Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(7):1726-1733. doi:10.7150/jca.26482 This issue Cite
Research Paper
P16 Methylation Leads to Paclitaxel Resistance of Advanced Non-Small Cell Lung Cancer
Zhaojun Liu1*, Hongmei Lin2,3*, Ying Gan1, Chenghua Cui1, Baozhen Zhang1, Liankun Gu1, Jing Zhou1, Guangying Zhu2,3 ![]() , Dajun Deng1
, Dajun Deng1 ![]()
1. Key Laboratory of Carcinogenesis and Translational Research, Division of Etiology,
2. Key Laboratory of Carcinogenesis and Translational Research, Department of Radiation Oncology, Peking University Cancer Hospital and Institute, Beijing, 100142;
3. National Clinical Research Center for Respiratory Diseases, Department of Radiation Oncology, China-Japan Friendship Hospital, Beijing, 100029, China
*Equal contribution
Received 2018-4-4; Accepted 2019-1-12; Published 2019-4-5
Citation:
Liu Z, Lin H, Gan Y, Cui C, Zhang B, Gu L, Zhou J, Zhu G, Deng D. P16 Methylation Leads to Paclitaxel Resistance of Advanced Non-Small Cell Lung Cancer. J Cancer 2019; 10(7):1726-1733. doi:10.7150/jca.26482. https://www.jcancer.org/v10p1726.htm
Other stylesAbstract
Paclitaxel-based chemotherapy is widely used as the first-line treatment for non-small cell lung cancer (NSCLC). However, only 20%-40% of patients have shown sensitivity to paclitaxel. This study aimed to investigate whether P16 methylation could be used to predict paclitaxel chemosensitivity of NSCLC. Advanced NSCLC (N=45) were obtained from patients who were enrolled in a phase-III randomized paclitaxel-based clinical trial. Genomic DNA samples were extracted from the biopsies prior to chemotherapy. P16 methylation was detected using MethyLight. The association between P16 methylation and the sensitivity of paclitaxel in cell lines was determined by in vitro assay using a P16-specific DNA demethylase (P16-TET) and methyltransferase (P16-Dnmt). The total response rate of the low-dose paclitaxel-based chemo-radiotherapy was significantly lower in P16 methylation-positive NSCLCs than that in the P16 methylation-negative NSCLCs (2/15 vs. 16/30: adjusted OR=0.085; 95%CI, 0.012-0.579). Results revealed that P16 demethylation significantly decreased paclitaxel resistance of lung cancer H1299 cells (IC50 values decreased from 2.15 to 1.13 µg/ml, P<0.001). In contrast, P16-specific methylation by P16-Dnmt significantly increased paclitaxel resistance of lung cancer HCC827 cells and gastric cancer BGC823 cells (IC50 values increased from 18.2 to 24.0 ng/ml and 0.18 to 0.81 µg/ml, respectively; P=0.049 and <0.001, respectively). The present results suggest that P16 methylation may lead to paclitaxel resistance and be a predictor of paclitaxel chemosensitivity of NSCLC.
Keywords: P16, methylation, paclitaxel, resistance, non-small cell lung cancer
Introduction
Non-small cell lung carcinoma (NSCLC) is one of the most diagnosed cancers in China (1). Approximately 70%-80% of NSCLC patients are not eligible for radical surgery at the time of diagnosis, due to late diagnoses at stages III or IV. Although combination chemotherapy treatments with 2-3 drugs and radiotherapy are the main methods of treating locally advanced NSCLC, no standard regimen has been established at present (2,3). Because of the low response rates and the toxic side effects of chemotherapy and chemo-radiotherapy, outcomes for patients with advanced NSCLC remain poor (4,5).
Paclitaxel-based chemotherapy is widely used as the first-line treatment for many cancers, including NSCLC. It has produced significant and incremental survival benefits (6,7). However, only 20%-40% of patients have shown sensitivity to this treatment (8). Most of the patients had either primary resistance to the drug or acquired resistance after repeated treatments. Therefore, identification of the relevant response predictors will highly benefit clinical practice by stratifying patients into suitable treatment options that are currently available (9). Although several molecules, including TUBB3, MDR1, and P16 proteins, are related to paclitaxel resistance (10-12), a clinical predictor for paclitaxel-based treatment is not yet available.
The P16 gene is a cell cycle regulator involved in the inhibition of G1 phase progression. Methylation of the CpG islands near the transcription start site of the P16 gene not only silences P16 transcription but also represses transcription of the lncRNA ANRIL (13-15). Aberrant P16 methylation occurs frequently in NSCLC and gastric cancer and is associated with their development and progression (16-19). Although it has been reported that P16 methylation is correlated with treatment responses to fluorouracil-based therapy in breast tumors, colorectal cancer, and gastric cancer (20-22), the feasibility of using P16 methylation to predict paclitaxel resistance has not previously been reported.
To determine if the P16 methylation is related to paclitaxel chemosensitivity in the advanced NSCLC, in the present study, we analyzed the association between P16 methylation and paclitaxel resistance in patients with NSCLC who were enrolled in a randomized phase III clinical trial (23). The direct effects of P16 methylation on the sensitivity of cancer cells to paclitaxel were also studied. The results indicate, for the first time, that P16 methylation may directly decrease the paclitaxel sensitivity of cancer cells and might be used as a biomarker for predicting paclitaxel resistance of NSCLC.
Materials and Methods
Patients and sample collection
A multicenter phase III randomized 2-arm study was conducted to assess the effects of low-dose paclitaxel therapy (45 mg/m2 per week for 6 weeks) combined with 60-71 Gy radiotherapy for 134 patients with locally advanced NSCLC at clinical stage III from March 2006 to February 2013 at Peking University Cancer Hospital and Institute and reported according to the CONSORT guidelines (23). Biopsy tissues of NSCLC were collected prior to chemotherapy and used for histological and cytological diagnoses. Among the 134 NSCLC patients who were enrolled in the clinical trial, 119 were from Peking University Cancer Hospital. Formalin fixed paraffin embedded (FFPE) NSCLC biopsies from 52 patients were available for P16 methylation detection.
The primary endpoint of the study was the response rate, which was assessed by the RECIST criteria (24). The responses were categorized as complete response (CR), partial response (PR), stable disease (SD), and progressive disease (PD). Patients with CR and PR were defined as responders, whereas patients with SD and PD were defined as non-responders. The secondary endpoints were recurrence-free survival and overall survival (RFS and OS).
These studies were approved by the Institutional Review Boards (IRB) at the Peking University Cancer Hospital and Institute (RCOG-0701), and all patients gave written informed consent to participate in the study. The study was performed in accordance with the ethical standards of the Declaration of Helsinki. The clinical trial was registered with the Chinese Clinical Trial Registry (ChiCTR-TRC-10000786).
Cell lines and culture
P16-methylated lung cancer cell line H1299, was kindly provided by Dr. Chengchao Shou at Peking University Cancer Hospital and Institute. P16-unmethylated lung cancer cell line HCC827 was purchased from the National Infrastructure of Cell Line Resource (Beijing). P16-unmethylated gastric cancer cell line BGC823 was kindly provided by Dr. Yang Ke at the same Institute. The cells were cultured in RPMI1640 medium supplemented with 10% FBS and maintained at 37 °C in humidified air with 5% CO2.
DNA extraction and bisulfite modification
Genomic DNA was extracted from the biopsies, and subjected to bisulfite treatment using the EpiTect Plus FFPE Bisulfite Kit (cat. no. 59144, Qiagen) according to the manufacturer's instructions. The modified DNA was stored at -20 °C before use.
Detection of P16 methylation using the MethyLight assay, DHPLC, MSP, and sequencing
The primer and probe sequences used in the 70 bp MethyLight assay have been described previously (Fig. 1A) (25). The P16 methylation levels were determined using an Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) and normalized to the internal control COL2A1 gene (collagen type II, alpha 1 gene). The relative copy number (RCN) of the methylated-P16 CpG islands was calculated according to the formula [2-ΔCt, (ΔCt=Ctmethylated-p16 - CtCOL2A1)]. When the Ct value for COL2A1 was ≤29.3, the tested sample was defined as P16 methylation informative [26]. The methylation status of the P16 CpG islands in cancer cells was detected with denatured high performance liquid chromatography (DHPLC) as described (19,27). Methylation-specific PCR (MSP; 150/151-bp) was also used to detect methylated-P16 and unmethylated-P16 alleles as described (28).
The 150 bp fragment of the P16 exon-1 CpG island, containing with the entire 70 bp MethyLight amplicon, was amplified from bisulfite-templates using the CpG-free primer set (forward, 5'-ggggagtagtatggagttt-3'; reverse, 5'-aaccctctacccacctaaa-3') at annealing temperature 55 ºC for 40 cycles, and clone-sequenced (Fig. 1A).
Construction of vectors and transfection
To induce demethylation of P16 CpG islands, an engineered P16-specific dioxygenase (P16-TET) pTRIPZ vector (29) was constructed by fusing a SP1-like engineered seven-zinc finger protein (7ZFP) capable of specifically binding the 21 bp fragment (5'-GAG GAA GGA AAC GGG GCG GGG-3', including a Sp1-binding site) within the human P16 promoter with the catalytic domain (CD: 1418aa-2136aa) of TET1 (NM_030625.2), and then inserted into the expression controllable pTRIPZ vector carrying a Tet-on' switch (Open Biosystem, USA) to control the expression of the P16-TET. The P16-TET stably transfected H1299 cells were treated with the doxycycline (final conc. 0.25 µg/ml) for 7 days for induction of demethylation of the P16 CpG islands. Similarly, the engineered P16-specific methyltransferase (P16-Dnmt) was constructed by fused the 7ZFP with the catalytic domain (approximately 608-908aa) of mouse Dnmt3a, and integrated into the pcDNA3.1 vector (for transient transfection) and the pTRIPZ vector (for stable transfection) to induce methylation of the P16 CpG islands as previously described (14). The BGC823 cells stably transfected with P16-Dnmt or pTRIPZ empty control vectors were induced with 0.25 µg/ml doxcycline for 14 days, and then paclitaxel sensitivity was assessed using the cell viability assay, as shown below.
Extraction of RNA and quantitative RT-PCR (qRT-PCR)
Cells with the density about 70% were harvested. Total RNA was extracted by TRIzol (Invitrogen). The cDNA was reverse-transcribed using the ImProm-IITM Reverse Transcription System (A3800;Promega). The expression level of the P16 gene was analyzed by quantitative RT-PCR as previously described (14). Power SYBR Green PCR Master Mix (Fermentas, Canada) was used in qRT-PCR analyses (ABI-7500FAST). The average copy numbers of P16 mRNA per cell was calculated based on the average Ct value of target gene and Alu reference [2-(Cttarget_gene-CtAlu)] (30).
Figure 1

The methylation status of the P16 CpG islands in NSCLC tissues. (A) Locations of 70 bp and 150 bp amplicons, primers, and probes used in MethyLight and bisulfite-sequencing (BS), respectively. (B) Bisulfite sequencing results for three representative NSCLC tissue samples (#1 and #2, P16 methylation-positive; #3, P16 methylation-negative, by MethyLight). Each line represented a clone; red dot, methylated CpG site; green dash, C-deletion.
Western blot
Protein levels were detected using Western blot as previously reported (14). The following antibodies were used: P16 (Proteintech), GAPDH (OriGene), MYC (Sigma).
Cell viability assay
Cell proliferation was analyzed using the Cell Counting Kit-8 (CCK-8). Briefly, the stably transfected cells were seeded into 96-well plates at 1,000 cells per well, with three wells for each treatment. Following the adhesion of cells to the wells, the cells were treated with paclitaxel (30 mg/5 ml, Haikou Pharmaceutical Factory Co., Ltd) at various concentrations (final concentration, 0.03, 0.3, 3.0, and 30.0 µg/ml for H1299 and BGC823 cells; 0.03, 0.3, 3.0, and 30.0 ng/ml for HCC827 cells). The control reagent was 0.9% NaCl. The viable cell numbers were assessed 48 h after treatment. The half maximal inhibitory concentration (IC50) was calculated using the SPSS16.0 software.
Statistical analysis
All statistical analysis was conducted with SPSS 16.0 software. The associations between gene methylation status and clinicopathologic characteristics and clinical response were assessed using Pearson's chi-square test unless the smallest expected value is <5; then, Fisher's exact test was used. The forward binary logistic regression analysis was used to perform multivariate analyses. The Kaplan-Meier survival curve and log-rank test were used to describe RFS or OS. All P-values were two-sided, and P < 0.05 was considered statistically significant.
Results
Characteristics of the eligible NSCLC patients from the clinical trial
Among the 52 available NSCLC biopsies, 45 were P16 methylation informative (COL2A1 Ct value ≤29.3) as assessed by the MethyLight analysis, and were therefore eligible for the association analysis. Among the 45 patients who received low-dose paclitaxel-based therapy, 18 patients were complete or partial responders (40.0%) and 36 patients (80.0%) died during the follow-up (range 7.4 to 77.4 months; median, 32.0 months). The median RFS and OS values for these patients were 9.9 and 21.0 months, respectively. The clinicopathological characteristics of these 45 patients are listed in Table 1.
A receiver operating characteristic (ROC) curve for P16 methylation was made based on the specificities and the sensitivities of response to determine the appropriate cutoff value (>2.07×10-4) to define P16 methylation-positive. The area under the curve is 0.652 (95% CI: 0.49-0.82, P=0.086, Fig. 2). Consequently, 15 NSCLC patients were P16 methylation-positive, and 30 NSCLC patients were P16 methylation-negative. The results of bisulfite-sequencing confirmed the MethyLight analysis: densely methylated CpG sites were observed in some P16 alleles in two representative P16 methylation-positive samples #1 and #2, and not observed in the P16 methylation-negative sample #3 (Fig. 1B). A C-deletion in P16 exon-1 coding region was also identified. The P16 methylation-positive rate in lung squamous carcinomas was significantly higher than that in lung adenocarcinomas or in large cell carcinomas (46.2% vs. 17.6% or 0.0%, P=0.031). Higher P16 methylation-positive rate was observed in NSCLC patients who were >60 years old than in those from patients <60 years old (52.6% vs. 19.2%, P=0.019) (Table 1).
Table 1
Clinicopathological characteristics and paclitaxel responses of NSCLC patients with and without P16 methylation from the clinical trial
| N | P16 Methylation | P- value | ||
|---|---|---|---|---|
| Negative cases (%) | Positive cases (%) | |||
| (All) | 45* | 30 (66.7) | 15 (33.3) | |
| Gender | 1.000 | |||
| Male | 38 | 25 (65.8) | 13 (34.2) | |
| Female | 7 | 5 (71.4) | 2 (28.6) | |
| Age | 0.019 | |||
| <60y | 26 | 21 (80.8) | 5 (19.2) | |
| ≥60y | 19 | 9 (47.4) | 10 (52.6) | |
| Pathology type | ||||
| Squamous carcinoma | 26 | 14 (53.8) | 12 (46.2) | 0.031 |
| Adenocarcinoma | 17 | 14 (82.4) | 3 (17.6) | |
| Large cell carcinoma | 2 | 2 (100) | 0 | |
| Clinical stage | 0.454 | |||
| IIIA | 9 | 5 (55.6) | 4 (44.4) | |
| IIIB | 36 | 25 (69.4) | 11 (30.6) | |
| Therapy response | 0.004 | |||
| Total response | 18 | 16 (53.3) | 2 (13.3) | |
| Complete response | 1 | 0 | 1 | |
| Partial response | 17 | 16 (94.1) | 1 (5.9) | |
| No response | 27 | 14 (46.7) | 13 (86.7) | |
| Stable | 18 | 9 (50.0) | 9 (50.0) | |
| Progression | 9 | 5 (55.6) | 4 (54.4) | |
* 23 cases from Arm-1 and 22 cases from Arm-2 in the clinical trial
Figure 2
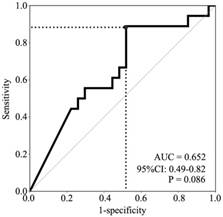
ROC curve for prediction of paclitaxel responses by different P16 methylation levels. The area under the curve is 0.652 (95% CI: 0.49-0.82, P =0.086). Grey dot, cutoff point of the relative copy number of methylated-P16, 2.07×10-4.
Table 2
Analysis of the clinical features and clinical responses using multivariate analysis
| OR | 95%CI | Adjusted P value | |
|---|---|---|---|
| P16 methylation | 0.085 | 0.012 - 0.579 | 0.013 |
| Sex | 0.267 | 0.032 - 2.213 | 0.221 |
| Age | 1.541 | 0.344 - 6.910 | 0.572 |
| Pathology type | 0.470 | 0.118 - 1.868 | 0.284 |
| Clinical stage | 1.776 | 0.298 -10.589 | 0.528 |
P16 methylation was associated with NSCLC resistance to low-dose paclitaxel
Further analysis showed that P16 methylation was significantly associated with a poor response to low-dose (45 mg/m2 per week for 6 weeks) paclitaxel-based treatment (Table 1). The total response rate of the P16 methylation-positive patients was significantly lower than that of the P16 methylation-negative patients: 13.3% (2/15) vs. 53.3% (16/30) (P=0.004) (Table 1). After adjusting for age, sex, pathology, and clinical stage, the differences in the response rates between the P16 methylation-positive and P16 methylation-negative patients were significant according to multivariate analyses (adjusted odds ratio= 0.085, 95% confidence interval [CI]: 0.012-0.579, P= 0.013; Table 2). Furthermore, the Kaplan-Meier analysis showed that the OS of the P16 methylation- positive patients was shorter than that of the P16 methylation-negative patients, but the difference was not statistically significant (Fig. S1A). No significant differences in RFS values were observed (Fig. S1B). These results indicate that P16 methylation is a potential biomarker for predicting resistance to paclitaxel-based therapy for NSCLC patients.
Figure 3
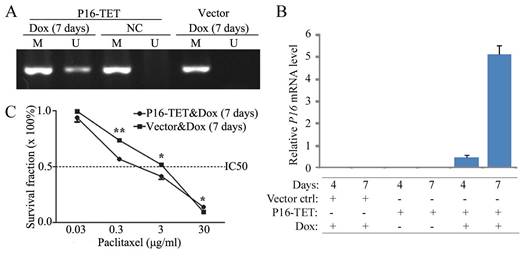
Effects of P16-specific demethylation by P16-TET on the survival of lung cancer H1299 cells treated with different doses of paclitaxel. (A) The results of methylation-specific PCR (MSP) to detect demethylated-P16 alleles in P16-TET stably transfected cells treated with doxycycline for 7 days. (B) The results of quantitative RT-PCR to detect the P16 mRNA level. (C) The results of MTT assay to detect the viability of P16-TET stably transfected cells. NC, negative control; *, P<0.05; **, P<0.01.
P16 demethylation sensitized NSCLC cancer cells to paclitaxel treatment
To study whether P16 demethylation sensitize cells to paclitaxel, an expression controllable P16-TET pTRIPZ vector was used to stably transfected H1299 cells, a P16 homogenously methylated lung cancer cell line. P16 demethylation and expression were detected in the P16-TET H1299 cells treated with doxycycline (Dox) for 7 days, but not detected in negative control (Fig. 3A and 3B). The paclitaxel IC50 values of these P16-TET H1299 cells were significantly lower than that of vector control cells (1.13 vs 2.15 µg/ml, P<0.001; Fig. 3C).
P16 methylation causes paclitaxel resistance in cancer cells
To further validate the causality between P16 methylation and paclitaxel resistance, we induced P16 methylation in another lung cancer cell line HCC827 through transient transfection with the P16-Dnmt vector and the pcDNA3.1 empty control vector. Methylated-P16 alleles were detected by MSP in the HCC827 cells 48 hours after P16-Dnmt transfection (Fig. 4A). Decrease of P16 protein level was induced by P16 methylation in these cells (Fig. 4B). As expected, the paclitaxel IC50 value of the P16-Dnmt cells increased from 18.2 to 24.0 ng/ml (P=0.049; Fig. 4C).
As described in the Discussion section, through re-analyzing our recently published data (22), P16 methylation was also significantly associated to paclitaxel response of patients with gastric cancer. To investigate whether P16 methylation directly leads to paclitaxel resistance in gastric cancer cells, we stably transfected BGC823 cells with the P16-specific methyltransferase P16-Dnmt as previously described (14). An obvious peak for methylated P16 alleles was detected on the 14th doxycycline-treatment days in DHPLC analysis (Fig. 5A). Significant downregulation of P16 expression was also detected (Fig. 5B and C). The results of the cell viability analysis showed that the P16-methylated BGC823 cells were more resistant to paclitaxel toxicity than were the control cells. The paclitaxel IC50 value of the P16-Dnmt cells significantly increased from 0.18 to 0.81 µg/ml (P<0.001; Fig. 5D).
Discussion
Paclitaxel-based chemotherapy regimens have been widely used to treat NSCLC (6,7). However, because of drug resistance, only 20%-40% patients have been responsive to the regimens (8). Although multiple studies have attempted to find biomarkers that could be used to predict which patients may benefit from this regimen, no feasible biomarker is available in clinical practice yet. In the present study, we observed that NSCLC patients who were P16 methylation-positive had consistently and significantly lower paclitaxel response rates than P16 methylation-negative patients. Our results showed that P16-specific methylation directly increased paclitaxel resistance in cancer cells. To our knowledge, this is the first study showing that P16 methylation could be used to predict paclitaxel resistance in NSCLC patients.
As a CDK4 inhibitor, the P16 protein is a cell cycle regulator involved in the inhibition of G1 phase progression through the CDK4-CCND1-Rb pathway (31). The activities of cell cycle regulation genes such as P53, P16 and P14, which arrest the cell cycle and/or apoptosis, are closely associated with chemosensitivity to cancer treatments (32,33). Inhibition of CDK4 by CINK4 or palbociclib could enhance paclitaxel sensitivity in lung adenocarcinoma and ovarian cancer cells (34,35). Therefore, increased activity of CDK4 by P16 methylation is supposed to reduce paclitaxel sensitivity. In consistent with this hypothesis, we found that P16 methylation by P16-Dnmt directly increased the paclitaxel IC50 value of BGC823 cells, whereas reactivation of P16 demethylation by P16-TET directly decreased the IC50 in H1299 cells. These results suggest that epigenetic inactivation of the P16 gene may cause paclitaxel resistance and reactivation of this gene may sensitize cancer cells to paclitaxel treatment.
Figure 4
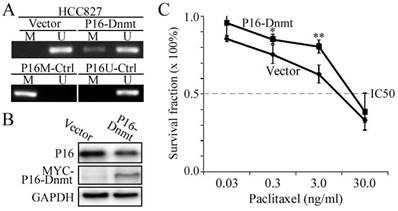
Effects of P16-specific methylation by P16-Dnmt on the survival of lung cancer HCC827 cells treated with different doses of paclitaxel. (A) The results of methylation-specific PCR (MSP) to detect methylated-P16 alleles in P16-Dnmt transiently transfected cells for 48 hours. (B) The results of Western blot to detect P16 protein. (C) The results of MTT assay to detect the viability of HCC827 cells. NC, negative control; *, P<0.05; **, P<0.01.
Figure 5
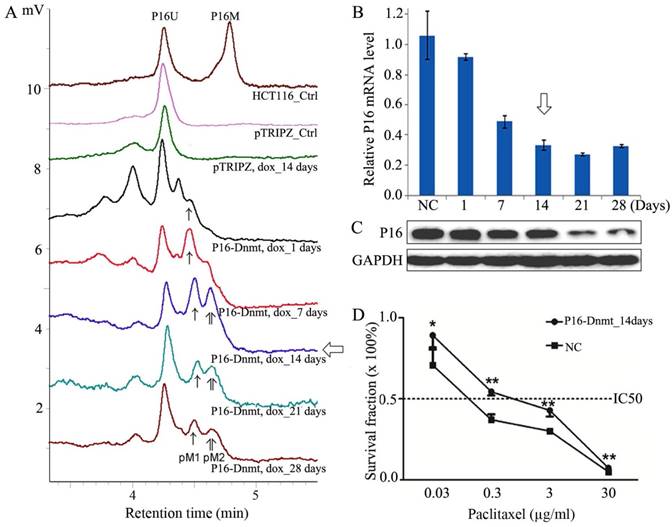
Effects of P16-specific methylation on the survival of cancer cell lines treated with different doses of paclitaxel. (A) DHPLC chromatograms for the detection of fully methylated (P16M), partially methylated (pM), and unmethylated (P16U) P16 CpG islands (392 bp) in BGC823 cells stably transfected with a P16-specific DNA methyltransferase P16-Dnmt or the pTRIPZ control vector. P16-Dnmt expression was induced using doxycycline (dox) for different durations. (B, C) Quantitative RT-PCR and Western blot that detected the P16 expression levels in BGC823 cells. (D) MTT assay to detect the viability of P16-Dnmt stably transfected-BGC823 cells treated with various concentrations of paclitaxel. P16-Dnmt expression in these cells were pre-induced with doxycycline for 14 days. NC, negative control. *, P<0.05; **, P<0.01.
The long non-coding RNA (lncRNA) ANRIL is transcribed from the antisense-strand of P15/P14/P16 gene. It was reported that enforced ANRIL overexpression increased paclitaxel resistance of A549 cells, a P15/P14/P16 gene homogenously deleted NSCLC cell line (36). We recently found P16 methylation (or demethylation) not only inactivated (or reactivated) the transcription of the P16 gene, but also repressed (or enhanced) ANRIL transcription in different cancer cells (15). Unlike enforced ANRIL overexpression, P16 reactivation by P16-TET restores ANRIL expression at the endogenous level (about 10% of P16 mRNA level). These phenomena imply that it is reactivation of endogenous P16, but not ANRIL (if any), that mainly contribute to paclitaxel resistance.
Previous studies, including our own, have shown that P16 methylation is correlated with treatment responses to 5-fluorouracil in locally advanced breast cancer, colorectal cancer, gastric cancer, and other cancers (20-22). When we re-analyzed our previously published retrospective study on P16 methylation and 5-fluorouracil among 134 gastric cancer patients (22), we found that P16 methylation was strongly correlated with paclitaxel (and 5-fluorouracil) resistance; 44.9% of the P16 methylation-negative patients (N=49) responded to paclitaxel whereas none of the P16 methylation-positive patients (N=11) responded to paclitaxel (P=0.005). However, among the remaining 74 gastric cancer patients who received cisplatin and 5-fluorouracil treatments, no significant differences in responsivity were observed between the P16 methylation-positive and P16 methylation-negative patients (4/13 [30.8%] vs. 27/61 [44.3%], respectively; P=0.731). This indicates that P16 methylation may be a paclitaxel response predictor for cancers in different organs. A large-scale multicenter clinical trial is necessary to determine whether P16 methylation is beneficial as a predictor of paclitaxel-based treatments for gastric cancer and other cancers.
The P16 gene is weakly expressed in normal cells and frequently inactivated by DNA methylation in many cancers (13,16-19, 37,38). Because of the limited number of available biopsies, we did not analyze the potential of using P16 immunostaining to predict paclitaxel resistance. P16 functions as a tumor suppressor in normal cell nuclei. However, most P16 proteins are aberrantly located in the cytoplasm of cancer cells. Because of the abnormal compartmentalization of P16 in cancer cells and the P16 methylation-induced inactivation of both P16 and ANRIL, P16 methylation and P16 should be considered two different biomarkers.
No significant difference in OS and PFS were seen between patients with or without P16 methylation. The small case number of our study may account for this phenomenon. In addition, it should be noted that patients received radiotherapy at the same time. The present study only provided an important clue on induction of resistance to paclitaxel by P16 methylation. To validate the application feasibility of using P16 methylation as a biomarker to predict paclitaxel resistance, additional clinical trials with specifically defined treatment regimens and larger sample sizes are necessary.
In NSCLC and gastric cancer patients, there were consistent, but not significant, differences in OS durations between P16 methylation-positive and P16 methylation-negative patients suggesting that paclitaxel-based treatments result in more short-term benefits than long-term benefits among the P16 methylation-negative patients. The short-term benefits may be especially useful in decision-making for patients who need to receive neoadjuvant chemotherapy to reduce cancer staging.
In conclusion, the present study demonstrated, for the first time, that P16 methylation is significantly correlated with the clinical response to paclitaxel-based chemotherapy and may be a potential biomarker to predict paclitaxel resistance in advanced NSCLC. P16 methylation may directly induce paclitaxel resistance and could be a therapy target for re-sensitizing refractory cancers to paclitaxel-based chemotherapy.
Abbreviations
NSCLC, non-small cell lung carcinoma; IC50, the half maximal inhibitory concentration; DHPLC, denatured high performance liquid chromatography; FFPE, formalin fixed paraffin embedded.
Supplementary Material
Supplementary figure.
Acknowledgements
This work was supported by the Education Ministry Innovative Research Team Program [grant number IRT13003 to GZ and DD] and the National Natural Science Foundation of China [grant number 91640108 to DD].
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115-132
2. Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J, Zhu J, Johnson DH, E.C.O. Group. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med. 2002;346:92-98
3. Pozzo C, Barone C. Is there an optimal chemotherapy regimen for the treatment of advanced gastric cancer that will provide a platform for the introduction of new biological agents? Oncologist. 2008;13:794-806
4. Gao G, Chu H, Zhao L, Gui T, Xu Q, Shi J. A meta-analysis of paclitaxel-based chemotherapies administered once every week compared with once every 3 weeks first-line treatment of advanced non-small-cell lung cancer. Lung Cancer. 2012;76:380-386
5. Zeng H, Zheng R, Guo Y, Zhang S, Zou X, Wang N, Zhang L, Tang J, Chen J, Wei K, Huang S, Wang J, Yu L, Zhao D, Song G, Shen Y, Yang X, Gu X, Jin F, Li Q, Li Y, Ge H, Zhu F, Dong J, Guo G, Wu M, Du L, Sun X, He Y, Coleman MP, Baade P, Chen W, Yu XQ. Cancer survival in China, 2003-2005: a population-based study. Int J Cancer. 2015;136:1921-1930
6. Xue C, Hu Z, Jiang W, Zhao Y, Xu F, Huang Y, Zhao H, Wu J, Zhang Y, Zhao L, Zhang J, Chen L, Zhang L. National survey of the medical treatment status for non-small cell lung cancer (NSCLC) in China. Lung Cancer. 2012;77:371-375
7. Belani CP, Ramalingam S, Perry MC, LaRocca RV, Rinaldi D, Gable PS, Tester WJ. Randomized, phase III study of weekly paclitaxel in combination with carboplatin versus standard every-3-weeks administration of carboplatin and paclitaxel for patients with previously untreated advanced non-small-cell lung cancer. J Clin Oncol. 2008;26:468-473
8. Socinski MA. Update on taxanes in the first-line treatment of advanced non-small-cell lung cancer. Curr Oncol. 2014;21:e691-703
9. Cottini F, Lautenschlaeger T. Predictors of biomarkers guiding targeted therapeutic strategies in locally advanced lung cancer. Cancer J. 2013;19:263-271
10. Ohashi T, Yoshimasu T, Oura S, Kokawa Y, Kawago M, Hirai Y, Miyasaka M, Aoishi Y, Kiyoi M, Nishiguchi H, Honda M, Okamura Y. Class III Beta-tubulin Expression in Non-small Cell Lung Cancer: A Predictive Factor for Paclitaxel Response. Anticancer Res. 2015;35:2669-2674
11. Vaidyanathan A, Sawers L, Gannon AL, Chakravarty P, Scott AL, Bray SE, Ferguson MJ, Smith G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br J Cancer. 2016;115:431-441
12. Arima Y, Hayashi N, Hayashi H, Sasaki M, Kai K, Sugihara E, Abe E, Yoshida A, Mikami S, Nakamura S, Saya H. Loss of p16 expression is associated with the stem cell characteristics of surface markers and therapeutic resistance in estrogen receptor-negative breast cancer. Int J Cancer. 2012;130:2568-2579
13. Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sidransky D. 5' CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995;1:686-692
14. Cui C, Gan Y, Gu L, Wilson J, Liu Z, Zhang B, Deng D. P16-specific DNA methylation by engineered zinc finger methyltransferase inactivates gene transcription and promotes cancer metastasis. Genome Biology. 2015;16:252
15. Gan Y, Ma W, Wang X, Qiao J, Zhang B, Cui C, Liu Z, Deng D. Coordinate Transcription of ANRIL and P16 Genes Silenced by DNA Methylation. Chinese Journal of Cancer Research. 2018;30:93-103
16. Vaissière T, Hung RJ, Zaridze D, Moukeria A, Cuenin C, Fasolo V, Ferro G, Paliwal A, Hainaut P, Brennan P, Tost J, Boffetta P, Herceg Z. Quantitative analysis of DNA methylation profiles in lung cancer identifies aberrant DNA methylation of specific genes and its association with gender and cancer risk factors. Cancer Res. 2009;69:243-252
17. Nikolaidis G, Raji OY, Markopoulou S, Gosney JR, Bryan J, Warburton C, Walshaw M, Sheard J, Field JK, Liloglou T. DNA methylation biomarkers offer improved diagnostic efficiency in lung cancer. Cancer Res. 2012;72:5692-5701
18. Sun Y, Deng DJ, You WC, Bai H, Zhang L, Zhou J, Shen L, Ma JL, Xie YQ, Li JY. Methylation of p16 CpG islands associated with malignant transformation of gastric dysplasia in a population-based study. Clin Cancer Res. 2004;10:5087-5093
19. Luo DY, Zhang BZ, Lv LB, Xiang SY, Liu YH, Ji JF, Deng DJ. Methylation of CpG islands of p16 associated with progression of primary gastric carcinomas. Lab Invest. 2006;86:591-598
20. Klajic J, Busato F, Edvardsen H, Touleimat N, Fleischer T, Bukholm I, Børresen-Dale AL, Lønning PE, Tost J, Kristensen VN. DNA methylation status of key cell-cycle regulators such as CDKNA2/p16 and CCNA1 correlates with treatment response to doxorubicin and 5-fluorouracil in locally advanced breast tumors. Clin Cancer Res. 2014;20:6357-6366
21. Han SW, Lee HJ, Bae JM, Cho NY, Lee KH, Kim TY, Oh DY, Im SA, Bang YJ, Jeong SY, Park KJ, Park JG, Kang GH. Methylation and microsatellite status and recurrence following adjuvant FOLFOX in colorectal cancer. Int J Cancer. 2013;132:2209-2216
22. Wang M, Li Y, Gao J, Li Y, Zhou J, Gu L, Shen L, Deng D. p16 Methylation is associated with chemosensitivity to fluorouracil in patients with advanced gastric cancer. Med Oncol. 2014;31:988
23. Lin H, Chen Y, Shi A, Pandya KJ, Yu R, Yuan Y, Li J, Li H, Wang Y, Xia T, Feng L, Ma H, Geng J, Zhu G. Phase 3 randomized low-dose paclitaxel chemoradiotherapy study for locally advanced non-small cell lung cancer. Front Oncol. 2016;6:260
24. Therasse P, Eisenhauer EA, Verweij J. RECIST revisited: a review of validation studies on tumour assessment. Eur J Cancer. 2016;42:1031-1039
25. Eads CA, Lord RV, Wickramasinghe K, Long TI, Kurumboor SK, Bernstein L, Peters JH, DeMeester SR, DeMeester TR, Skinner KA, Laird PW. Epigenetic patterns in the progression of esophageal adenocarcinoma. Cancer Res. 2001;61:3410-3418
26. Liu Z, Zhou J, Gu L, Deng D. Significant impact of amount of PCR input templates on various PCR-based DNA methylation analysis and countermeasure. Oncotarget. 2016;7:56447-56455
27. Deng DJ, Deng GR, Smith MF, Zhou J, Xin HJ, Powell SM, Lu YY. Simultaneous detection of CpG methylation and single nucleotide polymorphism by denaturing high performance liquid chromatography. Nucleic Acids Res. 2002;30:13e
28. Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821-9826
29. Gan Y, Li PY, Han X, Qin SS, Cui CH, Liu ZJ, Zhou J, Gu LK, Lu ZM, Zhang BZ, Deng DJ. Hydroxymethylated-P16 Allele Is Transcription-Inactive. BioRxiv. doi: 10.1101/405522
30. Zheng X, Zhou J, Zhang B, Zhang J, Wilson J, Gu L, Zhu B, Gu J, Ji JF, Deng DJ. Critical evaluation of Cbx7 downregulation in primary colon carcinomas and its clinical significance in Chinese patients. BMC cancer. 2015;15:1172
31. Serrano M, Hannon GJ, Beach D. A new regulatorymotif in cell-cycle control causing specific inhibition of cyclinD/CDK4. Nature. 1993;366:704-707
32. Higashiyama M, Miyoshi Y, Kodama K, Yokouchi H, Takami K, Nishijima M, Nakayama T, Kobayashi H, Minamigawa K, Nakamura Y. p53-regulated GML gene expression in non-small cell lung cancer. a promising relationship to cisplatin chemosensitivity. Eur J Cancer. 2000;36:489-495
33. Longley DB, Johnston PG. Molecular mechanisms of drug resistance. J Pathol. 2005;205:275-292
34. Zhang XH, Cheng Y, Shin JY, Kim JO, Oh JE, Kang JH. A CDK4/6 inhibitor enhances cytotoxicity of paclitaxel in lung adenocarcinoma cells harboring mutant KRAS as well as wild-type KRAS. Cancer Biol Ther. 2013;14:597-605
35. Gao Y, Shen J, Choy E, Mankin H, Hornicek F, Duan Z. Inhibition of CDK4 sensitizes multidrug resistant ovarian cancer cells to paclitaxel by increasing apoptosiss. Cell Oncol (Dordr). 2017;40:209-218
36. Xu R, Mao Y, Chen K, He W, Shi W, Han Y. The long noncoding RNA ANRIL acts as an oncogene and contributes to paclitaxel resistance of lung adenocarcinoma A549 cells. Oncotarget. 2017;8:39177-39184
37. Liu HW, Liu XW, Dong GY, Zhou J, Liu Y, Gao Y, Liu XY, Gu LK, Sun Z, Deng DJ. P16 methylation as an early predictor for cancer development from oral epithelial dysplasia: a double-blind multicentre prospective study. EbioMedicine. 2015;2:432-437
38. Deng D, Liu Z, Du Y. Epigenetic alterations as cancer diagnostic, prognostic, and predictive Biomarkers. In: (ed.) Herceg Z, Ushijima T. Epigenetics and Cancer. Elsevier Science publi. 2010:125-176
Author contact
![]() Corresponding authors: Prof. Guangying Zhu, Department of Radiation Oncology, Peking University Cancer Hospital and Institute, Beijing, 100142, China; Prof. Dajun Deng, Division of Etiology, Peking University Cancer Hospital and Institute, Fu-Chen-Lu #52, Haidian District, Beijing, 100142, China. Email: zgypu163com or dengdajunedu.cn
Corresponding authors: Prof. Guangying Zhu, Department of Radiation Oncology, Peking University Cancer Hospital and Institute, Beijing, 100142, China; Prof. Dajun Deng, Division of Etiology, Peking University Cancer Hospital and Institute, Fu-Chen-Lu #52, Haidian District, Beijing, 100142, China. Email: zgypu163com or dengdajunedu.cn
Citation styles
APA
Liu, Z., Lin, H., Gan, Y., Cui, C., Zhang, B., Gu, L., Zhou, J., Zhu, G., Deng, D. (2019). P16 Methylation Leads to Paclitaxel Resistance of Advanced Non-Small Cell Lung Cancer. Journal of Cancer, 10(7), 1726-1733. https://doi.org/10.7150/jca.26482.
ACS
Liu, Z.; Lin, H.; Gan, Y.; Cui, C.; Zhang, B.; Gu, L.; Zhou, J.; Zhu, G.; Deng, D. P16 Methylation Leads to Paclitaxel Resistance of Advanced Non-Small Cell Lung Cancer. J. Cancer 2019, 10 (7), 1726-1733. DOI: 10.7150/jca.26482.
NLM
Liu Z, Lin H, Gan Y, Cui C, Zhang B, Gu L, Zhou J, Zhu G, Deng D. P16 Methylation Leads to Paclitaxel Resistance of Advanced Non-Small Cell Lung Cancer. J Cancer 2019; 10(7):1726-1733. doi:10.7150/jca.26482. https://www.jcancer.org/v10p1726.htm
CSE
Liu Z, Lin H, Gan Y, Cui C, Zhang B, Gu L, Zhou J, Zhu G, Deng D. 2019. P16 Methylation Leads to Paclitaxel Resistance of Advanced Non-Small Cell Lung Cancer. J Cancer. 10(7):1726-1733.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.