Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(7):1642-1650. doi:10.7150/jca.28266 This issue Cite
Research Paper
A Comparative Study of Genetic Profiles of Key Oncogenesis-Related Genes between Primary Lesions and Matched Lymph Nodes Metastasis in Lung Cancer
Yuqiao Chen1, Beibei Mao2, Xiong Peng1, Yuan Zhou1, Kun Xia1, Hao Guo2, Hong Jiang3, Guo Wang4, Wei Zhuang1 ![]()
1. Department of Thoracic Surgery, Xiangya Hospital of Central South University, 410008 Changsha, Hunan, People's Republic of China.
2. Beijing Genecast Biotechnology Co. 100000 Beijing, People's Republic of China.
3. Department of Neurology, Xiangya Hospital, Central South University, 410008, Changsha, Hunan, People's Republic of China.
4. Department of Clinical Pharmacology, Xiangya Hospital, Central South University, Changsha 410008, Hunan, People's Republic of China.
Received 2018-7-2; Accepted 2019-1-10; Published 2019-3-3
Citation:
Chen Y, Mao B, Peng X, Zhou Y, Xia K, Guo H, Jiang H, Wang G, Zhuang W. A Comparative Study of Genetic Profiles of Key Oncogenesis-Related Genes between Primary Lesions and Matched Lymph Nodes Metastasis in Lung Cancer. J Cancer 2019; 10(7):1642-1650. doi:10.7150/jca.28266. https://www.jcancer.org/v10p1642.htm
Other stylesAbstract
Background: The genetic heterogeneity between primary and metastatic lesions has rarely studied.
Purpose: This study aims to compare the mutation profiling of key cancer-related genes in primary lung cancers and their matched lymph nodes (LN) metastasis.
Methods: The gene mutation profiling of both primary cancers and their matched LN metastasis was constructed using a hybridization capture-based massively parallel sequencing assay and the sequencing of 1,408 key cancer genes in 37 NSCLC patients.
Results: There are differences in the repertoire of somatic mutations and CNVs between primary lesions and metastatic lymphatic lesions in NSCLC. More mutations were discovered in the primary lesion than LN metastasis lesion, indicating that tumor mutation burden is higher in primary lesion than that in LN metastasis lesion. The copy number profiles were largely preserved through the progression of the metastasis in lung cancer individually. Regarding to the mutation context, more prevailing of signature 1 in LN metastasis lesion was observed than that of primary lesion, indicating that the spontaneous deamination of cytosine may contribute to the accumulation of mutations during tumor development and immigration. Furthermore, the LN metastasis exclusive somatic mutation (LME-SMs) genes were enriched in peptidyl-tyrosine phosphorylation, peptidyl-tyrosine modification, protein tyrosine kinase activity, and transmembrane receptor protein kinase. The KEGG pathway analysis revealed that these genes might participate in the PI3K-Akt signaling pathway, MAPK signaling pathway and Ras signaling pathway.
Conclusion: Since the heterogeneity of genetic profiling exists between primary lesion and LN metastasis lesion, multi-region lesion mutation profiling is required to fully understand the progression of NSCLC, and develop an appropriate therapeutic guide. Mutations in genes involved in cell proliferation, invasion and migration may be the genetic mechanism of metastasis.
Keywords: genetic heterogeneity, non-small cell lung cancer, metastasis, next generation sequencing
Introduction
Lung cancer is one of the most common malignant tumors in the world, and non-small cell lung cancer (NSCLC) accounts for more than 75% of all lung cancer cases[1, 2]. The identification of oncogenic mutations using variant sequencing techniques has been a routine clinical practice, which may guide selection of target agents and help to improve the clinical outcomes of lung cancer patients [3-7]. It is easy for cancer cells to develop resistance to targeting therapeutic drugs, which makes it hard to absolutely cure cancers [8, 9]. Intra-tumor genetic heterogeneity has been detected in previous studies using massively parallel sequencing analyses of multiple regions of primary lung cancer[10, 11], and this may be one of the main reasons for the resistance to targeted therapies. It is plausible that the metastatic process and the progression from primary to metastatic disease may result in genetic heterogeneity, along with molecular evolution [11].
The most common metastatic site of lung cancer is regional lymph nodes (LN). The spread of cancer cells from the primary tumor to regional lymph nodes is the main poor prognostic factor in NSCLC of stage I-III [12, 13]. The analysis of mutations of both the primary tumor and LN metastasis lesion is of great importance to understand the molecular evolutionary processes of lung cancer, and the process of lymphatic metastasis [14]. This would also help to develop a suitable therapeutic guide, and prolong the survival of patients with locally advanced NSCLC [15]. In the present study, genetic mutations in 1,408 key cancer genes were analyzed by hybridization capture-based massively parallel sequencing assay to define the extent of genetic heterogeneity between primary tumors and their respective LN metastasis and reflect the genetic evolutionary process of lymphatic metastasis in locally advanced NSCLC.
Materials and Methods
Patients and samples
This study was approved by the Local Ethics Committee of Xiangya Hospital of Central South University. All patients provided a written informative consent. A total of 37 patients pathologically diagnosed and confirmed with NSCLC (including 14 lung squamous cell carcinoma (LUSC) and 23 lung adenocarcinoma (LUAD)) and a LN metastasis lesion, but without distant metastasis, were enrolled into the present study. Blood samples, primary tumor tissues and metastasis LN were collected from each patient. Frozen sections were made from the freshly frozen tumor tissues and metastasis LN. One slide of each tissue sample was lightly stained with hematoxylin and eosin (H&E) to re-examine the presence of cancer cells. The tumor cell content was calculated by an independent lung pathologist. The histopathologic assessment confirmed that all collected tumor tissue samples had ≥10% viable tumor cell content. The baseline characteristics of the included patients are presented in Table 1.
Table 1
Baseline characteristics of the patients
| Characteristic | Number (%) |
|---|---|
| Age (year) | |
| ≥60 | 21(56.8%) |
| <60 | 16(43.2%) |
| Gender | |
| Male | 25(67.6%) |
| Female | 12(32.4%) |
| Smoking history | |
| Smoking | 24(64.9%) |
| Nonsmoking | 13(35.1%) |
| Histology | |
| LUAD | 23(62.2%) |
| Squamous | 14(37.8%) |
| Stage | |
| IIb | 11(29.7%) |
| IIIa | 21(56.8%) |
| IIIb | 5(13.5%) |
Targeted sequencing
Genomic DNA was extracted from two 4-μm fresh frozen tissue slices using a black PREP FFPE DNA Kit (Analytik Jena, GER). The DNA samples were quantified using a Qubit dsDNA HS Assay kit (Life Technologies, USA).
For sequencing, the genomic DNA was first sheared into 150-200 bp fragments using the Covaris M220 Focused-ultrasonicatorTM Instrument (Covaris, Massachusetts, USA). Next, the fragment DNA libraries were constructed using a KAPA HTP Library Preparation Kit (Illumina platforms) (KAPA Biosystems, Massachusetts, USA), according to manufacturer's instructions. Subsequently, the captured DNA fragments were sequenced using the Illumina Novaseq 6000 sequencing system, and the paired-end 150-bp was read using a custom capture panel (Genecast, Beijing, China). A total of 1,407 genes associated with cancer diagnosis and prognosis were identified. The raw sequencing data was de-multiplexed using the BCL2FASTQ software into individual paired-reads FASTQ files. The sequencing reads were eventually aligned against the hg19 reference genome using the BWA MEM software with default parameters sorted, and indexed with Samtools. The duplicated reads from tumor tissues and adjacent tissues were marked by the Picard MarkDuplicate tool, and were excluded from subsequent analyses. Further reads and mapping qualities were evaluated using FastQC and Samtools flagstat. The average unique sequence coverage was at least 900x for lymphocytes and 2400x for tumor tissue samples.
Single nucleotide variants (SNV) and Indel Calling
Paired sample variant calling on tumors were performed using matched adjacent tissues as the baseline reference. Varscan2 somatic calling was initially employed to detect somatic single nucleotide polymorphisms(SNPs) and Indels using loose parameters, while the calling results were subsequently filtered with custom and more stringent criteria. Especially in tumor tissue samples, merely bases with a depth of over 40x were considered. Both tumor somatic variants (SVs) were required to have at least one support read for each strand with a background P-value under 0.05. In addition, the minor allele frequency for the tumor was not less than 1%. Among the SVs, those with a P-value <0.05, a deeper depth of 100x, and an allele frequency of over 5% were filtered and termed as SVs in the present study.
Furthermore, common variants and potential background noises introduced by the platform were also excluded, in order to ensure the reliability of the data. In particular, SVs with a population frequency of over 0.5%, according to the ExAC database or 1,000 Genomes database, were excluded. In addition to these two databases, a sample pool based on 30 healthy Chinese donors was also used as a standard normal database to identify either prevalent germline mutations in the Chinese population or technical artifacts prevailing in the sequencing platform. Variants with prevalence of no less than 20% in the standard normal database were blacklisted in the present study, and excluded from the downstream analysis. In addition, variants with no less than 20% prevalence among a larger real-life patient sample population, which contains more than 5,000 samples in different cancer types, as tested by Genecast, Beijing, China on a Novaseq 6000 sequencer, were considered as sequencing-specific errors, and were also excluded from further analysis.
Copy number variation (CNV) calling
A negative control copy number baseline was constructed using all blood cell samples obtained from patients, and the CNV from FFPE samples was called for each patient using a CNV kit. A software package CNV kit that applies both sequencing target region reads and nonspecifically captured off-target reads was used to reduce the bias introduced by target region sequencing, and improve the somatic CNV detection resolution. During the software working procedure, three main sources of bias induced the extraneous variability of the sequencing read depth, which include GC content, target footprint size and spacing, and repetitive sequences, and these were evaluated and corrected. The thresholds of copy number ≥3 and ≤1.2 were used to categorize altered regions as CNV gains (amplification) and copy number losses (deletions).
Mutational signature analysis
6 categories of base substitutions exist, namely, C > T, C > A, C > G, T > C, T > G, and T > A. There will be a total of 96 substitution types, when the 5′ and 3′ flanking nucleotides of a specific mutated base are taken into consideration. The proportion of specific trinucleotide mutational signature in primary lesion and LN metastasis lesion was first compared in both LUAD and LUSC. Then, R package deconstructSigs [16] was used to extract the underlying specific trinucleotide mutational signature in every each sample, by using the 30 signatures documented by the COSMIC as reference, which can be obtained from COSMIC (https://cancer.sanger.ac.uk/cosmic/signatures). After that, the mean weights of different signatures in LN metastasis and primary tumor were calculated in both LUAD and LUSC.
Enrichment analysis of LN metastasis exclusive somatic mutations (LME-SMs)
Somatic variants that can be detected in the LN metastasis, but not in the primary tumor, were termed as LME-SMs. To study the functions of LME-SMs, Gene Ontology (GO) functional enrichment analysis and Kyoto Encyclopedia of Gene and Genomes (KEGG) pathway enrichment analysis was performed using R package clusterProfiler v3.6.0[17]. The GO functional analysis consisted of three categories: biological process (BP), cellular component (CC) and molecular function (MF). In the present study, an adjusted P value ≤0.1 was set as the cut-off criterion for the enrichment analysis.
Results
Mutational landscape of primary lung cancer and its synchronous lymphatic metastasis
In 23 patients with LUAD, a total of 116 and 84 mutations were discovered in the primary lesion and LN metastasis lesion, respectively. Among these, a total of 45 mutations were concordant, which could be both detected in the primary and metastasis lesions (Fig. 1A), leading to a concordance rate of 29.0% at the mutation site (45/155). TP53 and EGFR were the two most frequently mutated genes in both primary lesion (14/23 [61%) and 8/23 [35%], respectively) and LN metastases lesion (9/23 [39%] and 6/22 [26%], respectively) (Fig. 2A). In 14 patients with LUSC, a total of 74 and 37 mutations were discovered in the primary lesion and LN metastasis lesion respectively. Among these, a total of 17 mutations were concordant, which could be both detected in the primary and metastasis lesions (Fig. 1B), leading to a concordance rate of 18.1% (17/94). In LUSC, TP53 was the most frequently mutated gene in both primary lesion (11/22 [50%]) and LN metastases lesion (9/22 [41%]) (Fig. 2B). In conclusion, LUAD (29.0%) showed higher concordance rate in mutations between the primary and metastasis lesions than LUSC (18.1%). Besides, more mutations were discovered in the primary lesion than LN metastasis lesion in both LUAD and LUSC, indicating that tumor mutation burden is higher in the primary lesion than that in LN metastasis lesion.
Figure 1
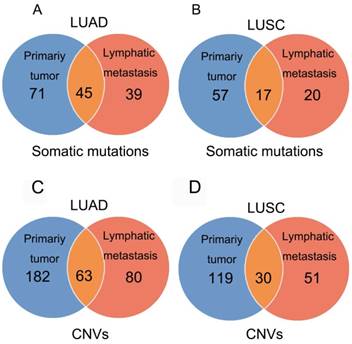
Mutations identified in primary tumors and metastatic lesions. The number of non-synonymous variants identified in primary tumors, metastatic lesions, and both tumor and metastatic lesions in LUAD (A) and LUSC (B). The number of amplifications and homozygous deletions in primary tumors, metastatic lesions, and both tumor and metastatic lesions in LUAD (C) and LUSC (D).
CNVs of primary lung cancer and its synchronous lymphatic metastasis
In 23 patients with LUAD, 245 and 143 CNVs were found in the primary lesions and LN metastasis lesions, respectively. Among these, a total of 19.4% (63/325) CNVs were concordant between primary tumors and their matched metastases (Fig. 1C). In addition, it was found that 44.1% of CNVs (63/143) detected in LN lesions were mainly preserved from the primary lesions. In 14 patients with LUSC, 149 and 81 CNVs were found in these primary lesions and LN metastasis lesions, respectively. Among these, a total of 15.0% (30/230) CNVs were concordant between primary tumors and their matched metastases (Fig. 1D). In addition, it was also found that 37% of CNVs (30/81) detected in LN lesions were mainly preserved from their primary lesions. Furthermore, the copy number profiles in each patient demonstrated that the copy number variants were quite similar between the primary tumor and its respective metastasis in both LUAD (Fig. 3A) and LUSC (Fig. 3B), suggesting that the copy number profiles were largely preserved through the progression of the lymphatic metastasis.
Mutation signature in primary lung cancers and LN metastases
Regarding the mutational context, a strong enrichment of C > T transitions was observed in the primary tumor and LN metastasis in bolt LUAD and LUSC, especially at the CpG dinucleotides, which is denoted as signature 1 documented in the COSMIC Mutational Signature Framework (Fig. 4A and 4B). The signature 1 is more dominant in LN metastasis than primary tumor, suggesting that the spontaneous deamination of cytosine contributes to the accumulation of mutations during tumor development and migration in both LUAD and LUSC(Fig. 4C and 4D) [18].
Functions of LME-SMs
LME-SM genes enriched in BP terms were mainly associated with peptidyl-tyrosine phosphorylation, and peptidyl-tyrosine modification in both LUAD and LUSC (Fig. 5A). LME-SM genes enriched in MF terms were mainly associated with protein tyrosine kinase activity, transmembrane receptor protein kinase activity, and transmembrane receptor protein kinase activity in both LUAD and LUSC (Fig. 5C). LME-SM genes enriched in CC terms were mainly associated with focal adhesion, cell-substrate adherent junction, cell-substrate adherent junction in LUSC. But no enrichment in CC terms was available in LUAD (Fig. 5B). Furthermore, the KEGG pathway enrichment analysis demonstrated that the LME-SM genes were mainly enriched in the PI3K-Akt signaling pathway, the MAPK signaling pathway, and the Ras signaling pathway in both LUAD and LUSC (Fig. 5D).
Discussion
To our knowledge, the current study represents the largest set of matched primary lung tumors and metastases so far to explore the genetic heterogeneity in the process of tumor migration and reflect the genetic evolutionary process of lymphatic metastasis in locally advanced NSCLC using large scale of key cancer genes. The results confirm that genomic heterogeneity exists between primary lesion and LN metastasis lesion, and may be the reason for the observed heterogeneous responses to therapy, and the eventual development of resistance to target agents in cancers. Mutations in genes related to cell migration and metastasis may explain the process of progression in NSCLC.
Regarding the mutational context, our results showed that the signature 1 documented in the COSMIC Mutational Signature Framework was both enriched in primary lesion and LN metastasis lesion in both LUAD and LUSC. But it was more prominent in LN metastasis lesion. The study by Chen et al. [19] showed that signature 1 was also strongly enriched in both 45 pairs of esophageal squamous cell carcinoma specimens and the matched dysplasia specimens. All these results suggest that the spontaneous deamination of cytosine may contribute to the accumulation of mutations during tumorigenesis and metastasis. But the mechanisms underlying the generation of mutation signature 1 remain unclear.
Figure 2
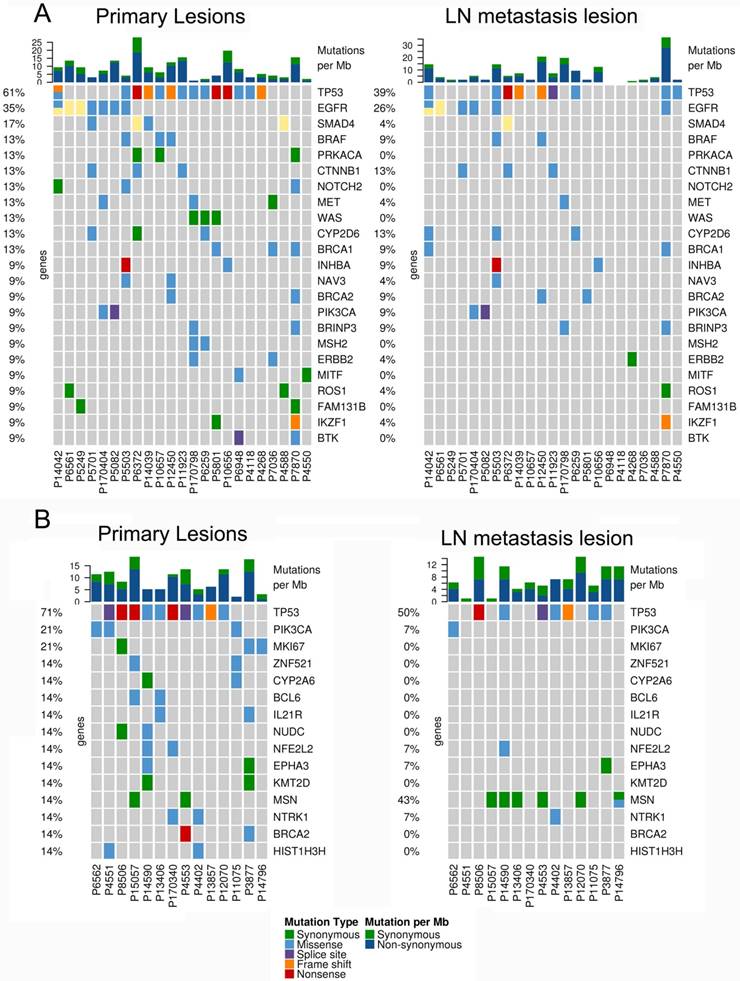
The mutational landscape of primary lung cancer and its matched LN metastasis in both LUAD (A) and LUSC(B).
Figure 3
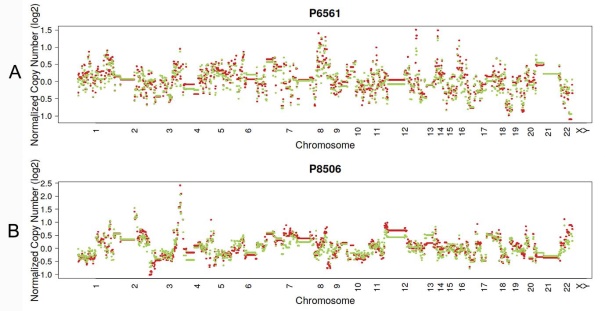
Typical presentation of copy number profiles of patient #6561 (A) and #8506 (B); the copy number profiles were largely preserved through the progression of the metastasis in both LUAD (patient #6561) and LUSC (#8506).
Figure 4
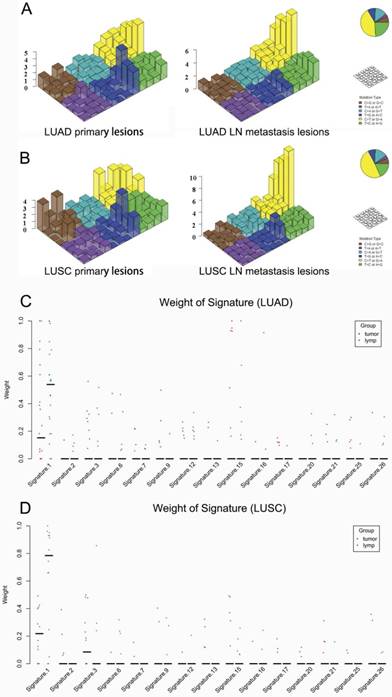
Mutational signatures in primary lung cancer lesions and LN metastatic lesions. 'Lego' plots exhibiting the frequency of specific trinucleotide mutational signature in primary lesion (left) and LN metastasis lesion (right) in both LUAD (A) and LUSC (B). The weight of decomposed signatures in each of primary tumor (purple dots) and LN metastasis (green dots) in both LUAD (C) and LUSC (D).
In the present cohort, TP53, EGFR, PIK3CA, BRAF, which have been known as cancer driver genes, were the most frequently mutated genes in primary tumors and LN metastases. EGFR mutation, as a targetable mutation, is of great significance in clinical practice in LUAD. Among the 23 LUAD patients enrolled in the present study, seven patients had EGFR targetable hotspot mutations (including L858R, 19del, and T790M) in the tumor sample, while six patients had EGFR mutations in LN samples. The discordance rate in mutations between tumor and LN samples was 13.0% (3/23). For patient P7870, a sensitive EGFR mutation L858R was detected in the metastasis LN tissue, but not in the primary tumor, which implies that mutational profiling in metastatic samples would be of more value. For patient P5082 and P5249, EGFR T790M mutations and EGFR 21del were only detected in primary tumors, respectively. These results were consistent with the observations reported by Park et al. [20]and Wang et al.[21], in which a considerable discrepancy(11.9% and 14.5%, respectively) in EGFR mutations generates between primary tumors and metastatic LN, suggesting that genetic heterogeneity originates at the molecular level during the process of metastasis. Furthermore, a considerable proportion of mutations were found to be different between primary tumors and metastatic lesions, which is in agreement with the observation reported by Um et al., in which a considerable proportion of NSCLCs exhibited unique mutations in metastatic LN, supporting that clonal evolution occurs at the molecular level during the process of metastasis[22]. Since considerable tumor heterogeneity exists at the molecular level during the process of metastasis, multiple biopsies and sequencing strategies for different tumor regions are required.
It is noteworthy that more mutations were found in the primary tumor in both LUAD and LUSC, when compared to the paired LN metastasis, indicating that tumor mutation burden is higher in primary lesion than that in LN metastasis lesion. This was also observed by Kim et al.[23], in which there was a higher chance of non-synonymous mutation in primary tumors than in metastatic LNs. The mechanisms underlying the decrease in mutations in metastatic lesions remain unclear.
Figure 5
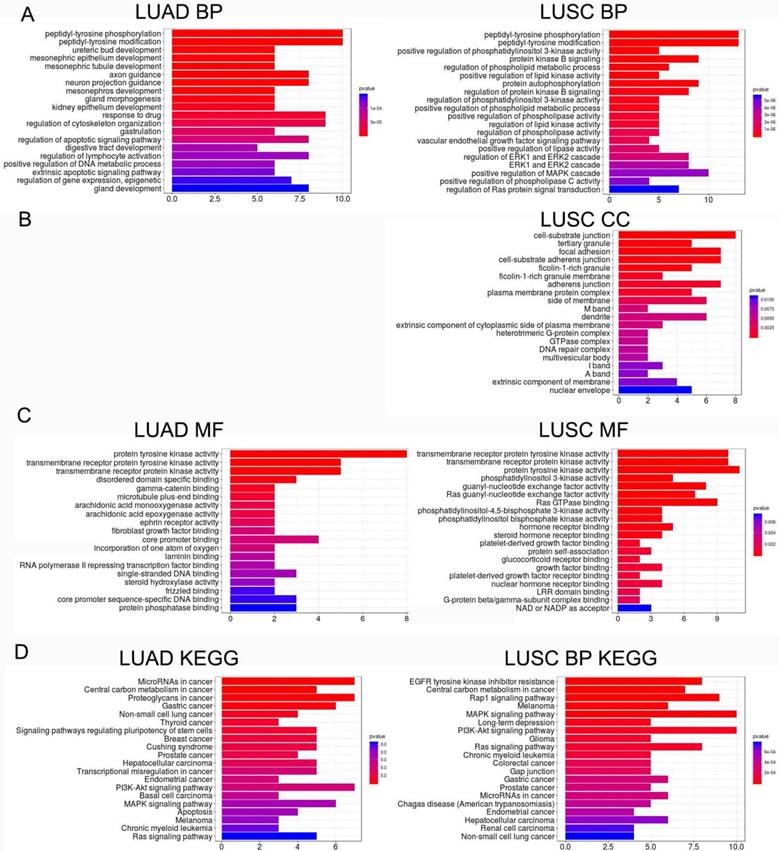
The functional analysis of LME-SM genes in both LUAD (left) and LUSC (right). (A) The Gene Ontology (GO) functional enrichment analysis of biological processes. (B) The cellular component. (C) The molecular function. (D) The Kyoto Encyclopedia of Gene and Genomes (KEGG) pathway enrichment analysis.
In the present study, the copy number profiles in the primary tumor and its respective metastasis lesion were quite similar in both LUAD and LUSC. This was consistent with the observation reported by Schrijver et al.[24], in which the copy number profiles were largely preserved through the progression of the metastasis in breast cancer. These results suggest that the copy number alteration, as a punctuated burst of evolution, is more likely to be preserved from its primary lesion.
It has been known that the PI3K-Akt, MAPK and Ras signaling pathways regulate cell proliferation, differentiation and migration, and are closely associated with carcinogenesis and disease progression [25, 26]. In our study, the KEGG pathway enrichment analysis demonstrated that LME-SM genes were enriched in the PI3K-Akt, MAPK and Ras signaling pathways in both LUAD and LUSC. These results imply that LN metastasis may be associated with the PI3K-Akt, MAPK and Ras signaling pathway in NSCLC. LUSCs are characterized histopathologically by the presence of intercellular bridges and high levels of proteins relevant to intercellular junctions, which distinguished from LUAD [27]. Previous studies indicated that altered genes that code for proteins involved in the formation of intercellular adhesion and cell-substrate adhesion were closely related to tumor migration and metastasis of LUSC [27, 28]. Our GO cellular component enrichment analysis revealed that the LME-SM genes were mainly associated with cell focal adhesion, cell-substrate adherent junction, cell-substrate adherent junction in LUSC but not LUAD, indicating that the malfunction of intercellular adhesion, cell-substrate adhesion and junction presented exclusively in LUSC but not LUAD.
The present study was limited by the relatively small sample size and limited capacity of targeted massively parallel sequencing, which may not uncover all the mutations involved in the progression, from primary lung cancer to metastatic disease. Whole exome sequencing and multi-region sampling of primary tumor and metastasis tumor tissues are required for the comprehensive understanding of the landscape of genetic alterations, and for guiding targeted therapy in locally advanced lung cancer.
In conclusion, there were differences in the repertoire of somatic mutations and CNVs, which even affected driver and potentially targetable genes in NSCLC. The genetic heterogeneity in cancer lesions from the same origin reminds people to develop multi-functional drugs to target metastatic lesions with different genetic profiles.
Abbreviations
NSCLC: non-small cell lung cancer; LME-SMs: lymphatic metastasis exclusive somatic mutations; LN: lymph nodes; CNV: Copy number variation; GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Gene and Genomes; BP: biological process; CC: cellular component; MF: molecular function.
Acknowledgements
The present study was funded by the Human Provincial Science and Technology Department (2017JJ2345) and the National Natural Science Foundation of China (No. 81673516).
Ethics approval and consent
This study has been approved by the ethics committee of Xiangya Hospital, Central South University. Informed consent was obtained from all individual participants included in the study.
Competing Interests
Hao Guo, and Beibei Mao are employees of Beijing Genecast Biotechnology Co., Beijing, China. The other authors declare no potential conflict of interest.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7-30
2. Hong QY, Wu GM, Qian GS, Hu CP, Zhou JY, Chen LA. et al. Prevention and management of lung cancer in China. Cancer. 2015;121(Suppl 17):3080-8
3. Tsao AS, Scagliotti GV, Bunn PA Jr, Carbone DP, Warren GW, Bai C. et al. Scientific Advances in Lung Cancer 2015. J Thorac Oncol. 2016;11:613-38
4. Zhou C, Wu Y-L, Chen G, Feng J, Liu X-Q, Wang C. et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. The Lancet Oncology. 2011;12:735-42
5. Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y. et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15:213-22
6. Shi Y, Zhang L, Liu X, Zhou C, Zhang L, Zhang S. et al. Icotinib versus gefitinib in previously treated advanced non-small-cell lung cancer (ICOGEN): a randomised, double-blind phase 3 non-inferiority trial. Lancet Oncol. 2013;14:953-61
7. Rimkunas VM, Crosby KE, Li D, Hu Y, Kelly ME, Gu TL. et al. Analysis of receptor tyrosine kinase ROS1-positive tumors in non-small cell lung cancer: identification of a FIG-ROS1 fusion. Clin Cancer Res. 2012;18:4449-57
8. Hsu PP, Shaw AT. Lung Cancer: A Wily Genetic Opponent. Cell. 2017;169:777-9
9. Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338-45
10. Andor N, Graham TA, Jansen M, Xia LC, Aktipis CA, Petritsch C. et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med. 2016;22:105-13
11. Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S. et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. New Engl J Med. 2017;376:2109-21
12. Goldstraw P, Chansky K, Crowley J, Rami-Porta R, Asamura H, Eberhardt WE. et al. The IASLC Lung Cancer Staging Project: Proposals for Revision of the TNM Stage Groupings in the Forthcoming (Eighth) Edition of the TNM Classification for Lung Cancer. J Thorac Oncol. 2016;11:39-51
13. Chen Y, Peng X, Zhou Y, Xia K, Zhuang W. Comparing the benefits of chemoradiotherapy and chemotherapy for resectable stage III A/N2 non-small cell lung cancer: a meta-analysis. World J Surg Oncol. 2018;16:8
14. Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J. et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. 2014;346:256-9
15. de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L. et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346:251-6
16. Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31
17. Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics: a journal of integrative biology. 2012;16:284-7
18. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV. et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415
19. Chen XX, Zhong Q, Liu Y, Yan SM, Chen ZH, Jin SZ. et al. Genomic comparison of esophageal squamous cell carcinoma and its precursor lesions by multi-region whole-exome sequencing. Nat Commun. 2017;8:524
20. Park S, Holmes-Tisch AJ, Shim YM, Kim J, Kim HS, Lee J. et al. Discordance of molecular biomarkers associated with epidermal growth factor receptor pathway between primary tumors and lymph node metastasis in non-small cell lung cancer. J Thorac Oncol. 2009;4:809-15
21. Wang S, Wang Z. Meta-analysis of epidermal growth factor receptor and KRAS gene status between primary and corresponding metastatic tumours of non-small cell lung cancer. Clin Oncol (R Coll Radiol). 2015;27:30-9
22. Um SW, Joung JG, Lee H, Kim H, Kim KT, Park J. et al. Molecular Evolution Patterns in Metastatic Lymph Nodes Reflect the Differential Treatment Response of Advanced Primary Lung Cancer. Cancer Res. 2016;76:6568-76
23. Kim EY, Cho EN, Park HS, Kim A, Hong JY, Lim S. et al. Genetic heterogeneity of actionable genes between primary and metastatic tumor in lung adenocarcinoma. BMC Cancer. 2016;16:27
24. Schrijver W, Selenica P, Lee JY, Ng CKY, Burke KA, Piscuoglio S. et al. Mutation Profiling of Key Cancer Genes in Primary Breast Cancers and Their Distant Metastases. Cancer Res. 2018;78:3112-21
25. Paik PK, Shen R, Won H, Rekhtman N, Wang L, Sima CS. et al. Next-Generation Sequencing of Stage IV Squamous Cell Lung Cancers Reveals an Association of PI3K Aberrations and Evidence of Clonal Heterogeneity in Patients with Brain Metastases. Cancer Discov. 2015;5:610-21
26. Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353-d61
27. Bonastre E, Brambilla E, Sanchez-Cespedes M. Cell adhesion and polarity in squamous cell carcinoma of the lung. J Pathol. 2016;238:606-16
28. Baronsky T, Ruhlandt D, Brückner BR, Schäfer J, Karedla N, Isbaner S. et al. Cell-Substrate Dynamics of the Epithelial-to-Mesenchymal Transition. Nano Lett. 2017;17:3320-6
Author contact
![]() Corresponding author: Wei Zhuang, Xiangya Hospital of Central South University, 410008 Changsha, Hunan, People's Republic of China. E-mail: zhuangweiedu.cn. Tel: +86-152-0089-9869, Fax: +86-731-8432-7623
Corresponding author: Wei Zhuang, Xiangya Hospital of Central South University, 410008 Changsha, Hunan, People's Republic of China. E-mail: zhuangweiedu.cn. Tel: +86-152-0089-9869, Fax: +86-731-8432-7623
Citation styles
APA
Chen, Y., Mao, B., Peng, X., Zhou, Y., Xia, K., Guo, H., Jiang, H., Wang, G., Zhuang, W. (2019). A Comparative Study of Genetic Profiles of Key Oncogenesis-Related Genes between Primary Lesions and Matched Lymph Nodes Metastasis in Lung Cancer. Journal of Cancer, 10(7), 1642-1650. https://doi.org/10.7150/jca.28266.
ACS
Chen, Y.; Mao, B.; Peng, X.; Zhou, Y.; Xia, K.; Guo, H.; Jiang, H.; Wang, G.; Zhuang, W. A Comparative Study of Genetic Profiles of Key Oncogenesis-Related Genes between Primary Lesions and Matched Lymph Nodes Metastasis in Lung Cancer. J. Cancer 2019, 10 (7), 1642-1650. DOI: 10.7150/jca.28266.
NLM
Chen Y, Mao B, Peng X, Zhou Y, Xia K, Guo H, Jiang H, Wang G, Zhuang W. A Comparative Study of Genetic Profiles of Key Oncogenesis-Related Genes between Primary Lesions and Matched Lymph Nodes Metastasis in Lung Cancer. J Cancer 2019; 10(7):1642-1650. doi:10.7150/jca.28266. https://www.jcancer.org/v10p1642.htm
CSE
Chen Y, Mao B, Peng X, Zhou Y, Xia K, Guo H, Jiang H, Wang G, Zhuang W. 2019. A Comparative Study of Genetic Profiles of Key Oncogenesis-Related Genes between Primary Lesions and Matched Lymph Nodes Metastasis in Lung Cancer. J Cancer. 10(7):1642-1650.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.