS wild type) and Calu-3 (EGFR and KRAS wild type) cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). H1650GR (gefitinib resistant) and HCC827IR (icotinib resistant) cells were generated in our laboratory from their parental cell line H1650 and HCC827 by exposing the cells to gradually increased concentrations of gefitinib and icotinib for 10 months and 1 year, respectively. EGFR and KRAS status of these cell lines were validated by direct sequencing. PC-9, HCC827, H1650, H1650GR, H1975, HCC827IR, A549 and H460 cells were cultured in RPMI 1640 medium, and H292 cell line was cultured in DMEM medium. These nine cell lines were supplemented with 10% fetal bovine serum (FBS), 100U/mL penicillin and 100μg/mL streptomycin. Calu-3 cells were cultured in MEM medium, supplemented with 10% FBS, 2mM L-glutamine and 1% non-essential amino acids. Icotinib was provided by Zhejiang Beta Pharma Inc (China). Chidamide (CS055) was provided by Shenzhen Chipscreen Biosciences Ltd (China).
Cell viability assay
Exponentially growing cells were seeded into 96-well plates at 1-5×103cells/well (according to the growth rate of individual cell line used). Twenty-four hours later, the growth medium was replaced with fresh media containing different concentration of chidamide and icotinib alone or in combination. After the cells grew for up to 72h, 20μL/well of CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI, USA) was added to the cells and further incubated for 3h at 37°C. The absorbance was measured at 490nm with a micro-plate reader (Bio-rad, Hercules, CA, USA) and cell survival rate was expressed as the absorbance relative to that of controls. All of the compounds were dissolved in dimethyl sulfoxide (DMSO). Each experiment was carried out in five replicated wells for each drug concentration and repeated at least thrice. The effect of drug on growth inhibition could be assessed as follows: (mean OD value of drug treated cells - mean OD value of blank cells) / (mean OD value of control cells- mean OD value of blank cells). IC50 (50% inhibitory effect) values were obtained by interpolation from the dose-response curves. Combination index (CI) was calculated by applying the equation CI = IC50Ac/IC50As + IC50Bc/IC50Bs, where IC50As and IC50Bs are the concentration of inhibitor A and B required to produce 50% effect when used as single agents, and IC50Ac and IC50Bc are the concentration of inhibitor A and B required to produce 50% effect when used in combination. CI = 1 indicates that effects of combined inhibitors are additive, while CI < 1 indicates that the combination is synergistic [25, 26].
Flow cytometric cell cycle and apoptosis assay
After treating NSCLC cells with chidamide or icotinib alone, or in combination for 48h, cells were washed with phosphate buffer saline (PBS) solution, and incubated in DNA staining solution and permeabilization solution for 30min at room temperature according to the kit instructions (MultiSciences (LIANKE) Biotech Co., Ltd) and then measured by FACS cytometry (BD Biosciences, NJ, USA). The DNA histograms were analyzed using ModFit LT cell cycle analysis software (Verify Software House).
Cell apoptosis was performed using Annexin V-FITC cell apoptosis kit (Keygen Biotech, Nanjing, China) according to the kit instructions. Briefly, after treating with chidamide or icotinib alone, or in combination for 48h, the cells from both suspension and adherence were collected and co-incubated with Annexin V-fluorescein isothiocyanate (FITC) and PI, then measured on a Becton-Dickinson FACScan instrument (BD Biosciences, NJ, USA) using Cell Quest software. This analysis allows discrimination between viable (annexin V-/PI-), early (annexin V+/PI-) and late (annexin V+/PI+) apoptotic cells, as well as necrotic and/or part of late apoptotic (annexin V-/PI+) cells.
RNA isolation and qRT-PCR
Total RNA from cultured cells was isolated with the RNeasy Mini Kit (Qiagen, Hilden, NRW, Germany) and then reverse-transcribed into cDNA using the High-capacity cDNA Reverse transcription Kits (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions. The cDNA samples were further amplified by quantitative real-time PCR using Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) with synthetic primers as follows: E-cadherin, 5'-TGCCCAGAAAATGAAAAAGG-3' and 5'-GTGTATGTGGCAATGCGTTC-3'; Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5'-GAAGGTGAAGGTCGGAGTC-3' and 5'-GAAGATGGTGATGGGATTTC-3'. The PCR conditions were set in a 20 μL reaction mixture containing cDNA (1.5μL), primers (10μM each, 0.4μL) and SYBR green mix (2×, 10μL). The RT-reaction was carried out at 95°C for 10min, followed by 40 cycles of 95°C for 15s, and then 60°C for 60s. All samples were analyzed in triplicate and repeated three times. The relative expression levels of E-cadherin were normalized to GAPDH calculating by F=2-ΔCt or F=2-ΔΔCt.
Protein extraction and Western blot
After treated with chidamide or icotinib alone, or in combination for 48h, cells were washed with ice-cold PBS solution and lysed on ice with RIPA lysis buffer supplemented with protease inhibitor and protein phosphatase inhibitor cocktail (Applygen Technologies Inc., Beijing, China). The lysates were centrifuged at 14,000 rpm for 30 min at 4˚C and the supernatant was collected. Equivalent amounts of protein were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membranes. Primary antibodies against E-cadherin, β-catenin, EGFR, p-EGFR, STAT3, p-STAT3, MAPK, p-MAPK, AKT, p-AKT, MET, p-MET, caspase-3, cleaved caspase-3, PARP, and cleaved PARP were obtained from Cell Signaling Technology (CST, USA). GAPDH (CST) was used as a loading control. The signal was visualized with a chemiluminescent HRP substrate kit (Millipore, Boston, MA).
In vivo studies
HCC827IR (EGFR Exon19del E746-A750 and MET amplification) and H1975 (EGFR Exon 21 L858R and Exon 20 T790M) cells were selected for the in vivo experiment due to MET amplification and T790M mutation were the main reasons for EGFR-TKI resistance. One × 107 HCC827IR and H1975 cells were re-suspended in 100μL of serum-free RPMI 1640 medium and inoculated into the subcutaneous tissue of the right flank of 4-week-old female Bagg Albino (BALB/c) athymic nude mice (Beijing HFK Bioscience Co., Ltd, China). Tumor volumes were measured every other day and calculated from the following formula: tumor volume (mm3) = (length × width2)/2. Chidamide and icotinib, both dissolved with 0.2% carboxymethyl cellulose (CMC) and 0.1% Tween 80. For HCC827IR cell xenograft model, once the average tumor volume reached approximately 100mm3, mice were randomized into three groups (n=8) and administered orally thrice weekly for 5 weeks with vehicle (0.2% CMC and 0.1% Tween-80), icotinib (50mg/kg) and chidamide (25mg/kg), respectively. For H1975 cell xenograft model, once the average tumor volume reached approximately 100mm3, mice were randomized into five groups and administered orally thrice weekly for 5 weeks with vehicle (0.2% CMC and 0.1% Tween-80), icotinib (50mg/kg), chidamide (25mg/kg), chidamide (12.5mg/kg) plus icotinib (50mg/kg) and chidamide (25mg/kg) plus icotinib (50mg/kg), respectively. We promise that the study was performed according to the international, national and institutional rules considering animal experiments, clinical studies and biodiversity rights. The study protocol was approved by Animal Care and Use Committee of Cancer Hospital, Chinese Academy of Medical Sciences.
Statistical analysis
Student's t-test was used for data analysis. A two-sided P value of 0.05 was considered statistically significant. All statistical analyses were performed by using Statistical Package for the Social Sciences Version 17.0 Software (SPSS, Inc., Chicago, Illinois).
Results
Anti-proliferative activity of chidamide alone or in combination with icotinib
Ten NSCLC cell lines with varying EGFR and KRAS mutation status were chosen to study, including six cell lines containing mutant EGFR, two containing mutant KRAS, and two containing wild-type EGFR and KRAS. Majority of these cell lines were adenocarcinoma (Table 1). MTS assays were first used to evaluate the anti-proliferative effects of chidamide and icotinib as single agents. Dose-dependent growth inhibitory effects of chidamide or icotinib were observed in NSCLC cells (Figure 1A, Figure 1B). HCC827, HCC827IR, and A549 cells were sensitive to chidamide, HCC827 and PC-9 cells were sensitive to icotinib. Table 1 summarized the IC50 of these two drugs. Because HCC827 and PC-9 cells were very sensitive to icotinib, they were eliminated in the following combined treatment experiments, and the other eight icotinib non-sensitive cells were treated with varying concentrations of icotinib either in the absence or presence of chidamide. Compared with icotinib alone, all cells treated with icotinib plus chidamide exhibited decreased viability (Figure 1C). CI values were all less than 1 (Table 1), indicating that there was a synergistic interaction between icotinib and chidamide in these cells examined, and the synergistic effect was most meaningful in H1975 cells. These data suggest that the HDACi, chidamide, can enhance the cell-killing effects of icotinib in resistant NSCLC cells.
Figure 1
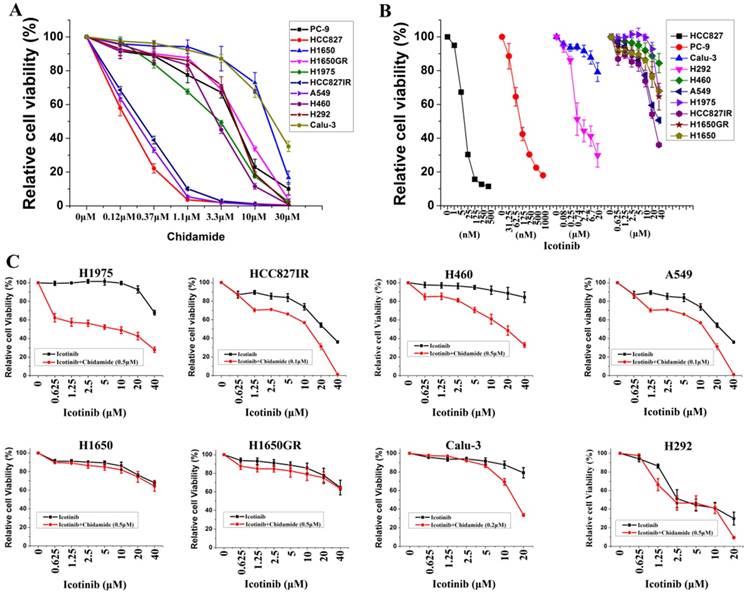
Effects of chidamide or combined with icotinib on the viability of NSCLC cells. A, representation pairs of cell viability curves of 10 NSCLC cell lines after treatment of serial dilutions of chidamide (72h); B, representation pairs of cell viability curves of 10 NSCLC cell lines after treatment of serial dilutions of icotinib (72h); C, chidamide synergistically increased the sensitivity of icotinib in eight icotinib non-sensitive NSCLC cells (72h). Experiment was repeated at least thrice. Control: DMSO.
Table 1
The IC50 values of chidamide and icotinib in NSCLC cell lines
| Cell line | Histology | Mutation status | Chidamide IC50±SD (μM) | Icotinib IC50±SD (μM) | |||
|---|---|---|---|---|---|---|---|
| EGFR mutational status | Other gene alternation | No Chidamide | Chidamidea | CI | |||
| HCC827 | AD | Exon 19 del (E746-A750) | / | 0.1549±0.019 | 0.0126±0.0001 | / | / |
| HCC827IR | AD | Exon 19 del (E746-A750) | MET amp | 0.227±0.027 | 25.115±2.240 | 7.58±0.13 (0.1) | 0.74 |
| PC-9 | AD | Exon 19 del (E746-A750) | / | 4.351±0.735 | 0.121±0.017 | / | / |
| H1650 | AD | Exon 19 del (E746-A750) | PTEN del | 14.510±1.362 | >100 | >100 (0.5) | 0.81 |
| H1650GR | AD | Exon 19 del (E746-A750) | PTEN del | 5.554±0.603 | >100 | >100 (0.5) | 0.89 |
| H1975 | AD | Exon 21 (L858R) T790M | / | 2.521±0.178 | 52.427±2.059 | 5.18±2.34(0.5) | 0.30 |
| A549 | AD | WT | KRAS (G12S) | 0.192±0.005 | 38.043±0.156 | 11.51±0.42 (0.1) | 0.82 |
| H460 | LC | WT | KRAS (Q61H) PIK3CA (E545K) | 2.848±0.192 | >100 | 17.89±4.2(0.5) | 0.32 |
| H292 | AD | WT | WT | 4.804±0.488 | 2.601±1.141 | 1.42±0.63(0.5) | 0.75 |
| Calu-3 | AD | WT | WT | 18.3±0.906 | >100 | 11.95±0.36 (0.2) | 0.54 |
Abbreviation: AD, adenocarcinoma; LC, large cell lung cancer; CI, combination index; SD, standard deviation.
Figure 2
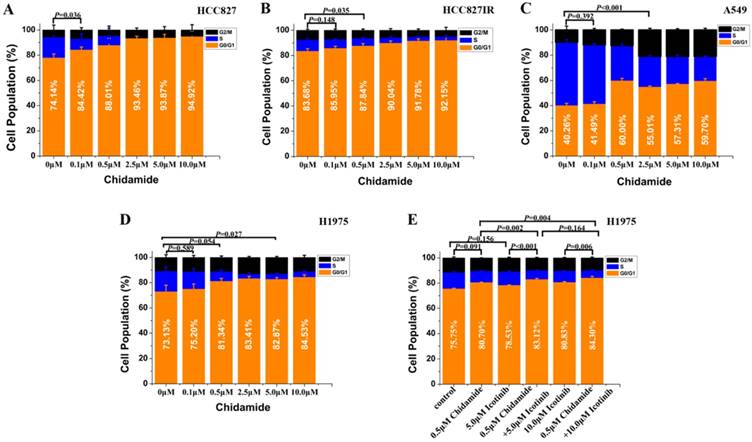
Effects of chidamide or combined with icotinib on the cell cycle distribution in NSCLC cells (48h). Experiment was repeated at least thrice. Control: DMSO.
Cell cycle effects of chidamide alone or in combination with icotinib
Flow cytometry was used to analyze the cell cycle distribution in HCC827, HCC827IR, A549 and H1975 cells exposed to chidamide, icotinib or chidamide plus icotinib for 48h. As shown in Figure 2, chidamide significantly altered the number of HCC827, HCC827IR and H1975 cells in the phase fractions with an increase in dose. In A549 cells, the cell fraction in G0/G1 phase did not change in a dose-dependent manner, but decrease of the cell fraction in S phase was observed with the increase dose of chidamide. Compared with icotinib or chidamide alone, co-treatment with icotinib and chidamide synergistically increased the cell fraction in G0/G1 phase of H1975 cells (P<0.05).
Effects of chidamide alone or in combination with icotinib on cell apoptosis
To investigate the mechanism of the cell death induced by chidamide or in combination with icotinib, changes of apoptosis after treatment were examined in HCC827, HCC827IR, A549, and H1975 cells. As illustrated in Figure 3A, chidamide significantly decreased the percent of viable cells in a dose-dependent manner, but this effect in H1975 cells was weaker than the other three cells. Results of western blot analysis of caspase-3 activation (cleaved caspase-3) and PARP cleavage (cleaved PARP), a key event that occurs upon caspase-3 activation during apoptosis [27], also showed that chidamide induced apoptosis of HCC827, HCC827IR, A549 and H1975 cells in direct relationship with the dosage (Figure 3B-3E). Moreover, apoptosis cells were markedly increased in H1975 cells with the co-treatment with chidamide and icotinib when compared with cells treated with either drug alone (P<0.05) (Figure 3F). Meanwhile, combined treatment with chidamide and icotinib significantly increased expression of cleaved caspase-3 and cleaved PARP in H1975 cells (Figure 3G). These findings suggest that chidamide or plus with icotinib could exert their inhibitory effects on NSCLC cells through triggering the apoptotic pathway.
Figure 3
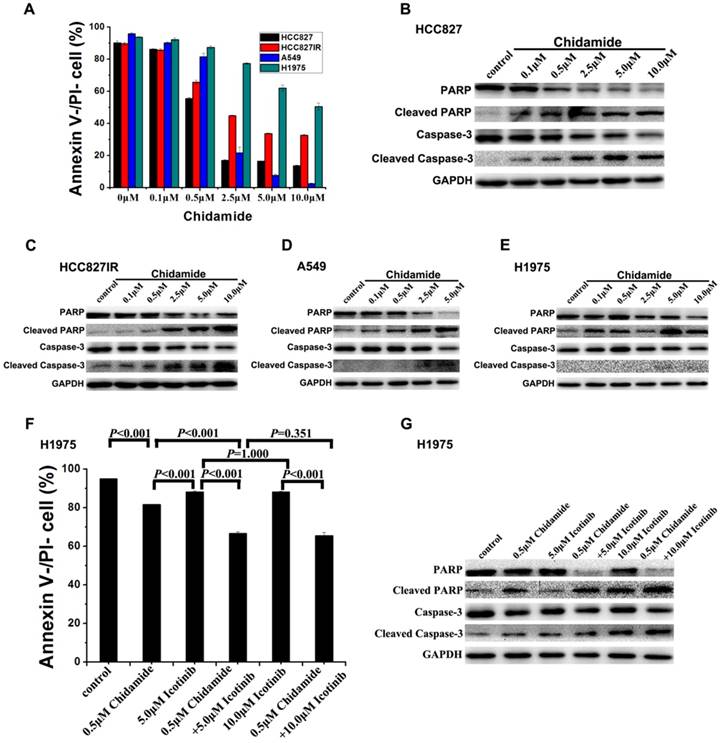
Effects of chidamide or combined with icotinib on the apoptosis in NSCLC cells. A, percentage of Annexin V-/PI- cells after treatment of serial dilutions of chidamide in HCC827, HCC827IR, A549 and H1975 cells (48h); B-E, expression and activation level of Caspase-3 and PARP after treatment of serial dilutions of chidamide in HCC827, HCC827IR, A549 and H1975 cells (48h); F, percentage of Annexin V-/PI- cells after treatment of chidamide or combined with icotinib in H1975 cells (48h). G, expression and activation level of Caspase-3 and PARP after treatment of chidamide or combined with icotinb in H1975 cells (48h). Experiment was repeated at least thrice. Control: DMSO.
Effects of chidamide alone or in combination with icotinib on the protein expression in receptor tyrosine kinase (PTK) signaling pathways
To elucidate the potential mechanisms involved in antitumor effects of chidamide alone or in combination with icotinib, western blot analysis was used to evaluate the effects on the protein expression and activation levels in PTK signaling pathways, including EGFR, MET and their downstream signaling pathways. As displayed in Figure 4A, with the concentration of chidamide increased, the expression levels of EGFR, p-EGFR, p-STAT3, AKT, p-AKT and MAPK had a tendency to decrease, but expression levels of STAT3 and p-MAPK were not significantly changed after chidamide treatment in HCC827 cells. In HCC827IR cells, chidamide tended to suppress the expression of EGFR, p-EGFR, MET, p-MET, p-AKT and p-MAPK in a dose-dependent manner, but no significant change was observed in expression levels of STAT3, p-STAT3, AKT and MAPK (Figure 4B). In A549 cells, EGFR, p-EGFR, p-STAT3, AKT, p-AKT, MAPK and p-MAPK were down-regulated with the increased dose of chidamide, however, STAT3 did not show the similar change (Figure 4C). In H1975 cells, chidamide alone had no obvious effects on the expression levels of these proteins (Figure 4D). But when combined with icotinib (5.0μM), AKT, especially p-AKT was slightly suppressed. And when icotinib concentration increased to 10.0μM, this effect became obvious, and p-EGFR was further down-regulated (Figure 4E).
Effects of chidamide alone or in combination with icotinib on the expression of E-cadherin and β-catenin
To further gain some insight into the molecular mechanism of the tumor inhibiting effect of chidamide alone or combined with icotinib, the expression levels of E-cadherin and β-catenin were further investigated. Western blot results showed that H460 and Calu-3 cells had no detectable expression of E-cadherin, H1975 cells had low level of E-cadherin expression, and the other seven cells had relatively high levels of E-cadherin expression before drug treatment. Meanwhile, with the increased dosage of chidamide, E-cadherin expression was significantly increased in H1975 cells, but no significant change was observed in chidamide sensitive cells. The results of qRT-PCR were in accordance with that of western blot analysis, suggesting that the protein expression of E-cadherin was regulated in mRNA level (Figure 5). As shown in Figure 5C-5F, a trend of down-regulation of the protein expression of β-catenin was observed in chidamide treated HCC827, HCC827IR, and H1975 cells. However, chidamide treatment had no effect on β-catenin expression in A549 cells. In H1975 cells, icotinib treatment had no obvious effect on β-catenin expression. But the expression of β-catenin was significantly decreased when combined with chidamide, which became more obvious with the increased icotinib concentration up to 10.0μM (Figure 5H).
Figure 4
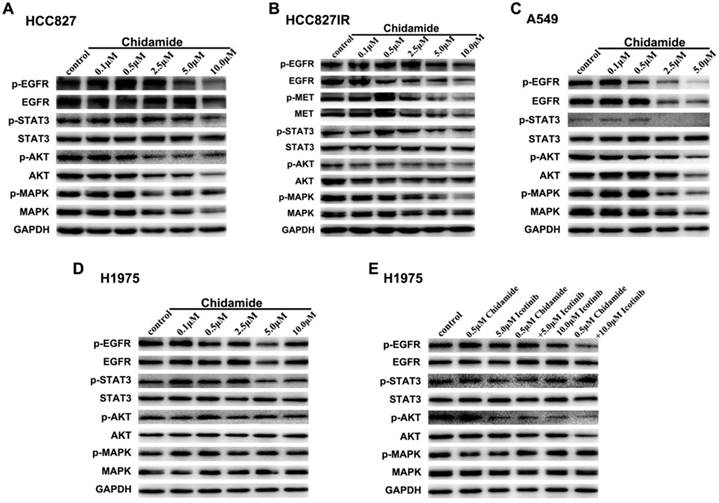
Effects of chidamide or combined with icotinib on the receptor tyrosine kinase (PTK) signaling molecules in NSCLC cells. A-D, chidamide inhibition of PTK signaling molecules in HCC827, HCC827IR, A549 and H1975 cells (48h). E, chidamide or combined with icotinib inhibition of PTK signaling molecules in H1975 cells (48h). Experiment was repeated at least thrice. Control: DMSO. GAPDH was used as a loading control.
Antitumor effects of chidamide alone or in combination with icotinib on EGFR-TKI resistant NSCLC cells in vivo
To further evaluate the antitumor effect of chidamide alone or combined with icotinib, athymic nude mice bearing established HCC827IR and H1975 tumor xenografts were treated by oral gavage with chidamide and icotinib, alone or in combination. For HCC827IR tumor xenografts, chidamide treatment significantly inhibited tumor growth when compared to the vehicle or icotinib-treated group (P<0.05, Figure 6A). For H1975 tumor xenografts, monotherapy with chidamide or icotinib resulted in only a marginal decrease in tumor volume, whereas co-administration with chidamide (no matter the dose) and icotinib led to a marked shrinkage of tumor (P<0.05, Figure 6C). Moreover, mice tolerated all treatments without significant weight loss or other apparent illness (Figure 6B and 6D). These findings are consistent with results in vitro.
Figure 5
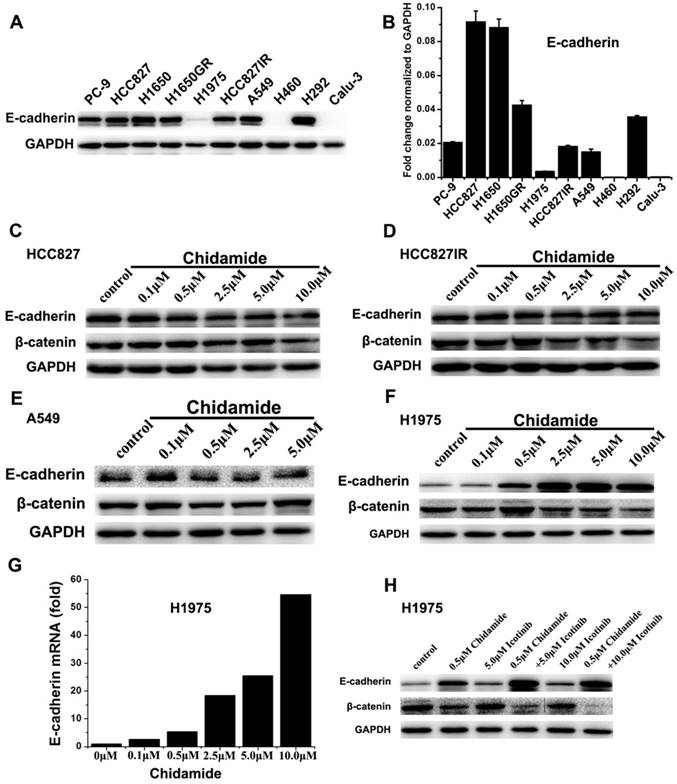
Effect of chidamide or combined with icotinib on E-cadherin and β-catenin expression in NSCLC cells. A, expression level of E-cadherin protein in 10 NSCLC cell lines; B, relative expression level of E-cadherin mRNA in 10 NSCLC cell lines (relative to GAPDH); C-F, expression of E-cadherin and β-catenin protein in HCC827, HCC827IR, A549 and H1975 cells after treatment of serial dilutions of chidamide (48h); G, expression of E-cadherin mRNA in H1975 cells after treatment of serial dilutions of chidamide (24h); H, expression of E-cadherin and β-catenin protein in H1975 cells after treatment of chidamide or combined with icotinib (48h). Experiment was repeated at least thrice. Control: DMSO.
Figure 6
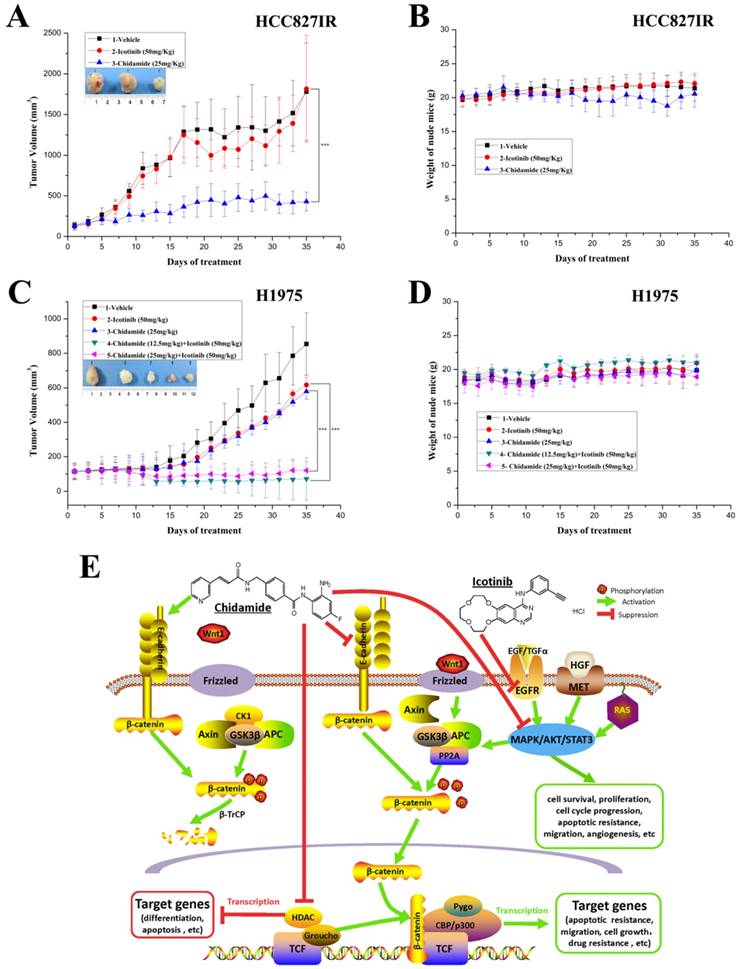
Antitumor activity in vivo and putative mechanisms of chidamide or in combination with icotinib in NSCLC. A and C, tumor growth curves showed that chidamide could significantly inhibit tumor growth in HCC827IR xenograft models compared to vehicle and icotinib; and chidamide plus icotinib could significantly inhibit tumor growth in H1975 xenograft models compared to vehicle, icotinib and chidamide, respectively; B and D, nude mice well tolerated the treatment; E, schematic illustration of the antitumor mechanisms of chidamide or combinated with icotinib in NSCLC. ***P<0.001.
Discussion
In the present study, we explored the antitumor activity of HDAC inhibitor chidamide alone or in combination with EGFR-TKI icotinib in NSCLC with varying mutation status in vitro and vivo. Our data indicated that chidamide had strong inhibitory effect on the growth of HCC827 cells (EGFR Exon19del E746-A750), HCC827IR cells (EGFR Exon 19del E746-A750 and MET amplification), and A549 cells (KRAS G12S). Co-treatment with chidamide and icotinib showed synergy in NSCLC cells non-sensitive to EGFR-TKI, and the synergistic effect was most meaningful in H1975 cells (EGFR Exon 21 L858R and Exon 20 T790M). Chidamide alone or in combination with icotinib induced cell cycle arrest and increased apoptotic cell fraction by inhibiting the activation of RAS/MAPK, PI3K/AKT and/or JAK/STAT pathways and inducing the activation of caspase-3 and PARP cleavage, respectively. Chidamide alone or in combination with icotinib downregulated the expression level of β-catenin (core molecular of Wnt signaling pathway) in HCC827, HCC827IR, and H1975 cells. Chidamide increased the sensitivity of H1975 cells to icotinib, at least in part, by restoring E-cadherin expression. Furthermore, chidamide monotherapy, and co-treatment with chidamide and icotinib showed antitumor activity against the EGFR-mutant, EGFR-TKI-resistant HCC827IR and H1975 tumor xenograft models in BALB/c nude mice, respectively. Most importantly, weight loss or other side effects were not observed in vivo study. These results suggest that HDAC inhibitor chidamide has potential therapeutic value in the treatment of NSCLC.
RAS/MAPK, PI3K/AKT, and JAK/STAT pathways, which stimulate mitosis, leading to cell proliferation and inhibition of apoptosis [28, 29], are three main downstream pathways of PTKs, including EGFR and MET [2, 30, 31] (Figure 6E). Hyperactive of these pathways have been associated with resistance to EGFR-TKIs in NSCLC. Previous studies reported that HDAC inhibitor chidamide could inhibit the PI3K/AKT and RAS/MAPK pathways, which resulted in cell cycle arrest and apoptosis in colon cancer cell [19]. In the current study, chidamide monotherapy mainly inhibited the PI3K/AKT pathway in HCC827 cells. While, in A549 cells the above three pathways were inhibited in different levels. In HCC827IR cells, chidamide down-regulated the expression and activation of EGFR and MET, and mainly inhibited the RAS/MAPK pathway. A recent study showed that HDAC inhibitor SAHA activated the phosphorylation of EGFR and increased the cell sensitivity to erlotinib in pancreatic cancer [32]. However, in the present study, chidamide monotherapy did not have obvious effect on signal pathway in H1975 cells, but when combined with a relatively high concentration of icotinib, the PI3K/AKT pathway was significantly inhibited, which resulted in arresting cell cycle and promoting apoptosis. All these data indicated that chidamide alone or in combination with icotinib showed its antitumor effect by inhibiting PTK signaling pathways in NSCLC with different genetic background.
EMT was closely associated with tumor metastasis, invasion, and therapeutic resistance [33]. E-cadherin, a prototypical EMT marker, usually acted as a tumor suppressor in epithelial cells. Its dysregulation affects the signaling pathways, which induce cell proliferation, motility, and metastatic potential, thereby influencing the survival of cancer patients [34, 35]. Multiple complex mechanisms are involved in the EMT progress. Evidences suggest that PI3K/AKT activation degrades E-cadherin and promotes cell invasion and migration, while inhibition of PI3K/AKT signaling pathway reduces EMT [36]. In NSCLC, loss expression of E-cadherin was not only correlated with a poor prognosis [37], but also associated with EGFR-TKI resistance [38], which was supposed to be caused by triggering the activation of PI3K/AKT signaling pathway [39]. Previous studies showed that restoring E-cadherin expression by HDAC inhibitor MS-275 increased the sensitivity to EGFR-TKI in lung cancer cells [14, 40]. In the present study, in H1975 cells, which has lower expression of E-cadherin, chidamide upregulated E-cadherin expression both in mRNA and protein level with a dose-dependent manner. The co-treatment of chidamide and icotinib inhibited the activation of the PI3K/AKT pathway, leading to cell cycle arrest and apoptosis. These results indicate that the synergistic effect of chidamide and icotinib in H1975 cells might be mediated, at least in part, by reversing EMT and inhibiting the PI3K/AKT pathway. Therefore, it would be important to consider the influence of EMT in the application of HDACi in the development of treatment against EGFR-TKI resistance.
Wnt signaling pathway is crucial in many physiological processes, including embryogenesis and development of cancer. It was demonstrated that this pathway correlated with tumorigenesis, drug resistance and prognosis in NSCLC [41, 42]. β-catenin is the core mediator in Wnt pathway and its overexpression is associated with EGFR-TKI resistance in NSCLC cells [43]. β-catenin functions in a dual manner depending on the intracellular localization [44]. In absence of Wnt signals, β-catenin acts in cell-cell adhesion by linking E-cadherin at the plasma membrane and free cytosolic β-catenin is targeted for proteasome breakdown by a multiprotein degradation complex [45]. In absence of nuclear β-catenin, various additional proteins with transcriptionally repressive properties, such as members of groucho/transducin-like enhancer of split (TLE) protein family, are recruited and serve as intermediary molecules to recruit HDAC, causing inhibition of target gene transcription by condensing chromatin [46]. However, in many cancer cells, the expression or activity of HDAC is elevated, which leads to dysregulated expression of target genes involved in a broad range of cell functions, such as tumor suppression, cell cycle, differentiation, and DNA repair [47]. Indeed, previous studies demonstrated that HDACi treatment affected various genes expressed in cancer cells, including transcriptional repressors and non-transcriptional proteins, resulted in induction of differentiation and apoptosis, inhibition of angiogenesis and invasion and, importantly, led to synergistic effects with other anti-cancer drugs [48].
On the contrary, when Wnt signaling cascade is activated, cytoplasmic β-catenin evades degradation, accumulates in the cytoplasm and finally translocates to the nucleus. And then, complexes with members of the T-cell factor (TCF)/lymphoid enhancer-binding factor (LEF) family of transcription factor, which are known to transcribe tumorigenesis-enhancing proteins, to recruit basal transcription machinery and transcription factors, lead to expression of various target genes, including cyclin D1 and c-Myc oncogenes [49, 50]. Multiple processes, including hepatocyte growth factor (the ligand of MET), E-cadherin and PKA (protein kinase A), influence β-catenin levels. It has been suggested that over-activation of EGFR/MET signaling pathways enhance nuclear accumulation of active β-catenin, and results in the activation of transcriptional targets [42, 51, 52]. The PI3K/AKT pathway positively stimulates the Wnt/β-catenin cascade by increasing intracellular β-catenin levels through AKT phosphorylation, which induces the movement of β-catenin into nucleus and then promotes its induction transcription through TCF/LEF. A simple reduction of cellular E-cadherin was shown to be sufficient to increase significantly free cellular β-catenin, and an abrogation of E-cadherin-mediated adhesion correlated with an increase of the transcription of β-catenin target genes [53] (Figure 6E). HDAC inhibitors downregulate RTK signaling, including EGFR and MET, by inhibiting activation of PI3K/AKT, RAS/MAPK and other pathways [54]. In the present study, chidamide lowered β-catenin expression and inhibited the activation of PI3K/AKT or RAS/MAPK pathway with different degree in HCC827 and HCC827IR cells. However, it did not significantly affect the expression of E-cadherin. These results provided a putative mechanism that in addition to the direct effect of HDAC enzymes inhibition, which resulted in increased histone acetylation, and thereby inducing an open chromatin conformation and transcription of previously dormant genes, mediate antitumor function. Chidamide also inhibited EGFR, MET or their downstream signaling pathways and played direct or indirect (by inhibiting the activation of Wnt/β-catenin signal transduction pathway) antitumor effect in HCC827 and HCC827IR cells (Figure 6E). Chidamide significantly upregulated E-cadherin expression in H1975 cells, while the combined treatment with chidamide and icotinib significantly downregulated β-catenin expression and inhibited the activation of PI3K/AKT pathway. Therefore, apart from above-mentioned mechanisms, chidamide and icotinib may exert its synergistic effect via E-cadherin/β-catenin pathway in H1975 cells (Figure 6E). In addition, chidamide had obvious effect of proliferation inhibition, cycle arrest and apoptosis induction on A549 cells, which harbors KRAS mutation. However, chidamide did not regulate its expression of E-cadherin and β-catenin. In consideration of the cross-talk between gene and protein, whether Wnt signaling pathway was involved in the antitumor effect of chidamide on A549 cells should be further explored. However, the direct effect of HDAC enzymes inhibition of chidamide on NSCLC cells with different genetic background, was not detected, which is a limitation in our study. It is worth to comprehensively investigating in the further study.
Chidamide, as a novel HDACi newly designed and synthesized in China, has shown inhibitory activity against several cancer types including leukemia [16], lymphoma [17], multiple myeloma [18], colon cancer [19], hepatocellular carcinoma [20], and pancreatic cancer [21]. Meanwhile, it has displayed synergistic effect with gemcitabine in pancreatic cancer cells [23], and with platinum in NSCLC [24]. Our results showed that chidamide alone or in combination with icotinib induced cell cycle arrest and apoptosis in NSCLC cells, both of which eventually result in cell proliferation inhibition. All these data indicate that chidamide alone or in combination with icotinib may benefit NSCLC patients, especially those who are resistant to EGFR-TKI therapy.
Although first-generation EGFR-TKI resistant NSCLC patients harboring EGFR T790M mutation or MET amplification have the choice of third-generation EGFR-TKI, such as AZD9291, or MET inhibitor, such as crizotinib, the economic burden of these drugs limits clinical application in current stage. The results presented in this study provide evidence that chidamide may be a potentially useful HDACi for the clinical treatment of NSCLC, especially for EGFR-TKI resistant patients harboring EGFR T790M mutation or MET amplification. However, the evidence is limited as only one cell line harboring EGFR T790M mutation and MET amplification was used respectively in the present study. Combination of PC (paclitaxel and carboplatin) and chidamide is currently being evaluated in a multi-center phase II clinical trial (NCT01836679), which aimed to explore the clinical benefits of this combination regimen for NSCLC patients with negative EGFR mutation and to set up a correlation analysis between the level of E-cadherin and the efficacy. We are looking forward that the results of the clinical trial can provide valuable reference for the clinical trial design and conduct of the combination treatment with chidamide and EGFR-TKI in NSCLC.
Acknowledgements
We would like to thank for Zhejiang Beta Pharma Inc and Shenzhen Chipscreen Biosciences Ltd for providing icotinib and chidamide, respectively.
This study was supported by National High Technology Research and Development Program of China (863 Program) (2011AA02A110), Chinese National Major Project for New Drug Innovation (2017ZX09304015), Beijing Municipal Science and Technology Commission Major Project (D141100000214003, D141100000214005), Beijing Key Laboratory of Clinical Study on Anticancer Molecular Targeted Drugs-2012 Ladder Program (Z121102009212055), National Natural Science Foundation of China (81871739, 81472752), Specialized Research Fund for the Doctoral Program of Higher Education (20131106130004) and Peking Union Medical College Graduate Students Innovation Found (2012-1002-054).
Part of this study was displayed as a poster at the European Lung Cancer Conference (ELCC 2015, April15-18; Geneva. No.49P), the World Conference on Lung Cancer (WCLC 2015, September 06-09, Denver. No.3090), the European Cancer Congress (ECCO 2015, September 25-29, Vienna. No. P251).
Authors' contributions
HXH, SYK and ZNN conceived and designed the study. ZNN, LCX, SWY, TD, YJR and WS performed the experiments. ZNN wrote the draft of the manuscript. HXH and SYK reviewed and edited the manuscript. All authors read and approved the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ahsan A. Mechanisms of Resistance to EGFR Tyrosine Kinase Inhibitors and Therapeutic Approaches: An Update. Adv Exp Med Biol. 2016;893:137-53
2. Huang L, Fu L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm Sin B. 2015;5:390-401
3. Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M. et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786-92
4. Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK. et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105:2070-5
5. Remon J, Moran T, Majem M, Reguart N, Dalmau E, Marquez-Medina D. et al. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer: a new era begins. Cancer Treat Rev. 2014;40:93-101
6. Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ. et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4:1046-61
7. Janne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS. et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372:1689-99
8. Planchard D, Loriot Y, Andre F, Gobert A, Auger N, Lacroix L. et al. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann Oncol. 2015;26:2073-8
9. Kim TM, Song A, Kim DW, Kim S, Ahn YO, Keam B. et al. Mechanisms of Acquired Resistance to AZD9291, a Mutation-Selective, Irreversible EGFR Inhibitor. J Thorac Oncol. 2015;10:1736-44
10. Eberlein CA, Stetson D, Markovets AA, Al-Kadhimi KJ, Lai Z, Fisher PR. et al. Acquired Resistance to the Mutant-Selective EGFR Inhibitor AZD9291 Is Associated with Increased Dependence on RAS Signaling in Preclinical Models. Cancer Res. 2015;75:2489-500
11. Yoon S, Eom GH. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med J. 2016;52:1-11
12. Khan AU, Krishnamurthy S. Histone modifications as key regulators of transcription. Front Biosci. 2005;10:866-72
13. Zhang W, Peyton M, Xie Y, Soh J, Minna JD, Gazdar AF. et al. Histone deacetylase inhibitor romidepsin enhances anti-tumor effect of erlotinib in non-small cell lung cancer (NSCLC) cell lines. J Thorac Oncol. 2009;4:161-6
14. Witta SE, Gemmill RM, Hirsch FR, Coldren CD, Hedman K, Ravdel L. et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006;66:944-50
15. Lee TG, Jeong EH, Kim SY, Kim HR, Kim CH. The combination of irreversible EGFR TKIs and SAHA induces apoptosis and autophagy-mediated cell death to overcome acquired resistance in EGFR T790M-mutated lung cancer. Int J Cancer. 2015;136:2717-29
16. Gong K, Xie J, Yi H, Li W. CS055 (Chidamide/HBI-8000), a novel histone deacetylase inhibitor, induces G1 arrest, ROS-dependent apoptosis and differentiation in human leukaemia cells. Biochem J. 2012;443:735-46
17. Li YY, Wang YF, Wang J, Ke XY. [Effect of chidamide on human B lymphoma cell lines and its mechanisms]. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2012;20:893-9
18. Xu L, Tang HL, Gong X, Xin XL, Dong Y, Gao GX. et al. Inducing effect of chidamide on apoptosis of multiple myeloma cells and its relerance to DNA damage response. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2015;23:450-4
19. Liu L, Chen B, Qin S, Li S, He X, Qiu S. et al. A novel histone deacetylase inhibitor Chidamide induces apoptosis of human colon cancer cells. Biochem Biophys Res Commun. 2010;392:190-5
20. Wang H, Guo Y, Fu M, Liang X, Zhang X, Wang R. et al. Antitumor activity of Chidamide in hepatocellular carcinoma cell lines. Mol Med Rep. 2012;5:1503-8
21. Zhao B, He T. Chidamide, a histone deacetylase inhibitor, functions as a tumor inhibitor by modulating the ratio of Bax/Bcl-2 and P21 in pancreatic cancer. Oncol Rep. 2015;33:304-10
22. Ning ZQ, Li ZB, Newman MJ, Shan S, Wang XH, Pan DS. et al. Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother Pharmacol. 2012;69:901-9
23. Qiao Z, Ren S, Li W, Wang X, He M, Guo Y. et al. Chidamide, a novel histone deacetylase inhibitor, synergistically enhances gemcitabine cytotoxicity in pancreatic cancer cells. Biochem Biophys Res Commun. 2013;434:95-101
24. Zhou Y, Pan DS, Shan S, Zhu JZ, Zhang K, Yue XP. et al. Non-toxic dose chidamide synergistically enhances platinum-induced DNA damage responses and apoptosis in Non-Small-Cell lung cancer cells. Biomed Pharmacother. 2014;68:483-91
25. Zhao L, Wientjes MG, Au JL. Evaluation of combination chemotherapy: integration of nonlinear regression, curve shift, isobologram, and combination index analyses. Clin Cancer Res. 2004;10:7994-8004
26. Bliesath J, Huser N, Omori M, Bunag D, Proffitt C, Streiner N. et al. Combined inhibition of EGFR and CK2 augments the attenuation of PI3K-Akt-mTOR signaling and the killing of cancer cells. Cancer Lett. 2012;322:113-8
27. Robertson JD, Orrenius S, Zhivotovsky B. Review: nuclear events in apoptosis. J Struct Biol. 2000;129:346-58
28. Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358:1160-74
29. Talapatra S, Thompson CB. Growth factor signaling in cell survival: implications for cancer treatment. J Pharmacol Exp Ther. 2001;298:873-8
30. Appleman LJ. MET signaling pathway: a rational target for cancer therapy. J Clin Oncol. 2011;29:4837-8
31. Garajova I, Giovannetti E, Biasco G, Peters GJ. c-Met as a Target for Personalized Therapy. Transl Oncogenomics. 2015;7:13-31
32. Park SJ, Kim SM, Moon JH, Kim JH, Shin JS, Hong SW. et al. SAHA, an HDAC inhibitor, overcomes erlotinib resistance in human pancreatic cancer cells by modulating E-cadherin. Tumour Biol. 2016;37:4323-30
33. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265-73
34. Lewis-Tuffin LJ, Rodriguez F, Giannini C, Scheithauer B, Necela BM, Sarkaria JN. et al. Misregulated E-cadherin expression associated with an aggressive brain tumor phenotype. PLoS ONE. 2010;5:e13665
35. Herzig M, Savarese F, Novatchkova M, Semb H, Christofori G. Tumor progression induced by the loss of E-cadherin independent of beta-catenin/Tcf-mediated Wnt signaling. Oncogene. 2007;26:2290-8
36. Byeon HK, Na HJ, Yang YJ, Ko S, Yoon SO, Ku M. et al. Acquired resistance to BRAF inhibition induces epithelial-to-mesenchymal transition in BRAF (V600E) mutant thyroid cancer by c-Met-mediated AKT activation. Oncotarget. 2017;8:596-609
37. Yang YL, Chen MW, Xian L. Prognostic and clinicopathological significance of downregulated E-cadherin expression in patients with non-small cell lung cancer (NSCLC): a meta-analysis. PLoS One. 2014;9:e99763
38. Thomson S, Buck E, Petti F, Griffin G, Brown E, Ramnarine N. et al. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005;65:9455-62
39. Barr S, Thomson S, Buck E, Russo S, Petti F, Sujka-Kwok I. et al. Bypassing cellular EGF receptor dependence through epithelial-to-mesenchymal-like transitions. Clin Exp Metastasis. 2008;25:685-93
40. Suda K, Tomizawa K, Fujii M, Murakami H, Osada H, Maehara Y. et al. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J Thorac Oncol. 2011;6:1152-61
41. Stewart DJ. Wnt signaling pathway in non-small cell lung cancer. J Natl Cancer Inst. 2014;106:djt356
42. Botting GM, Rastogi I, Chhabra G, Nlend M, Puri N. Mechanism of Resistance and Novel Targets Mediating Resistance to EGFR and c-Met Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer. PLoS ONE. 2015;10:e0136155
43. Fang X, Gu P, Zhou C, Liang A, Ren S, Liu F. et al. beta-Catenin overexpression is associated with gefitinib resistance in non-small cell lung cancer cells. Pulm Pharmacol Ther. 2014;28:41-8
44. Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009;28:151-66
45. Kimelman D, Xu W. beta-catenin destruction complex: insights and questions from a structural perspective. Oncogene. 2006;25:7482-91
46. Cavallo RA, Cox RT, Moline MM, Roose J, Polevoy GA, Clevers H. et al. Drosophila Tcf and Groucho interact to repress Wingless signalling activity. Nature. 1998;395:604-8
47. Walkinshaw DR, Yang XJ. Histone deacetylase inhibitors as novel anticancer therapeutics. Curr Oncol. 2008;15:237-43
48. Ocker M, Schneider-Stock R. Histone deacetylase inhibitors: signalling towards p21cip1/waf1. Int J Biochem Cell Biol. 2007;39:1367-74
49. Teng Y, Wang X, Wang Y, Ma D. Wnt/beta-catenin signaling regulates cancer stem cells in lung cancer A549 cells. Biochem Biophys Res Commun. 2010;392:373-9
50. Mazieres J, He B, You L, Xu Z, Jablons DM. Wnt signaling in lung cancer. Cancer Lett. 2005;222:1-10
51. Hoschuetzky H, Aberle H, Kemler R. Beta-catenin mediates the interaction of the cadherin-catenin complex with epidermal growth factor receptor. J Cell Biol. 1994;127:1375-80
52. Danilkovitch-Miagkova A, Miagkov A, Skeel A, Nakaigawa N, Zbar B, Leonard EJ. Oncogenic mutants of RON and MET receptor tyrosine kinases cause activation of the beta-catenin pathway. Mol Cell Biol. 2001;21:5857-68
53. Voronkov A, Krauss S. Wnt/beta-catenin signaling and small molecule inhibitors. Curr Pharm Des. 2013;19:634-64
54. Gallagher SJ, Tiffen JC, Hersey P. Histone Modifications, Modifiers and Readers in Melanoma Resistance to Targeted and Immune Therapy. Cancers (Basel). 2015;7:1959-82
Author contact
![]() Corresponding authors: Xiaohong Han, E-mail: hanxhac.cn; Yuankai Shi, E-mail: syuankaiac.cn
Corresponding authors: Xiaohong Han, E-mail: hanxhac.cn; Yuankai Shi, E-mail: syuankaiac.cn
Citation styles
APA
Zhang, N., Liang, C., Song, W., Tao, D., Yao, J., Wang, S., Ma, L., Shi, Y., Han, X. (2019). Antitumor activity of histone deacetylase inhibitor chidamide alone or in combination with epidermal growth factor receptor tyrosine kinase inhibitor icotinib in NSCLC. Journal of Cancer, 10(5), 1275-1287. https://doi.org/10.7150/jca.28570.
ACS
Zhang, N.; Liang, C.; Song, W.; Tao, D.; Yao, J.; Wang, S.; Ma, L.; Shi, Y.; Han, X. Antitumor activity of histone deacetylase inhibitor chidamide alone or in combination with epidermal growth factor receptor tyrosine kinase inhibitor icotinib in NSCLC. J. Cancer 2019, 10 (5), 1275-1287. DOI: 10.7150/jca.28570.
NLM
Zhang N, Liang C, Song W, Tao D, Yao J, Wang S, Ma L, Shi Y, Han X. Antitumor activity of histone deacetylase inhibitor chidamide alone or in combination with epidermal growth factor receptor tyrosine kinase inhibitor icotinib in NSCLC. J Cancer 2019; 10(5):1275-1287. doi:10.7150/jca.28570. https://www.jcancer.org/v10p1275.htm
CSE
Zhang N, Liang C, Song W, Tao D, Yao J, Wang S, Ma L, Shi Y, Han X. 2019. Antitumor activity of histone deacetylase inhibitor chidamide alone or in combination with epidermal growth factor receptor tyrosine kinase inhibitor icotinib in NSCLC. J Cancer. 10(5):1275-1287.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.