Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(9):2109-2127. doi:10.7150/jca.30410 This issue Cite
Review
BRCA Genes: The Role in Genome Stability, Cancer Stemness and Therapy Resistance
Ielizaveta Gorodetska1 ![]() , Iryna Kozeretska2, Anna Dubrovska3
, Iryna Kozeretska2, Anna Dubrovska3 ![]()
1. OncoRay-National Center for Radiation Research in Oncology, Faculty of Medicine and University Hospital Carl Gustav Carus, Technische Universität Dresden and Helmholtz-Zentrum Dresden-Rossendorf, Dresden, Germany;
2. Department of General and Medical Genetics, ESC "The Institute of Biology and Medicine", Taras Shevchenko National University of Kyiv, Kyiv, Ukraine;
3. OncoRay - National Center for Radiation Research in Oncology, Faculty of Medicine and University Hospital Carl Gustav Carus, Technische Universität Dresden, Germany; Helmholtz-Zentrum Dresden - Rossendorf, Institute of Radiooncology - OncoRay, Dresden, Germany; German Cancer Consortium (DKTK), Partner site Dresden, Germany; German Cancer Research Center (DKFZ), Heidelberg, Germany.
Received 2018-10-4; Accepted 2019-2-20; Published 2019-5-14
Abstract
Carcinogenesis is a multistep process, and tumors frequently harbor multiple mutations regulating genome integrity, cell division and death. The integrity of cellular genome is closely controlled by the mechanisms of DNA damage signaling and DNA repair. The association of breast cancer susceptibility genes BRCA1 and BRCA2 with breast and ovarian cancer development was first demonstrated over 20 years ago. Since then the germline mutations within these genes were linked to genomic instability and increased risk of many other cancer types. Genomic instability is an engine of the oncogenic transformation of non-tumorigenic cells into tumor-initiating cells and further tumor evolution. In this review we discuss the biological functions of BRCA1 and BRCA2 genes and the role of BRCA mutations in tumor initiation, regulation of cancer stemness, therapy resistance and tumor progression.
Keywords: BRCA1, BRCA2, genomic instability, cancer stem cells, cancer treatment
Introduction
Mutations in the tumor suppressor genes BRCA1 (breast cancer susceptibility gene 1) and BRCA2 (breast cancer susceptibility gene 2) predispose to different types of cancer. The association of BRCA1 and BRCA2 genes with breast and ovarian cancer susceptibility was first demonstrated over 20 years ago [1], [2]. Since then specific germline mutations in BRCA1 and BRCA2 genes were linked to increased risk of several additional types of human malignancies including prostate, colorectal, stomach and pancreatic cancers [3]-[5]. Mutations in BRCA1 and BRCA2 genes are associated with about 20% of familial breast and ovarian cancers [4], [6]. In contrast to the gynecological tumors, the reported contribution of BRCA1 and BRCA2 mutations to the hereditary of pancreatic, stomach and prostate cancer is marginal [3]-[5].
Both BRCA1 and BRCA2 proteins play a crucial role in the maintenance of genomic integrity through the process of precise DNA repair by homologous recombination [7], [8]. Loss of BRCA functions results in the genomic instability that eventually results in the oncogenic transformation of non-tumorigenic cells into tumor initiating cells, or cancer stem cells (CSCs) and further tumor evolution. During the last decade, a number of studies demonstrated that cancer cells within the same tumor substantially differ by degree of their tumor initiating ability. A CSC population possesses a long-term self-renewal capacity, as well as a potential to differentiate into other tumor cell types and initiate tumor growth. Given an extensive self-renewal and clonogenic potential of CSCs coupled to a high level of genomic instability attributed to tumor cells, CSCs might play a role as an engine of cancer evolution [9], [10]. Accordingly, intratumoral heterogeneity depends on the evolution of CSCs that is reflected in the number of emerging tumor clones.
Until recently, the role of DNA repair mechanisms and, in particular, BRCA proteins in regulation of CSC populations was not clear. Over the last years several seminal studies highlighted the role of BRCA proteins in the maintenance and evolution of the CSC populations. This review focuses on the cellular mechanisms deregulated in BRCA mutated cancers which appear to be important for CSC development, maintenance and therapy resistance.
BRCA genes, BRCA proteins and their clinical relevance
Tumor suppressor genes BRCA1 and BRCA2 were first linked to the breast and ovarian cancer susceptibility by Mick and colleagues in 1994 (BRCA1) [1] and by Wooster et al. in 1995 (BRCA2) [2]. Since then the number of studies demonstrated a role of BRCA genes in the different cellular processes regulating tumor development has grown dramatically. BRCA1 (17q21, chromosome 17: base pairs 43,044,294 to 43,125,482) is a 1,863 amino acids protein composed of 24 exons. It consists of several domains that are essential for its multiple functions. At the N-terminal region it carries zinc-binding finger domain RING (Really Interesting New Gene) which is essential for interaction of BRCA1 and BARD1 (BRCA1 Associated RING Domain protein 1) and formation of E3 ubiquitin ligase complex [11]. At the C terminus two phosphopeptide-binding BRCT (BRCA1 C-terminal) domains [12] are mediating interaction of BRCA1 with key partner proteins such as CtIP (C-terminal binding protein 1 (CtBP1) interacting protein), BRCA1 A Complex Subunit (ABRAXAS), and BRCA1 interacting protein C-terminal helicase 1 (BRIP1/ FACJ) [13]. The central part of BRCA1 is encoded by exons 11-13 and the mutations in these regions are frequently detected in breast cancer patients. These parts consist of two nuclear localization signals (NLS) and one coiled coil domain which is important for interaction with BRCA2 through partner and localizer of BRCA2 (PALB2) [14], [15] (Figure 1).
Schematic representation of functional domains within BRCA1 and BRCA2 proteins and the position of several founder mutations. BRCA1 is composed of 23 exons and BRCA2 includes 27 exons. Both genes encode large proteins: BRCA1 consists of 1,863 amino acids and BRCA2 of 3,418 amino acids. BRCA1 has a highly conserved zinc-binding RING (really interesting new gene) finger domain which is located close to the N-terminus. At the C-terminus, two BRCT (BRCA1 C-terminal) domains are located. The central part of BRCA1 consists of two NLS (nuclear localization signals) and one coiled coil domain. BRCA2 contains eight copies of a 20-30 amino acid repeat, termed BRC repeats. At the amino-terminus, BRCA2 has a TAD (transcriptional activation domain) domain and at the carboxyl-terminus two NLS and one TR2 domain. DNA-binding domain is located close to the C-terminal region and is composed of a conserved helical domain (H), three oligonucleotide binding (OB) folds and a tower domain (T). Domains are indicated by violet (BRCA1) and green (BRCA2) boxes. Domain names are shown above. Exons are indicated by braces. Positions of founder mutations are indicated beneath.
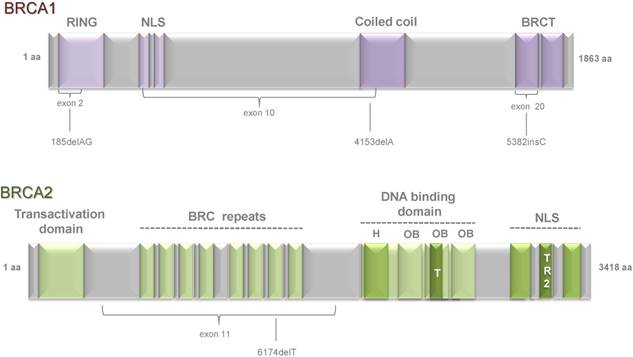
More than 1600 mutations were identified in BRCA1 gene including deletions, insertions, and many single nucleotide polymorphisms in the coding or noncoding sequences [16]. Some of them occur with high frequency in isolated groups and are called founder mutations. The most frequently identified BRCA1 mutations are located in the gene regions corresponding to the BRCT and RING domains as well as in the exons 11-13 encoding NLS essential for BRCA1 functions and binding sites for different BRCA1-interacting proteins including c-Myc, Rad50, pRb, Rad51, BRCA2 and PALB2 [17]. A founder mutation 185delAG in the region of RING and BRCT domains was found among Ashkenazi Jews [18], and 5382insC frameshift mutation arose in either Scandinavia or northern Russia [19]. A number of mutations in exon 11-13 of BRCA1 gene were associated with breast and ovarian cancer [20], [21]. The majority of the lesions in BRCA1 gene are frameshift insertions/ deletions, nonsynonymous truncations, and disruptions of splice site resulting in missense mutations or expression of non-functional proteins [22], [23]. In general, mutations in BRCA1 gene predispose to the different cancer types, such as breast and ovarian cancer in women, male breast cancer and prostate cancer. In addition, BRCA1 mutation carriers may be at high risk for the development of other types of cancer including colon, rectal, pancreatic cancer [24] as well as stomach cancer [25].
BRCA2 (13q12.3, chromosome 13: base pairs 32,315,479 to 32,399,671) is a large 3418 amino acids protein. It contains 27 exons and covers approximately 84.2 kb of genomic DNA. At the N-terminus BRCA2 contains a transcriptional activation domain (TAD). The middle part is encoded by exon 11 and contains eight conserved motifs termed BRC repeats that bind to RAD51 [26]. A DNA-binding domain is located in the carboxyl terminus of the BRCA2 protein and assembled of a conserved helical domain, three oligonucleotide binding (OB) folds and a tower domain (T), which facilitates BRCA2 binding to double-stranded DNA (dsDNA) and single-stranded DNA (ssDNA) [27]. The C terminus of BRCA2 contains two NLS and one TR2 domain (Figure 1).
As of today, more than 1800 mutations have been identified in BRCA2 [16]. These lesions mainly include frameshift insertions, deletions and nonsense mutations and result in premature truncation or non-functional protein [28]. The most frequently identified germline frameshift mutation 6174delT was found in exon 11 of BRCA2 gene in an Ashkenazi Jewish population [29]. Mutations in BRCA2 predispose not just to breast, ovarian and prostate cancer, but are also associated with malignant cutaneous and ocular melanoma, pancreatic, gall bladder, bile duct and stomach cancer [30].
The relationship between the domain functions of BRCA1/2 proteins and tumor development have been intensively investigated in animal models as reviewed elsewhere [13], [31], [32] although the results of these studies are still awaiting to be confirmed by clinical data. About 70-80% of the mutations in BRCA genes result in protein dysfunction or absence of protein product. These mutations were confirmed as clinically relevant and are associated with an increased risk for development of hereditary malignancies [33]. Many studies have also reported an association of BRCA1/2 mutations with tumor aggressiveness and poor clinical outcomes in cancer patients. Large number of studies demonstrated the association of BRCA1/2 mutations in prostate cancer patients with an increased rate of intermediate- and high risk disease [34]-[37]. Resent study of 603 sporadic pancreatic cancer patients in China revealed an association between germline missense variant rs1799966 (c.4837A>G[p.Ser1613 Gly]) within the BRCT domain of BRCA1 gene and lower overall patients' survival rates [38].
On the other hand, clinical studies of potential correlation between BRCA1 and BRCA2 mutations and outcome in breast cancer patients provide conflicting results. A resent prospective multi- hospital study for 2733 young women diagnosed with breast cancer including 388 patients with BRCA1/2 mutations showed no difference in overall survival for BRCA mutations carriers and non-carriers [39]. Moreover, analysis of the 558 patients with triple-negative breast cancer demonstrated that BRCA1/2 mutation carriers had better overall survival than non-carriers [39]. At the same time, loss of heterozygosity (LOH) at BRCA1 locus was associated with significantly shorter disease-free survival, distant metastasis-free survival, and overall survival in retrospective study of 202 Japanese patients with invasive breast cancer [40]. Another retrospective analysis of the pathogenic germline BRCA2 mutations in 458 Chinese individuals who had breast cancer showed that BRCA2 mutation status is associated with a higher rate of lymph node metastases at diagnosis and significantly worse outcomes including disease free survival and distant recurrence [41]. The clinical studies of ovarian cancer also provide conflicting data for the effect of BRCA gene mutations on patients' prognosis. A retrospective study for 53 patients with germline BRCA1 mutations showed that BRCA- mutation carriers have more favorable clinical outcome than sporadic cancers [42]. Another study which analyzed 151 ovarian cancer patients with germline BRCA1/2 mutations and 119 patients with sporadic ovarian cancer demonstrated that survival of the patients with familial ovarian cancer is worse compared to the patients with sporadic cases independently of the BRCA1/2 mutation status [43]. A resent meta-analysis based on 33 clinical studies showed that BRCA1 mutations can be associated with improved overall survival in ovarian cancer patients [44].
To further elucidate the effect of BRCA1/2 mutations on the prognosis of breast and ovarian patients, more prospective studies are warranted regarding the contribution of individual pathological mutations to the tumor progression and patients' response to therapy. Better understanding of the relationship between the pathophysiological role of BRCA mutations and tumor aggressiveness may be beneficial for a more individualized clinical management of the BRCA-related cancers.
Biological function of BRCA1 and BRCA2
BRCA genes as caretakers of genomic stability
BRCA1 and BRCA2 proteins play a crucial role in the process of DNA double strand break (DSB) repair by regulation of homologous recombination (HR) [45]. HR is a high fidelity mechanism of DNA repair with using of homologous template such as sister chromatids. Therefore, this process can be active in S and G2 phase of cell cycle when sister chromatids are available for HR. The homology-directed DNA repair process includes a few steps such as pre-synapsis, synapsis and post-synapsis [46], [47]. In the first step the Mre11-RAD50-Nbs1 (MRN) complex and its interacting partner C-terminal binding protein interacting protein (CtIP) with nuclease activity performs resection of the DSB ends to generate a 3′ - single strand (ss) DNA tail protected from degradation by replication protein A (RPA). Next, dependent on BRCA1 and BRCA2 proteins, RAD51-ssDNA filament invades homologous duplex DNA which serves as a template. This generates a D-loop (displacement loop) structure, which is a DNA structure where the strands of a double strand (ds) DNA are separated for a stretch by a third strand of DNA. The resulted nucleoprotein-filament searches for homologous DNA sequence on the sister chromatid and invades the duplex to form a joint molecule. Finally, during post-synapsis RAD51 dissociates from dsDNA and the 3′ end of the damaged DNA is elongated by DNA polymerases and followed by DNA ligation [48]. The resulted intermediate structures of DNA recombination called Holliday junctions are further resolved by the different mechanisms as discussed elsewhere [49] resulting in an error-free repair.
In addition to its role in HR-dependent DNA repair, BRCA1 also regulates the non-homologous end-joining (NHEJ) repair pathway. NHEJ is one of the main DNA repair pathways when the broken DNA ends are directly ligated without the need for a homologous template. HR is an error-free repair with a high fidelity at the site of correction, but it needs more time to complete. In contrast, NHEJ is an error-prone DNA reparation that causes mutations at the site of damage with a high frequency. However, it is relatively fast and the most common repair mechanism for DNA DSBs [50]. Classical (C) NHEJ predominates in G0 and G1 but can operate in all phases of the cell cycle. It consists of a few steps: break recognition, end-processing and ligation. First, DSB ends are recognized by the Ku70/Ku80 heterodimer, which recruits DNA-dependent protein kinase (DNA-PK) and other NHEJ proteins such as DNA polymerase, helicase and ligase. Then DNA-PK phosphorylates and recruits endonuclease Artemis, which further processes DSB ends. End ligation is facilitated by the XRCC4 (X-ray repair cross- complementing protein 4)/Lig4 [50].
In contrast to C-NHEJ mechanism, alternative (A)-NHEJ depends on other factors such as MRN complex, CtIP, and poly (ADP-ribose) polymerase-1 (PARP-1) that plays a role in the DNA lesion detection and recruitment of the protein to the sites of DNA damage [51], [52]. A-NHEJ functions as a backup repair pathway when C-NHEJ is compromised, and its mechanism is less defined than for C-NHEJ [53].
BRCA1 is involved in both, C-NHEJ and A-NHEJ pathways. BRCA1 interaction with the C-NHEJ factor Ku80 stabilizes the Ku heterodimer at DSB sites that is required for precise end-joining repair. In contrast, a growing body of evidence suggests that BRCA1 blocks A-NHEJ through phosphorylation of BRCA1 at S988 by checkpoint kinase 2 (Chk2) [54], [55], but the exact mechanism of this regulation is unknown.
Studies of BRCA genes revealed their interaction with HR factors such as RAD51 [56], [57] a central regulator of the strand exchange [58]. Recent studies demonstrated that BRCA1 promotes HR-dependent DNA repair by dephosphorylation of 53BP1 (p53-binding protein 1) [59] that consequently results in the repair pathway switch from NHEJ to HR. Interaction between BRCA1, CtIP and MRN complex has been shown to be important for activation of HR by the mechanisms involving CDK (cyclin-dependent kinase)-mediated phosphorylation of CtIP at Ser327 [60]. BRCA1 is important for BRCA2 recruitment to the sites of DNA DSBs during HR, and association between these two proteins is mediated through interaction with PALB2/FANCN (Fanconi anemia, complementation group N) protein [15], [61].
The role of BRCA2 in repair of DSBs has also been extensively studied. It was demonstrated that BRCA2 plays an essential role in HR by the mechanisms involving recruitment of RAD51 to the sites of DSBs [62]. The loss of BRCA2 leads to genomic instability and tumorigenesis [56]. This can be, in part, explained by the role of BRCA2 in regulation of the intracellular localization and DNA binding ability of RAD51 recombinase. After detection of DSB by the MRN complex, the consequent protein phosphorylation and ubiquitination events recruit BRCA1 and CtIP proteins to the site of DNA DSB. Together with Exo1 and DNA2-BLM (Bloom syndrome protein) exonucleases, this complex triggers DNA end resection and mobilizes BRCA2 to the sites of DNA DSBs. BRCA2 promotes HR by the displacement of RPA and recruitment of RAD51 recombinase on the sites of DNA damage. BRCA2 directly binds to RAD51 through its BRC repeats and TR2 domain and therefore facilitates the loading of RAD51 on ssDNA and a search of homologous DNA template [63]-[65]. BRCA2 may function as a complex with RAD51 paralogs such as XRCC2 and XRCC3 to facilitate an assembly of RAD51 with ssDNA [66].
BRCA2 plays a protective role for the maintenance of genomic stability upon replication stress. It has been characterized as a regulator of the stalled DNA replication fork by loading and stabilization of polymerized RAD51 onto DNA through binding to its BRC repeats [67]. At the same time BRCA2 can prevent formation of chromosomal aberrations during replication stalling by inhibition of MRE11 nuclease [68] (Figure 2). BRCA2 is also recruited by 3′-repair exonuclease 2 (TREX-2) complexes for processing of R-loops, the structures formed during transcription and composed of a DNA-RNA hybrid and associated ssDNA [69]. BRCA2 can protect telomere integrity via loading of RAD51 on telomeres during S/G2 phase that is evidenced by the accumulation of telomere dysfunction-induced foci and telomere shortening in Brca2- but not Brca1- deficient mice [70].
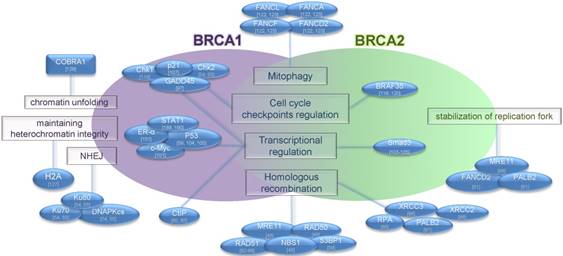
Functional features of BRCA proteins. The BRCA1 protein has multiple functions in different cellular processes, including DNA repair, transcriptional activation, cell cycle regulation and chromatin remodeling. BRCA2 plays a role in transcriptional and cell cycle regulation, DNA repair, mitophagy and stabilization of replication fork. BRCA proteins and interacting partners are shown. Functions of proteins are shown in rectangles. Interacting partners are shown in blue ovals.
BRCA1 as a regulator of oxidative stress
BRCA1 also acts as protector of genome stability from oxidative stress caused by chemically reactive molecules called reactive oxygen species (ROS). Normally they are formed as a side product of the oxygen metabolism and play an important role in cell signaling and homeostasis [71]. However, under stress conditions, such as nutrient deficiency, metal toxicity, chemotherapy as well as UV- and X-ray radiation, ROS levels can increase dramatically that result in substantial damage to the cellular structures [72]. It was shown that loss of BRCA1 increases cellular ROS, and BRCA1 overexpression reduces ROS levels [73], [74]. While the mechanisms of this regulation remain poorly understood, one study revealed that BRCA1 can protect vascular smooth muscle cells from H2O2-induced ROS production, in part, by downregulation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase subunits Nox1 and p47phox [75]. BRCA1 mutant mice which are also heterozygous for a p53-null mutation exhibited high levels of ROS and were more sensitive to oxidative stress-induced lethality [76]. Of note, this mouse model also displayed an increased expression of Redd1 (regulated in development and DNA damage response 1), which is an inhibitor of mTORC1 and an inducer of ROS production [77], [78].
BRCA1-BARD1 ubiquitin ligase activity
BRCA1 possesses an E3 ubiquitin ligase activity causing a posttranslational modification of target molecules by covalent coupling of a small (76 amino acids) ubiquitin protein [79]. Ubiquitination regulates different cellular processes including protein activity, degradation and intracellular localization [79]. Ubiquitination process depends on three main enzymatic components: ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3) [80]. E3 ligase activity of BRCA1 depends on heterodimerization with BARD1 via its N-terminal RING finger domain and formation of the BRCA1-BARD1 ligase complex [11], [81]. Deficiency of either BRCA1 or BARD1 in this heterodimeric complex reduces its stability and prevents its ubiquitin E3 ligase activity [81]. BRCA1 is reported to catalyze ubiquitination of a plethora of proteins including cell cycle regulators Cyclin B and Cdc25C [82], IGF-1 receptor [83], nucleophosmin/B23 (NPM) [84], CtIP [85], polymerase (RNA) II (DNA directed) polypeptide H (POLR2H) [70], transcription factor 776 IIE (TFIIE) [87], histones H2A, H2B, H3, H4, and H2AX [81], as well as γ-tubulin [88], estrogen receptor α (ERα) [89] and BRCA1 itself [90]. BRCA1- dependent ubiquitination differentially regulates stability of target proteins. For example, ubiquitination of Cyclin B and Cdc25C results in their accelerated degradation [82]. In contrast, BRCA1-BARD1 mediated ubiquitination of NPM results in NPM stabilization rather than degradation [84]. Similarly, BRCA1- mediated ubiquitination of CtIP is not associated with proteasomal pathways, but with induced binding of CtIP to chromatin and foci formation after DNA damage [85]. BRCA1-dependent ubiquitination also does not destabilize another target protein, RNA Polymerase II Subunit H (POLR2H). POLR2H is polyubiquitinated by BRCA1-BARD1 complex after UV irradiation, and tumor cell lines stably expressing a mutant form of POLR2H which is resistant to polyubiquitination exhibited a high sensitivity to UV irradiation [86]. The BRCA1-dependent protein ubiquitination is also a mechanism of inhibition of mRNA synthesis. In particular, it prevents association of the transcription factors TFIIE and TFIIH, and thus blocks the initiation of transcription [87]. BRCA1- BARD1 complex ubiquitinates the nucleosome core histones H2A, H2B, H3 and H4 as well as the phosphorylated form of γH2AX that co-localizes in nuclear foci with BRCA1 at sites of DNA damage [81]. The ubiquitination of γH2AX by BRCA1-BARD1 at DNA breaks plays an important role in HR- dependent DNA repair by promoting 53BP1 repositioning and accumulation of the SWI/SNF- related matrix-associated actin-dependent regulator of chromatin (SMARCAD1) protein at the sites of DNA damage [81] [91]. BRCA1-BARD1 dependent ubiquitination of γ-tubulin might protect cells from amplification of centrosomes and aneuploidy [92]. Of importance, BRCA1-BARD1 ubiquitinates ERα, and its abrogation results in ERα accumulation [89], [93]. The cancer-related BRCA1 mutations have been shown to abrogate ERα ubiquitination that can partially explain tissue specificity of BRCA-dependent tumor development [89]. Moreover, BRCA1-BARD1 can be self-ubiquitinated that plays a role in DNA repair by increasing BRCA1 binding to DNA [90] [94].
The large number of BRCA1 mutations occur in N-terminal RING domain [17] [95], which is responsible for the E3 ubiquitin ligase activity of BRCA1 [11]. A growing body of evidence suggests that mutations in the BRCA1 RING domain, which inactivate BRCA1 ubiquitin protein ligase activity, may predispose to cancer development [95] [89] [96]. Study of Nelson and Holt showed that BRCA1 protein with mutation in the RING domain fails to co-localize with BRCA1-interacting proteins BARD1 and BACH1, which are essential for DNA repair [96]. Another study by Hashizume et al. showed that BRCA1-BARD1 heterodimeric complex can be inactivated by RING finger mutation which results in the loss of ubiquitin ligase activity [11]. Taken together, the high rate of cancer-related mutations in specific domains of BRCA1 such as RING might explain the critical role of these domains for BRCA1 tumor suppressor activity.
BRCA-dependent transcriptional regulation
Both BRCA1 and BRCA2 proteins have been shown to be involved in transcriptional regulation [97], [98]. First studies that revealed a potential role of BRCA1 in transcriptional regulation employed a DNA plasmid construct encoding a BRCA1 C-terminal fragment (aa 1528-1863) fused to the yeast GAL4 (galactose-responsive transcription factor) DNA- binding domain (GAL4-BRCA1). This recombinant protein activated gene transcription in both mammalian and yeast cells [99]. Of importance, BRCA1 mutations found in breast and ovarian patients markedly decreased this transcriptional activity [99].
Since this seminal study, further investigations demonstrated that although BRCA1 protein can directly bind to DNA via its DNA binding domain it is not a sequence-specific transcriptional factor, but rather a co-regulator of a broad range of other transcriptional factors [100]. BRCA1 modulates activity of different transcription regulators including OCT-1, c-Myc, ERα, p53, Smad3 and others which were reviewed previously [101]. For example, BRCA1 interaction with ERα regulates transcription of VEGF (vascular endothelial growth factor) in breast cancer [102]. BRCA1 C-terminal region has been shown to stimulate transcription of the p53 target gene MDM2 (Mouse double minute 2 homolog) in breast cancer cells [100]. BRCA1 has been shown to synergize with Smad3 in induction of Smad3-specific promoter [103]. Some other studies have also shown the role of BRCA2 in transcriptional regulation, for example, by forming a complex with p53 and Smad3 [104], [105].
Regulation of cell cycle progression
Early studies discovered that BRCA1 becomes hyperphosphorylated during the late G1 and S phases of the cell cycle and is transiently dephosphorylated shortly after M phase that suggests its involvement in the regulation of cell cycle progression [106]. When introduced into mammalian or yeast cells, BRCA1 suppresses cell division [107], [108]. In response to DNA damage, BRCA1 is phosphorylated by several DNA-damage response kinases such as ATM, ATR and Chk1 at the DNA damage checkpoints enabling cell to repair DNA prior to mitotic entry and to survive after DNA damage [109]-[112]. BRCA1 inhibits G1/S cell cycle transition by induction of p21WAF1/CIP1, which is a cyclin-dependent kinase inhibitor [107]. In addition to the p21-mediated mechanism, BRCA1-induced G1 arrest depends on the presence of Rb protein. BRCA1 binds to the hypophosphorylated form of pRb and induces its dephosphorylation through cyclin dependent kinase 2 (CDK2) inhibition which results in the accumulation of cells in G1 or G2 phase of the cell cycle [113], [114]. According to the current view, BRCA1 induced G1/S arrest may occur through a number of distinct pathways that mainly involve ATM, ATR, BARD1, RB, p53, p21 and their downstream effectors [115]. Furthermore, BRCA1 loss results in defective S-, G2/M- and spindle checkpoints. Together with abnormal centrosome duplication and defective DNA damage repair, this leads to genetic instability in BRCA1 deficient cells [115]. BRCA1 has been characterized as an activator of G2/M checkpoint in response to anti-microtubule agents such as Taxol and Vincristine [97]. It has been demonstrated that BRCA1 regulates G2/M checkpoint by activation of the checkpoint kinase 1 (Chk1) in response to the DNA lesions [116]. Furthermore, BRCA1 interacts with mediator of DNA damage checkpoint protein 1 (MDC1). MDC1 acts together with γH2AX to recruit DNA repair proteins such as 53BP1, BRCA1 and MRN to the sites of DNA breaks and to induce cell arrest in S and G2/M phase of the cell cycle [117]. BRCA1 also plays a role in the induction of a spindle checkpoint which occurs during the cell division and prevents the aberrant separation of the sister chromatids. The cell cycle arrest during the mitotic phase depends on the presence of Mad2 protein which expression is regulated by BRCA1 [118]. A growing body of evidence suggests that BRCA2 protein may also contribute to the regulation of the cell cycle progression. Marmorstein et al. has demonstrated that histone H3 phosphorylation at Ser28 and Ser10, which plays a role in mitotic chromosome condensation, is conterminous with BRAF35/BRCA2 complexes on mitotic chromosomes. By using the microinjection of anti-BRAF35 or anti-BRCA2 antibodies in HeLa cell nuclei, this study demonstrated that BRCA2/BRAF35 complex is important for cell cycle progression since antibody microinjection delayed cell entry into mitosis [119], [120]. The recent studies demonstrated that BRCA2-deficient cells have a high sensitivity to the anti-cancer DNA binding drug S23906, which is attributed not only to the lack of HR dependent DNA repair but also to the defective S-phase checkpoint. [121].
The role of BRCA proteins in autophagy
Both BRCA1 and BRCA2 proteins were shown to play an essential role in the process of mitophagy, which is a specialized autophagy pathway that occurs to defective mitochondria and is important for mitochondrial quality control. Knockdown of BRCA1 and BRCA2 inhibited the clearance of mitochondria by mitophagy after treatment with oligomycin and antimycin A (two specific inhibitors of mitochondrial respiration) or with PARP inhibitor AZD2281 [122]- [124]. Tang et al. showed that small interfering (si) RNA-mediated knockdown of BRCA1 led to induction of pro-survival autophagy response under endoplasmic reticulum (ER) stress and serum starvation. They also demonstrated that inhibition of autophagy in BRCA1 deficient cells resulted in their sensitization to chemotherapeutic drugs [125]. Notably, induction of the protective autophagy depends on the BRCA1 functional activity. In contrast to ovarian cells without BRCA1 mutations, cells carrying BRCA1 mutations were able to induce pro-survival autophagy in response to chemotherapeutic agents [125]. This study suggested a role of BRCA1 as a differential regulator of chemotherapy- induced tumor cell death depending on its mutational status.
BRCA-mediated chromatin remodeling and epigenetic regulation of gene expression
BRCA1 plays a role in the epigenetic regulation of gene expression by chromatin remodeling. BRCA1 and its ubiquitin E3 ligase activity are required for the maintenance of gene silencing in the constitutive heterochromatin structure via ubiquitination of histone H2A. Modulation of the BRCA1 levels or its functions through BRCA1 knockout or expression of a pathogenic BRCA1 mutant with a lack of ubiquitin ligase activity (T37R) as well as expression of shRNA against BARD1 resulted in the transcriptional activation at tandemly repeated DNA regions [126], [127]. BRCA1 can directly bind to the BRG1 subunit of the SWItch/Sucrose Non-Fermentable (SWI/SNF) chromatin remodeling complex. Notably, the p53-mediated BRCA1-dependent gene transcription can be completely abrogated in the presence of the dominant negative BRG1 mutant. This study suggested that BRCA1-containing chromatin remodeling complexes can play a role in breast and ovarian carcinogenesis [128]. Ye et al. identified and characterized a novel cofactor of BRCA1 (COBRA1) and demonstrated its direct interaction with endogenous BRCA1. This study showed that COBRA1 can stimulate chromatin decondensation and is recruited to the chromosome site by specific subdomains in the BRCT1 domain of BRCA1 protein. Notably, this finding suggests that only 50-aa of the COOH-terminal part of BRCT1 domain is sufficient for inducing a large-scale chromatin unfolding [129].
A number of studies demonstrated a role of BRCA1 in the epigenetic regulation of oncogenic microRNAs (miR) [130]. Expression of miR-155 can be suppressed by the BRCA1-HDAC2 (Histone diacetylase 2) complex that is recruited to its promoter and keeps H2A and H3 histones deacetylated. Loss of BRCA1 or treatment with HDAC inhibitors results in the disruption of the BRCA1-HDAC2 complex and activation of miR-155 expression. This mechanism of BRCA1-dependent negative control of the oncogenic microRNA (oncomiR) expression by decreasing the acetylation of H2A and H3 may contribute to the BRCA1-mediated suppression of tumor growth [131]. Another study reported that miR-151-5p, which targets chromatin regulator SMARCA5, was upregulated in BRCA-related and sporadic breast cancers with high expression of PARP [132]. This study suggests that BRCA1-mediated upregulation of miR-151-5p could be a potential mechanism druggable by PARP inhibitors in cancer patients carrying germline BRCA1 mutations. A link between genetic lesions in the BRCA1 gene and epigenetic mechanisms of breast cancer development was also demonstrated by Li and coauthors who found that BRCA1 mutations are associated with an epigenetic silencing of phosphatidylethanolamine N-methyltransferase (PEMT) gene, which is important for the synthesis of choline, a nutrient associated with breast cancer development. This epigenetic repression is mediated by DNA hypermethylation in the PEMT gene promoter around -132 site and accompanied by increase of repressive chromatin mark H3K9me as well as loss of active chromatin signature H3K9ac [133]. Another study from the same group revealed BRCA1 as a positive regulator of sirtuin 1 (SIRT1), which is a nicotinamide adenine dinucleotide (NAD)-dependent histone deacetylase, an epigenetic enzyme which is upregulated in many types of tumors [134] [135]. BRCA1 deficiency or mutations lead to increased NAD levels which in turn inhibit SIRT1 expression but induce SIRT1 activity. This observation proposed that the shifting of a balance between BRCA1- and SIRT1-dependent biological processes may contribute to the cellular transformation and tumor development [134]. Recent studies also demonstrated the role of BRCA1 in regulation of the polycomb-repressive complex 2 (PRC2), which is one of key chromatin modifiers involved in the maintenance of transcriptional silencing [136]. It was demonstrated that BRCA1 can directly bind to one of the components of PRC2, EZH2 (enhancer of zeste homolog 2). BRCA1 binds EZH2 in the region important for its interaction with oncogenic long non-coding RNA (lncRNA) HOTAIR, which is required for the PRC2 occupancy on chromatin followed by the H3K27 tri-methylation and heterochromatin formation [121]. Loss of BRCA1 expression results in EZH2 re-targeting and epigenetic silencing of tumor suppressor genes in human breast cancer cells. Taken together, these results suggested that BRCA1 can contribute to the development of aggressive breast cancer phenotype through inhibition of PRC2 complex.
Regulation of BRCA 1 and BRCA2 turnover
The regulation of BRCA1 protein level is tightly controlled during the cell cycle in a post-translational manner [106], [138]. In addition, BRCA2 is becoming ubiquitinated and degraded during tumorigenesis which contributes to genomic instability and development of non-familial cancers [13], [139]. A number of proteins have been identified as regulators of BRCA1/BRCA2 stability including cysteine protease Cathepsin S (CTSS) which interacts with the BRCT domain of BRCA1 and activates its proteolytic degradation [140], E2 ubiquitin-conjugating enzyme E2T (UBE2T) [139], E3 ubiquitin ligases Herc2 and F-box-protein 44 (FBXO44) which ubiquitinate and downregulate BRCA1 protein [141], [142]. Interaction with BARD1 protein results in the reduction of proteasome-sensitive ubiquitination and stabilization of BRCA1 expression [143]. On the other hand, AKT-dependent phosphorylation of BRCA1 in response to estrogen and IGF-1 receptor signaling prevents proteasomal degradation and increases BRCA1 protein level [144]. A recent study by Kim et al. showed that Fyn-related kinase (Frk)/Rak also directly phosphorylates BRCA1 and by this positively regulates BRCA1 protein stability [129]. In addition to the regulation at the protein level, BRCA1/2 has been shown to be exposed to a complex post-transcriptional regulatory program. For example, the 3'UTR of BRCA2 mRNA physically interacts with miR-19a and miR-19b resulting in concomitant decrease of mRNA and protein levels of BRCA2 [146]. Expression of BCR-ABL1 oncoprotein in chronic myeloid leukemia cells is associated with BRCA1 downregulation. A study showed that this downregulation of BRCA1 is governed by TIRA (TIA1 cytotoxic granule-associated RNA-binding protein-like 1) protein which suppresses BRCA1 mRNA translation by the binding to AU-Rich Element (ARE) sites in 3'UTR of BRCA1 mRNA [147]. The same study also described HuR (Hu antigen R) mRNA-binding protein which can complex with TIRA and BRCA1 mRNA but increases mRNA stability and translation [147]. BRCA1 was shown to be epigenetically silenced when UHRF1 (ubiquitin- like, containing PHD and RING finger domains 1) is overexpressed. This study showed that UHRF1 is involved in regulation of BRCA1 transcription by multiple ways including accumulation of the repressive histone marks on the BRCA1 promoter and inhibition of the binding of key transcription factors such as MyoD (Myogenic Differentiation 1), CREB- binding protein (CBP) and p300 [148].
Tissue specific tumor development upon BRCA1 and BRCA2 mutations
A number of studies have demonstrated that tumor suppressor role of BRCA1 and BRCA2 genes is tissue-specific and BRCA1/2 mutations mainly contribute to breast and ovarian cancer development, however the exact mechanisms of this selectivity remains obscure [149]. Research over the last decade shed light on the sex hormone-driven growth of BRCA-mutated breast cancer cells and on the mechanism of mutual regulation of BRCA1/2 and hormone levels. It is known that starting from puberty period breast epithelium begins to proliferate rapidly in response to estrogen, therefore tissue-specificity of the BRCA1-related carcinogenesis can be attributed to its estrogen dependence [150]. Indeed, estrogen receptor alpha (ERα) activates the BRCA1 promoter and increases expression of BRCA1 protein [151]. Studies by Gorrini and coauthors demonstrated that estrogen protects BRCA1-deficient cells from reactive oxygen species-induced death by activation of the PI3K/AKT signaling pathway, upregulation of NRF2 (nuclear factor erythroid 2-related factor 2)-dependent transcriptional program and, consequently, increased expression of the anti-oxidant genes [152], [153], [138]. According to these findings, an upregulated local concentration of estrogen would selectively supports survival and expansion of breast cancer cells with BRCA1 mutations [149]. On the other hand, BRCA1 affects the hormone functions in multiple ways including repression of estrogen-dependent gene transcription [155], [156], activating ERα expression [157] and regulating the level and activity of progesterone receptor (PR) [158], [159]. Expression of PR, especially PRA isoform is markedly increased in the mammary glands of Brca1 knock-out mice that might provide additional explanation for the tissue specific tumor development upon BRCA mutations [159], [160]. By employing the cre-mediated excision of exon 11 of Brca1 gene, Xu and co-authors have shown that Brca1 is critical for regulation of mammary gland development in mice, and conditional mutation of Brca1 results in abnormal ductal development and tumor formation [161]. The clinical findings also demonstrated that carriers of BRCA1 and BRCA2 mutations have abnormal levels of estrogen and PR that might increase the risk for breast cancer development [162], [163].
Paradoxically, more than 90% of breast tumors with BRCA1 mutations have a lack of ERα expression; however the mechanism of this negative feedback loop has long time remained elusive [164]. To address this question, Hosey and colleagues analyzed the relationship between BRCA1 mutations and ERα expression in preclinical models and tumor tissues and discovered that BRCA1 directly regulates promoter activity of ESR1 gene encoding ERα [157]. This study provided an insight into the interaction between BRCA1 and ERα levels, although this relationship might be much more complex than previously anticipated. Whereas ER expression is downregulated in tumor cells, mutations of BRCA1 and BRCA2 genes were not correlated with changed levels of ER. Of note, this study showed that BRCA mutations in normal tissues were associated with profound attenuation of estrogen-dependent gene expression including progesterone receptor isoform B (PRB) and marked predominance of PRA suggesting a role of PRA in the initiation of breast tumor growth [165]. Bramley et al. showed that normal breast epithelial cells carrying BRCA1/2 mutations have markedly increased proliferation in response to the upregulated estrogen levels, and treatment with anti-estrogen therapies such as tamoxifen (TAM) and fulvestrant (FUL) can prevent this proliferative activity [166].
In contrast to BRCA1 hereditary tumors which usually lack ER expression, BRCA2 hereditary tumors are usually ER positive. The study of Malone et al. shed light on the link between BRCA2 and ER showing that BRCA2 protein is expressed in normal breast tissues and in ER positive breast cancers but not in ER negative breast cancer tissues [167]. Moreover, a high estrogen level induces the cyclin- dependent kinase 2 (Cdk2) mediated phosphorylation and stabilization of BRCA2 protein that results in an increased DNA repair and improved breast cancer cell survival after irradiation [167].
The role of BRCA proteins in the regulation of cancer stem cells
A high intratumoral morphological and histological heterogeneity was first documented over a century ago by German pathologist Rudolf Virchow [168]. These findings were followed by a zooming into cellular and molecular levels that revealed a substantial diversity in the mutation burden, gene expression, tumorigenic properties and therapy resistance among the cells of each individual tumor [169]-[176]. In 1994, John Dick and coworkers experimentally demonstrated that only CD34+CD38- acute myeloid leukemia (AML) cells isolated from patients by fluorescence activated cell sorting (FACS) were able to self-renew, differentiate into other cell populations and to initiate new tumors in mice [177], [178]. These experiments provided functional evidence that tumors are hierarchically organized and consist of cells with different self-renewal, tumorigenicity and differentiation potential. According to this view, a population of cancer stem cells (CSCs) resides on top of the intratumoral hierarchy and is the only cell subset which maintains tumor growth. During the last decades identification and characterization of CSC populations was also conducted for other tumor entities including breast cancer [179], glioblastoma [180], colon cancer [181], prostate cancer [182] and other types of cancer, and a number of markers which can be used for the detection, isolation and monitoring of CSCs such as CD133, CD44, aldehyde dehydrogenase (ALDH) etc. have been identified [9], [183]. On the other hand, the identification and characterization of CSC specific markers and targeted therapies remain challenging since a growing body of evidence suggests that stemness is a more transient feature than initially believed, and therefore the current experimental approaches for CSC analysis need further development [184], [185]. Nevertheless, there is no doubt that CSCs are drivers of tumor growth and a unit of tumor evolution due to their ability to self-renew and pass favorable heritable mutations to indefinite number of the next cell generations [9], [10]. Furthermore, given the clinical importance of CSCs, a number of studies are focused on the identification of the druggable signaling pathways which can be used for targeting CSCs such as JAK (Janus kinases)-STAT (Signal Transducer and Activator of Transcription proteins) signaling, Hedgehog, Wnt, Notch, PI3K (Phosphatidylinositol-4,5-bisphosphate 3-kinase)/AKT (Protein kinase B) pathways etc. [186].
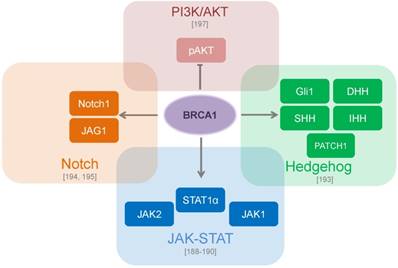
As important regulators of genome stability and gene expression, BRCA1/2 plays an important role in CSCs development and evolution and can be employed in different CSC signaling mechanisms. JAK-STAT signaling plays a role in the maintenance of CSCs in solid tumors and hematological cancers [187]. The BRCA1 protein binds and activates JAK1 and JAK2, and induction of BRCA1 expression results in the constitutive activation of STAT3 [188] and upregulation of JAK1, STAT1 and STAT2 gene expression [189], [190]. The key role of Hedgehog (Hh) signaling during embryonic development is also recapitulated in cancer progression as it is involved in the maintenance of CSCs in many types of tumors [191], [192]. Luca et al. have shown that BRCA1 depletion increased growth of prostate xenograft tumors in mice, induced expression of the epithelial-mesenchymal transition markers such as vimentin, fibronectin and Snail and led to reduced expression of the Hh effector Gli1. However, no modulation of other Hh pathway genes was observed [193]. A recent study by Buckley et al. also determined the role of BRCA1 in Notch signaling - another key developmental pathway implicated in breast CSC regulation [194]. These finding demonstrated that BRCA1 activates transcription of Notch receptors and ligands, and knockdown of BRCA1 results in decreased Notch activity, enhanced tumor sphere formation and upregulated ALDH activity [194]. This study along with previous findings revealed that BRCA1 governs mammary epithelial cell fate by transcriptional regulation of a panel of basal (e.g. cytokeratins KRT5, KRT17, p-cadherin) and luminal (e.g. ERα, KRT18) markers, and that Notch signaling is also implicated in this regulation [191], [195]. These findings demonstrated that inhibition of the BRCA1/p63/Notch signaling results in the enrichment of CSC populations [191]. Previous studies demonstrated an important role of PI3K/AKT pathway in the maintenance of breast CSCs [196]. The loss of BRCA1 constitutively activates PI3K/AKT signaling through up-regulation of AKT phosphorylation at the critical amino acid residue T308 which is important for AKT activation [197]. In accordance with this data, knockdown of BRCA1 sensitizes breast cancer cells to PI3K/AKT pathway inhibitors [197] (Figure 3).
CSC signaling pathways which are regulated by BRCA proteins. BRCA1 directly binds to JAK1 and JAK2 and leads to constitutive activation of STAT3 [188, 189, 190]. BRCA1 modulates genes involved in the Hedgehog pathway such as SHH, IHH, DHH, Gli1 and PATCH1 [193]. BRCA1 transcriptionally upregulates the Notch ligand JAG1 (Jagged1) with further activation of the Notch pathway [194, 195]. The loss of BRCA1 constitutively activates PI3K (Phosphatidylinositol-4,5-bisphosphate 3-kinase)/AKT (Protein kinase B) pathway through down-regulation of phospho-AKT [197].
Growing evidence supports a critical role of CSCs in tumor resistance to different types of conventional therapy including chemotherapy and radiation therapy as reviewed previously [198]-[200]. One of the key mechanisms of CSC resistance to radiotherapy and DNA-reactive chemotherapy is activation of the DNA repair pathways, such as HR and NHEJ. It has been shown that some of the DNA damage checkpoint and repair genes including Brca1 are highly upregulated in CSC populations in a mouse model of mammary tumor that closely mimics human breast cancer [201]. In line with these data, genes which contribute to DNA repair, such as BRCA1, Exo1, Mre11, Rad51 were upregulated in CD133+ human lung CSCs [202]. It has been also demonstrated that DNA repair genes including BRCA1 were highly expressed in the invasive pancreatic cells as well as in metastatic pancreatic tissue specimens [203]. Taken together these findings could potentially suggest that subpopulations of tumor-initiating cells possessing a high potency of DNA repair have better chances to withstand the harsh conditions during tumor dissemination.
Taken together, these data demonstrate that BRCA1 may play an important role in tumor initiation, maintenance of CSC population and its therapy resistance. In the next chapter we review the role of BRCA proteins for the CSC regulation in different tumor entities.
The role of BRCA proteins for the CSC maintenance in the different tumor entities
Breast cancer
Breast cancer is the second most common cancer overall and the most common malignant tumor among women, with an incidence rate of more than 1.5 million new cases per year [204]. Mutations in BRCA1 gene are associated with a high cancer incidence, and mean cumulative risks for development of breast cancer by the age of 70 years was estimated to be more than 50% for BRCA1 mutation carriers [205].
Breast tumor initiating cells were first found and isolated by Al-Hajj et al. in 2003 by using CD44 and CD24 surface marker expression. A population of CD44+CD24-/low cells isolated from human breast cancer specimens possesses a high tumor forming capacity in NOD/SCID mice [179]. The role of BRCA1 in mammary stem cell maintenance and development was intensively investigated during the last ten years. The increased ALDH (aldehyde dehydrogenase) activity was shown to be a marker of breast CSCs [207]. Using ALDEFLUOR assay, Ginestier et al. showed that the ALDH+ population, but not the ALDH- cell subset isolated from the normal mammary epithelium was able to generate mammospheres in non-adherent conditions upon serial passages as well as to form the ductal structure and to differentiate in vivo. At the same time ALDH+ population isolated from breast tumor specimens displayed a high tumorigenic potential in mice models [207]. BRCA1 plays a role in self-renewal of progenitor cells which was demonstrated by serial passaging of BRCA1-deficient normal mammary epithelial cells where the number of mammospheres increased with number of passage [208]. The knockdown of BRCA1 expression by siRNA lentiviruses in normal mammary cells resulted in increase of ALDH+ population and blockage of the ER-/ALDH+ stem cell/progenitor differentiation in vitro and in vivo [208]. The study of Liu et al. employed in vitro and in vivo examination of the breast tissue samples with and without BRCA1 germ-line mutations. This study showed that BRCA1 is required for the differentiation of ER-/ALDH1+ mammary stem cell/progenitor cells into ER+/ALDH1- luminal cells. According to this finding, 5 out of 13 breast tissue samples with BRCA1 mutation exhibited accumulation of ALDH1+ acini compared to the control specimens without BRCA1 mutations, and these ALDH1+ lobules have low expression of luminal marker CK18 and ER [208]. Heerma van Voss et al. compared the expression of the stem cell maker ALDH1 in 32 normal breast tissues from the BRCA1 mutation carriers with prophylactic mastectomies to 32 mammoplasty control samples. In contrast to the results of Liu et al. there were no differences between carriers and non-carriers regarding the ALDH1 positive epithelial cell population which may be due to the fact that Liu and co-authors analyzed a small and non-age matched cohort. Interestingly, this study found a significantly higher expression of ALDH1 in normal stroma of the BRCA1 mutation carriers [209]. One year later Heerma van Voss et al. published a paper comparing expression of ALDH1 in epithelium and stroma of tissues obtained from BRCA1 related breast cancers versus sporadic breast cancers. Compared to sporadic controls, expression of ALDH1 in tumors was higher in BRCA1-mutated breast cancers suggesting that BRCA1 related breast tumors have increased CSC population. The authors assumed that ALDH1 could serve as biomarker of BRCA1 mutations and potential therapeutic target in BRCA1-mutant breast cancer [210]. Kubista et al. investigated the role of BRCA1 protein for the differentiation of normal murine mammary epithelial cell line HC11, C2C12 mouse myoblasts and N1E-115 mouse neuroblastoma cells. During in vitro differentiation, BRCA1 protein becomes upregulated with simultaneous decrease of cell proliferation in all three cell lines. Of note, this study demonstrated that ectopic expression of BRCA1 induced differentiation of mammary epithelial cells in vitro, and knockdown of BRCA1 expression attenuated this process. At the same time, high level of BRCA1 has no effect on muscle differentiation that might suggest tissue-specificity of this BRCA1 function. [211]. Liu et al. analyzed the effect of PARP inhibitor (PARPi ) Olaparib on BRCA1-mutant and BRCA1-wild-type triple-negative breast cancer cell lines. After PARPi treatment, the number of viable cells becomes lower in BRCA1-mutant cell lines compared to the wild-type BRCA1 cell lines, whereas CSCs in these cell lines were resistant to PARPi treatment. One of the binding proteins of BRCA1, RAD51, was found to mediate this resistance suggesting that targeting of RAD51 may increase tumor sensitivity to PARPi [179]. Taken together, these data suggest that BRCA1 is a negative regulator of breast CSCs. Further investigations in this field are warranted to understand the molecular mechanism of this regulation that may be beneficial for the development of novel biomarkers for tumor diagnostics and therapy.
Ovarian cancer
Ovarian cancer is the leading cause of deaths among all gynecological malignancies and accounts for more than 200,000 new cases and 150,000 deaths in women worldwide annually [204]. From 10% to 12% of women with ovarian cancer carry mutations in the BRCA1 or BRCA2 gene [213]. Ovarian CSCs are often isolated from the ascites of ovarian cancer patients [214], [215], [216]. Unfortunately, the data describing the impact of BRCA1 on ovarian cancer stemness is quite limited. A seminal study of Meng et al. showed that ALDH1A1 knockdown resulted in increase of BRCA1 expression, and that high ALDH expression correlates to stem cell-like properties, activation of DNA repair and resistance to platinum drugs [217]. Other study by Lim et al. demonstrated that inhibition of vascular endothelial growth factor receptor 3 (VEGFR3) downregulated BRCA1 and BRCA2 gene expression, attenuated CD133+ stem-like cell population, induced sensitivity to cisplatin and resulted in growth arrest. This study suggested that inhibition of VEGFR3 could be a promising approach to treat chemotherapy resistant ovarian cancers, half of which has a reversion of BRCA function [218]. Despite the data on BRCA1- dependent regulation of ovarian CSCs is still limited, these two studies paved the road for further investigation of this clinically important topic.
Prostate cancer
Prostate cancer (PCa) is the second most common cancer and the fifth leading cause of death from cancer in men, accounting for 1.1 million cases worldwide in 2012 [204]. BRCA1 and BRCA2 genes were shown to be associated with an increased risk of prostate cancer development [24], [219]. Prostate cancer cells are highly heterogeneous at the phenotypical, functional and tumorigenic levels that reflect their genetic and epigenetic heterogeneity [183], [220]. Prostate cancer is characterized by a high level of mutations in DNA repair genes including BRCA genes that results in genomic instability, clonal selection and tumor progression [221]. BRCA2 protein was shown to be involved in the progression of prostate tumorigenesis in Brca2 deleted mice [222]. BRCA2 deficiency contributes to the prostate cancer cell migration and invasion by PI3K/AKT pathway activation, mitogen-activated protein kinase/ extracellular regulated kinase (MAPK/ERK) activation and matrix metalloproteinase (MMP)-9 secretion [223]. Using genomic and methylation profiles of localized PCa from BRCA2 mutation carriers, Taylor et al. showed that germline BRCA2-mutant PCa is genotypically similar to metastatic castration-resistant prostate cancer (mCRPC) and frequently harbor mutations in key signaling pathways such as WNT/APC, mTOR, ATR, MYCN leading to genomic instability, tumor progression and metastasis [37]. Taken together, these data can explain the aggressiveness of PCa tumors with mutated BRCA genes and adverse prognosis for affected patients.
Hematopoiesis and Fanconi anemia
Fanconi anemia (FA) is cancer susceptibility syndrome associated with predisposition to different diseases, such as myelodysplasia, leukemia, bone marrow failure and certain solid tumors such as oropharyngeal, gastrointestinal and gynecological cancers [224], [225]. FA patients' cells are sensitive to the DNA cross-linking induced by chemical drugs such as mitomycin C or by ionizing irradiation. In response to DNA damage, FANCD2 protein, which is mutated in Fanconi's anemia, becomes monoubiquitynated and co-localizes with BRCA1 [225] and BRCA2 [226] at the sites of DNA lesions suggesting that this interaction is important for DNA-damage response network.
Hematopoiesis is a process of blood cell formation by hematopoietic stem cells (HSCs) [227]. Recent findings revealed a role of BRCA1 gene in this process. In the study published by Vasanthakumar et al. the authors generated a conditional mouse model with Mx1-Cre-mediated deletion of Brca1 gene to check its role in hematopoiesis. As a result, Brca1-/- mice exhibited macrocytic anemia and low count of white blood cells already at 1 month of age. The karyotyping of Brca1-/- bone marrow showed presence of various cytogenetic abnormalities that indicates genomic instability. Some Brca1-/- mice developed different hematologic malignancies, such as lymphomas, acute myeloid leukemia, and erythroleukemia by age of 4,5 to 6 months. Brca1-/- bone marrow cells showed a lower capacity to form hematopoietic colonies in vitro. Taken together, these data have demonstrated that Brca1 deficiency leads to bone marrow disorders and susceptibility to hematologic malignancies similar to the human FA [228]. Mgbemena et al. developed a mouse model with homozygous Brca1 null mutation in the hematopoietic system. Experiments showed that lack of Brca1 can attenuate HSCs and population of hematopoietic progenitor cells. This study also demonstrated that HSCs with Brca1 deficiency have limited reconstructive capacity upon serial transplantation compared to wild-type HSCs. The influence of human germline BRCA1 mutation on hematopoiesis was investigated in mice with Brca1 5382insC mutation. Of note, Brca1 5382insC genotype was associated with more severe hematopoietic defects than null allele suggesting that the mutated BRCA1 protein is more harmful to hematopoietic stem and progenitor cells than BRCA1 deficiency [229]. Taken together, the results of the above described studies show that function of BRCA genes is essential for the maintenance of HSCs and for normal hematopoiesis.
Perspectives for the treatment of patients with BRCA mutations
Recent studies propose that 55-65% of BRCA1 mutation carriers, and about 45% of BRCA2 mutation carriers will develop breast cancer by the age of 70 years [204] [230]. Prophylactic surgery in BRCA carriers i.e. salpingo-oophorectomy and mastectomy are proven to be beneficial for reduction the risk of breast and ovarian cancer development [231], [232]. The carriers of BRCA1 mutations are more likely to develop triple negative breast cancer (TNBC). TNBC refers to breast cancers which are negative for the expression of hormone epidermal growth factor receptor 2 (HER-2), estrogen receptors (ER), and progesterone receptors (PR). Since growth of these types of tumors does not depend on the signaling from these receptors, conventional treatments like hormone therapy and drugs that target estrogen, progesterone, and HER-2 are not effective in TNBC patients [233]. At the same time TNBC tumors are highly heterogeneous that is associated with their high resistance to chemotherapy [234]. The deficiency of BRCA1 and BRCA2 proteins in tumor cells is associated with a higher sensitivity to DNA- damaging agents [16], [235], [236]. As a result, BRCA1/2-deficient TNBC cancers are more sensitive to standard chemotherapy and have a higher level of immune cell activation compared to TNBC with functional BRCA1/2 proteins [237].
One of the most common therapies used for treatment of breast and prostate cancer are taxanes such as paclitaxel and docetaxel. Taxanes are microtubule targeting agents which block cell growth and induce apoptosis. Recent studies revealed that carriers of BRCA1 mutation with hormone-negative cancers (n=20) have less sensitivity to taxane therapy than non-BRCA1 mutation carriers (n=19). In contrast to this, hormone positive breast cancers have similar clinical outcome after taxane treatment in BRCA1- mutation carriers (n = 11) and sporadic breast cancer (n = 61) [238]. The retrospective analysis of the breast cancer patients (n = 317) with and without BRCA mutations treated with neo-adjuvant anthracycline- taxane regimens demonstrated that BRCA1 status is independently associated with a higher pathologic complete response (pCR) [239]. A high pCR rate after neo-adjuvant anthracycline-taxane therapy was also demonstrated in BRCA1/2-mutated triple-negative breast cancer (n = 53) [240]. Mutations in BRCA2 have a negative impact on the response rate to docetaxel in metastatic castration resistant prostate cancer (mCRPC) (n = 53) although no correlation was found between BRCA1/2 protein expression and response to treatment [241]. Another large multi-institutional retrospective comparison of mCRPC patients (n = 390) with and without germline mutations in DNA repair genes (including BRCA2, ATM, CHEK2, BRCA1, PALB2, RAD51D and others) showed no difference between mutation carriers and non-carriers in their therapy response, progression-free survival after docetaxel or anti-androgen treatment and overall survival [242].
Platinum-based compounds are another mainstay of anti-cancer treatment. Platinum drugs such as cisplatin induce DNA platination and inter-strand DNA crosslinking leading to the accumulation of DNA DSBs and cell death. A retrospective study carried out on a cohort of young women with BRCA1-mutated breast cancer (n = 102) showed a higher pCR after treatment with neo-adjuvant cisplatin therapy compared to pCR for other types neo-adjuvant chemotherapy [243]. A pilot study showed that patients with metastatic breast cancer and BRCA1 mutations (n = 20) are highly sensitive to cisplatin chemotherapy [244]. The resent multicenter phase II clinical trial for treatment of metastatic TNBC (n = 86) confirmed efficiency of platinum monotherapy for these patients, especially for BRCA1/2 mutation carriers (n = 11) [245]. Ovarian cancer patients with germline BRCA1/2 mutations (n = 112) are more response to first-line chemotherapy including platinum-based treatment and have better progression-free survival (PFS) and overall survival (OS) compared to the patients with sporadic ovarian carcinoma (n = 222) [246]. Another study also demonstrated that germline and somatic mutations in homologous recombination genes including BRCA1 and BRCA2 were found to be highly predictive for improved overall survival and responsiveness to platinum-based treatment in patients with ovarian, fallopian tube and peritoneal cancer (n = 390) [247]. Of note, some patients with breast and ovarian BRCA-mutated cancers which were previously sensitive to platinum therapy can become resistant to treatment due to reversion of BRCA mutations that restore BRCA1/2 protein function [233], [234], [235].
A growing body of evidence demonstrated a great potential of PARP inhibitors for treatment of breast and ovarian cancer with BRCA mutations. PARP is important enzyme in DNA damage repair machinery, and activation of PARP is one of the early DNA damage responses. PARP detects and rapidly binds to DNA strand breaks and catalyzes poly-ADP- ribosylation of target proteins using NAD+ as substrate. After formation of long, branched polymers, PARP dissociates from DNA ends and facilitates further DNA repair processes [251]. PARP inhibitors block the repair of DNA damage resulting in persistence of DNA lesions, cell cycle arrest and subsequent apoptosis. BRCA-mutated cells are unable to repair DSBs properly and PARP inhibition leads to the accumulation of DNA breaks and cell death [252]. Nonfunctional BRCA1 or BRCA2 sensitizes cells to PARP inhibition, leading to chromosomal instability and cell cycle arrest [253]. Clinical trial showed that treatment with PARP inhibitors improve clinical outcome in patients with advanced, BRCA-mutated breast and ovarian cancers [254]-[256], [257], [16]. A few PARP inhibitors have been approved by FDA (Food and Drug Administration) for BRCA-mutated cancer such as Olaparib, (breast and ovarian cancer), Rucaparib (ovarian cancer), Niraparib (ovarian cancer, regardless of BRCA mutation status), and a number of PARP inhibitors are under preclinical and clinical development [16], [256]-[258].
The important role of BRCA1/2 in DNA DSB repair makes it an attractive biomarker for radiation therapy, which is one of the mainstay modalities together with surgery and chemotherapy for treatment of many types of tumors [259]. The curative potential of ionizing radiation (IR) depends on induction of DSBs which results in accumulation of DNA damage and tumor cell death [260], [261]. Preclinical studies using different in vitro and in vivo tumor models demonstrated that BRCA1 deficiency can lead to radiosensitivity [262], [263]. On the other hand, clinical studies are rather contradictive, and many of them demonstrated that BRCA deficient cancers are the same sensitive to irradiation as sporadic cancers [262], [263]. An improved design, complete follow-up data and better standardizing of the future studies might help to elucidate the role of BRCA mutations in tumor radioresponse.
Taken together, experimental data and clinical trials show that BRCA mutation status can provide important clinical information for patients' response to the different types of conventional therapies and for selection of more personalized cancer treatment.
Conclusions
Experimental and clinical data discussed in this review suggest that BRCA genes play a pivotal role in a number of biological processes including maintenance of the genomic integrity, regulation of the oxidative stress and protein stability, modulation of gene transcription, cell cycle progression and therapy resistance. Given its multifaceted functions as tumor suppressor, BRCA1/2 mutations contribute to the development of different types of malignancies including breast, ovarian and prostate cancers. On the other hand, due to reduced DNA repair capacity, BRCA-mutated cancers are also more responsive to the distinct types of treatment such as platinum-based compounds and PARP inhibitors, which induce accumulation of DNA DSBs. However, acquired resistance to this treatment due to reversion of BRCA mutations that restore BRCA1/2 protein function is an urgent clinical challenge and requires careful analysis of restored allele frequencies during the course of treatment. Of note, the development of BRCA reversion mutations during anticancer treatment demonstrates that BRCA deficiency is critical at the initial stage of tumorigenesis and is less important for the maintenance of the established tumors. Indeed, accumulating in vivo evidence suggests that BRCA proteins might contribute to the normal developmental processes as well as to the regulation of CSCs and tumor initiation in different tumor entities. Development of the combination approaches with targeted therapy for other cancer-related pathways, therapeutic targeting of CSC or immunotherapy may overcome the acquired resistance associated with BRCA reversion mutation and improve treatment efficiency.
Acknowledgements
The authors acknowledge their colleagues, especially Alexander Schulz and Vasyl Lukiyanchuk for critical reading the manuscript. This work was in part supported by German Academic Exchange Service (DAAD) and the Federation of European Biochemical Societies (FEBS) for Ielizaveta Gorodetska. Work in Anna Dubrovska lab is partially supported by grants from Deutsche Forschungsgemeinschaft (DFG) (273676790, 416001651 and 401326337), from Wilhelm Sander-Stiftung (2017.106.1), BMBF (Grant-No. 03Z1NN11) and DLR Project Management Agency (01DK17047).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Miki Y, Swensen J, Shattuck-Eidenset D. et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66-71
2. Wooster R, Bignell G, Lancaster J. et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378(6559):789-792
3. Cavanagh H, Rogers KMA. The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Hered Cancer Clin Pract. 2015;13:16
4. Jae Myoung Noh, Doo Ho Choi, Hyejin Baek. et al. Associations between BRCA Mutations in High-Risk Breast Cancer Patients and Familial Cancers Other than Breast or Ovary. J Breast Cancer. 2012;15(3):283-287
5. Mersch J, Jackson MA, Park M. et al. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. 2015;121(2):269-275
6. Easton DF. How many more breast cancer predisposition genes are there? Breast Cancer Res. 1999;1:14-17
7. Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481(7381):287-294
8. Jiang Q, Greenberg RA. Deciphering the BRCA1 Tumor Suppressor Network. J Biol Chem. 2015;290(29):17724-17732
9. Peitzsch C, Tyutyunnykova A, Pantel K. et al. Cancer stem cells: The root of tumor recurrence and metastases. Semin Cancer Biol. 2017;44:10-24
10. Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275-291
11. Hashizume R, Fukuda M, Maeda I. et al. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J Biol Chem. 2001;276(18):14537-14540
12. Manke IA, Lowery DM, Nguyen A. et al. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 2003;302(5645):636-639
13. Silver DP, Livingston DM. Mechanisms of BRCA1 tumor suppression. Cancer Discov. 2012;2(8):679-684
14. Zhang F, Fan Q, Ren K. et al. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol Cancer Res. 2009;7:1110-1118
15. Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci USA. 2009;106(17):7155-7160
16. Godet I, Gilkes DM. BRCA1 and BRCA2 mutations and treatment strategies for breast cancer. Integr Cancer Sci Ther. 2017:4 (1). doi: 10.15761/ICST.1000228
17. Clark SL, Rodriguez AM, Snyder RR. et al. Structure-Function Of The Tumor Suppressor BRCA1. Comput Struct Biotechnol J. 2012:1 (1). doi: 10.5936/csbj.201204005
18. Laitman Y, Feng BJ, Zamir IM, at al. Haplotype analysis of the 185delAG BRCA1 mutation in ethnically diverse populations. Eur J Hum Genet. 2013;21(2):212-216
19. Hamel N, Feng BJ, Foretova L. et al. On the origin and diffusion of BRCA1 c.5266dupC (5382insC) in European populations. Eur J Hum Genet. 2011;19(3):300-306
20. Gayther SA, Warren W, Mazoyer S. et al. Germline mutations of the BRCA1 gene in breast and ovarian cancer families provide evidence for a genotype-phenotype correlation. Nat Genet. 1995;11(4):428-433
21. Thompson D, Easton D, Breast Cancer Linkage Consortium. Variation in BRCA1 cancer risks by mutation position. Cancer Epidemiol Biomark Prev. 2002;11(4):329-336
22. Thompson D, Easton D. The genetic epidemiology of breast cancer genes. J Mammary Gland Biol Neoplasia. 2004;9(3):221-236
23. Karami F, Mehdipour P. A comprehensive focus on global spectrum of BRCA1 and BRCA2 mutations in breast cancer. BioMed Res Int. 2013;2013:928562
24. Thompson D, Easton D, Breast Cancer Linkage Consortium. Cancer Incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002;94(18):1358-1365
25. Brose MS, Rebbeck TR, Calzone KA. et al. Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J Natl Cancer Inst. 2002;94(18):1365-1372
26. Chatterjee G, Jimenez-Sainz J, Presti T. et al. Distinct binding of BRCA2 BRC repeats to RAD51 generates differential DNA damage sensitivity. Nucleic Acids Res. 2016;44(11):5256-5270
27. Yang H, Li Q, Fan J. et al. Holloman WK BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA-ssDNA junction. Nature. 2005;433(7026):653-657
28. Fackenthal JD, Olopade OI. Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat Rev Cancer. 2007;7(12):937-948
29. Walsh T, Mandell JB, Norquist BM. et al. Genetic Predisposition to Breast Cancer Due to Mutations Other Than BRCA1 and BRCA2 Founder Alleles Among Ashkenazi Jewish Women. JAMA Oncol. 2017;3(12):1647-1653
30. Breast Cancer Linkage Consortium. Cancer risks in BRCA2 mutation carriers. 1999; J Natl Cancer Inst. 91(15): 1310-1316.
31. Cassidy LD, Liau SS, Venkitaraman AR. Chromosome instability and carcinogenesis: insights from murine models of human pancreatic cancer associated with BRCA2 inactivation. Mol Oncol. 2014;8(2):161-168
32. Moynahan ME. The cancer connection: BRCA1 and BRCA2 tumor suppression in mice and humans. Oncogene. 2002;21(58):8994-9007
33. Calò V, Bruno L, La Paglia L. et al. The Clinical Significance of Unknown Sequence Variants in BRCA Genes. Cancers. 2010;2(3):1644-1660
34. Castro E, Eeles R. The role of BRCA1 and BRCA2 in prostate cancer. Asian J Androl. 2012;14(3):409-414
35. Bancroft EK, Page EC, Castro E. et al. Targeted prostate cancer screening in BRCA1 and BRCA2 mutation carriers: results from the initial screening round of the IMPACT study. Eur Urol. 2014;66(3):489-499
36. Walker R, Louis A, Berlin A. et al. Prostate cancer screening characteristics in men with BRCA1/2 mutations attending a high-risk prevention clinic. Can Urol Assoc J. 2014;8:E783-788
37. Taylor RA, Fraser M, Livingstone J. et al. Germline BRCA2 mutations drive prostate cancers with distinct evolutionary trajectories. Nat Commun. 2017;8:13671
38. Zhu Y, Zhai K, Ke J. et al. BRCA1 missense polymorphisms are associated with poor prognosis of pancreatic cancer patients in a Chinese population. Oncotarget. 2017;8(22):36033-36039
39. Copson ER, Maishman TC, Tapper WJ. et al. Germline BRCA mutation and outcome in young-onset breast cancer (POSH): a prospective cohort study. Lancet Oncol. 2018;19(2):169-180
40. Okada S, Tokunaga E, Kitao H. et al. Loss of heterozygosity at BRCA1 locus is significantly associated with aggressiveness and poor prognosis in breast cancer. Ann Surg Oncol. 2012;19(5):1499-1507
41. Wang YA, Jian JW, Hung CF. et al. Germline breast cancer susceptibility gene mutations and breast cancer outcomes. BMC Cancer. 2018;18(1):315
42. Rubin SC, Benjamin I, Behbakht K. et al. Clinical and pathological features of ovarian cancer in women with germ-line mutations of BRCA1. N Engl J Med. 1996;33(19):1413-1416
43. Pharoah PD, Easton DF, Stockton DL. et al. Survival in familial, BRCA1-associated, and BRCA2-associated epithelial ovarian cancer. United Kingdom Coordinating Committee for Cancer Research (UKCCCR) Familial Ovarian Cancer Study Group. Cancer Res. 1999;5(4):868-871
44. Huang YW. Association of BRCA1/2 mutations with ovarian cancer prognosis. Medicine (Baltimore). 2018;97(2):e9380
45. Venkitaraman AR. Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science. 2014;343(6178):1470-1475
46. San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229-257
47. West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol. 2003;4(6):435-445
48. van Gent DC, Hoeijmakers JH, Kanaar R. Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet. 2001;2(3):196-206
49. Matos J, West SC. Holliday junction resolution: Regulation in space and time. DNA Repair (Amst). 2014;19(100):176-181
50. Mao Z, Bozzella M, Seluanov A. et al. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair. 2008;7(10):1765-1771
51. Dueva R, Iliakis G. Alternative pathways of non-homologous end joining (NHEJ) in genomic instability and cancer. Transl Cancer Res. 2013;2(3):163-177
52. Frit P, Barboule N, Yuan Y. et al. Alternative end-joining pathway(s): bricolage at DNA breaks. DNA Repair. 2014;17:81-97
53. Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181-211
54. Zhuang J, Zhang J, Willers H. et al. Checkpoint kinase 2-mediated phosphorylation of BRCA1 regulates the fidelity of nonhomologous end-joining. Cancer Res. 2006;66(3):1401-1408
55. Zhang J, Willers H, Feng Z. et al. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol Cell Biol. 2004;24(2):708-718
56. Moynahan ME, Chiu JW, Koller BH. et al. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4(4):511-518
57. Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7(2):263-272
58. Krejci L, Altmannova V, Spirek M. et al. Homologous recombination and its regulation. Nucleic Acids Res. 2012;40(13):5795-5818
59. Isono M, Niimi A, Oike T. et al. BRCA1 Directs the Repair Pathway to Homologous Recombination by Promoting 53BP1 Dephosphorylation. Cell Rep. 2017;18(2):520-532
60. Chen L, Nievera CJ, Lee AY. et al. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J Biol Chem. 2008;283(12):7713-7720
61. Zhang F, Ma J, Wu J. et al. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol. 2009;19(6):524-529
62. Yuan SS, Lee SY, Chen G. et al. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res. 1999;59(15):3547-3551
63. Fradet-Turcotte A, Sitz J, Grapton D. et al. BRCA2 functions: from DNA repair to replication fork stabilization. Endocr Relat Cancer. 2016;23(10):T1-T17
64. Davies AA, Masson JY, McIlwraith MJ. et al. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell. 2001;7(2):273-282
65. Zhao W, Vaithiyalingam S, San Filippo J. et al. Promotion of BRCA2-Dependent Homologous Recombination by DSS1 via RPA Targeting and DNA Mimicry. Mol Cell. 2015;59(2):176-187
66. Orelli BJ, Bishop DK. BRCA2 and homologous recombination. Breast Cancer Res. 2001;3(5):294-298
67. Lomonosov M, Anand S, Sangrithi M. et al. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev. 2003;17(24):3017-3022
68. Schlacher K, Christ N, Siaud N. et al. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145(4):529-542
69. Bhatia V, Barroso SI, García-Rubio. et al. MLBRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature. 2014;511(7509):362-365
70. Badie S, Escandell JM, Bouwman P. et al. BRCA2 acts as a RAD51 loader to facilitate telomere replication and capping. Nat Struct Mol Biol. 2010;17(12):1461-1469
71. Devasagayam TP, Tilak JC, Boloor KK. et al. Free radicals and antioxidants in human health: current status and future prospects. J Assoc Physicians India. 2004;52:794-804
72. Tudek B, Winczura A, Janik J. et al. Involvement of oxidatively damaged DNA and repair in cancer development and aging. Am J Transl Res. 2010;2(3):254-284
73. Martinez-Outschoorn UE, Balliet R, Lin Z. et al. BRCA1 mutations drive oxidative stress and glycolysis in the tumor microenvironment. Cell Cycle. 2012;11(23):4402-4413
74. Saha T, Rih JK, Rosen EM. BRCA1 Down-Regulates Cellular Levels of Reactive Oxygen Species. FEBS Lett. 2009;583(9):1535-1543
75. Lovren F, Pan Y, Quan A. et al. BRCA1 shields vascular smooth muscle cells from oxidative stress. J Thorac Cardiovasc Surg. 2014;147(6):1946-1955
76. Cao L, Xu X, Cao LL. et al. Absence of full-length Brca1 sensitizes mice to oxidative stress and carcinogen-induced tumorigenesis in the esophagus and forestomach. Carcinogenesis. 2007;28(7):1401-1407
77. Qiao S, Dennis M, Song X. et al. A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to control stress-induced autophagy and sustain exercise capacity. Nat Commun. 2015;6:7014
78. Ellisen LW, Ramsayer KD, Johannessen CM. et al. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol Cell. 2002;10(5):995-1005
79. Mansour MA. Ubiquitination: Friend and foe in cancer. Int J Biochem Cell Biol. 2018;101:80-93
80. Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004;1695(1-3):55-72
81. Mallery DL, Vandenberg CJ, Hiom K. Activation of the E3 ligase function of the BRCA1/BARD1 complex by polyubiquitin chains. EMBO J. 2002;21(24):6755-6762
82. Shabbeer S, Omer D, Berneman D. et al. BRCA1 Targets G2/M Cell Cycle Proteins for Ubiquitination and Proteasomal Degradation. Oncogene. 2013;32(42):5005-5016
83. Razandi M, Pedram A, Rosen EM. BRCA1 inhibits membrane estrogen and growth factor receptor signaling to cell proliferation in breast cancer. Mol Cell Biol. 2004;24(13):5900-5913
84. Sato K, Hayami R, Wu W. et al. Nucleophosmin/B23 is a candidate substrate for the BRCA1-BARD1 ubiquitin ligase. J Biol Chem. 2004;279(30):30919-30922
85. Yu X, Fu S, Lai M. et al. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev. 2006;20(13):1721-1726
86. Wu W, Nishikawa H, Hayami. et al. BRCA1 ubiquitinates RPB8 in response to DNA damage. Cancer Res. 2007;67(3):951-958
87. Horwitz AA, Affar el B, Heine GF. et al. A mechanism for transcriptional repression dependent on the BRCA1 E3 ubiquitin ligase. Proc Natl Acad Sci USA. 2007;104(16):6614-6619
88. Starita LM, Parvin JD. Substrates of the BRCA1-dependent ubiquitin ligase. Cancer Biol Ther. 2006;5(2):137-141
89. Eakin CM, Maccoss MJ, Finney GL. et al. Estrogen receptor alpha is a putative substrate for the BRCA1 ubiquitin ligase. Proc Natl Acad Sci USA. 2007;104(14):5794-5799
90. Chen A, Kleiman FE, Manley JL. et al. Autoubiquitination of the BRCA1*BARD1 RING ubiquitin ligase. J Biol Chem. 2002;277(24):22085-22092
91. Densham RM, Garvin AJ1, Stone HR. et al. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat Struct Mol Biol. 2016;23(7):647-655
92. Starita LM, Machida Y, Sankaran S. et al. BRCA1-dependent ubiquitination of gamma-tubulin regulates centrosome number. Mol Cell Biol. 2004;24(19):8457-8466
93. Dizin E, Irminger-Finger I. Negative feedback loop of BRCA1-BARD1 ubiquitin ligase on estrogen receptor alpha stability and activity antagonized by cancer-associated isoform of BARD1. Int J Biochem Cell Biol. 2010;42(5):693-700
94. Simons AM, Horwitz AA, Starita LM. et al. BRCA1 DNA-binding activity is stimulated by BARD1. Cancer Res. 2006;66(4):2012-2018
95. Ruffner H, Joazeiro CA, Hemmati D. et al. Cancer-predisposing mutations within the RING domain of BRCA1: Loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proc Natl Acad Sci USA. 2001;98(9):5134-5139
96. Nelson AC, Holt JT. Impact of RING and BRCT domain mutations on BRCA1 protein stability, localization and recruitment to DNA damage. Radiat Res. 2010;174(1):1-13
97. Mullan PB, Quinn JE, Gilmore PM. et al. BRCA1 and GADD45 mediated G2/M cell cycle arrest in response to antimicrotubule agents. Oncogene. 2001;20(43):6123-6131
98. Vidarsson H, Mikaelsdottir EK, Rafnar T. et al. BRCA1 and BRCA2 bind Stat5a and suppress its transcriptional activity. FEBS Lett. 2002;532(1-2):247-252
99. Monteiro AN, August A, Hanafusa H. Evidence for a transcriptional activation function of BRCA1 C-terminal region. Proc Natl Acad Sci USA. 1996;93(24):13595-13599
100. Nadeau G, Boufaied N, Moisan A. et al. BRCA1 can stimulate gene transcription by a unique mechanism. EMBO Rep. 2000;1(3):260-265
101. Yi YW, Kang HJ, Bae I. BRCA1 and Oxidative Stress. Cancers. 2014;6(2):771-795
102. Kawai H, Li H, Chun P. et al. Direct interaction between BRCA1 and the estrogen receptor regulates vascular endothelial growth factor (VEGF) transcription and secretion in breast cancer cells. Oncogene. 2002;21(50):7730-7739
103. Dubrovska A, Kanamoto T, Lomnytska M. et al. TGFbeta1/Smad3 counteracts BRCA1-dependent repair of DNA damage. Oncogene. 2005;24(14):2289-2297
104. Preobrazhenska O, Yakymovych M, Kanamoto T. et al. BRCA2 and Smad3 synergize in regulation of gene transcription. Oncogene. 2002;21(36):5660-5664
105. Marmorstein LY, Ouchi T, Aaronson SA. The BRCA2 gene product functionally interacts with p53 and RAD51. Proc Natl Acad Sci USA. 1998;95(23):13869-13874
106. Ruffner H, Verma IM. BRCA1 is a cell cycle-regulated nuclear phosphoprotein. Proc Natl Acad Sci USA. 1997;94(14):7138-7143
107. Somasundaram K, Zhang H, Zeng YX. et al. Arrest of the cell cycle by the tumour-suppressor BRCA1 requires the CDK-inhibitor p21WAF1/CiP1. Nature. 1997;389(6647):187-190
108. Humphrey JS, Salim A, Erdos MR. et al. Human BRCA1 inhibits growth in yeast: potential use in diagnostic testing. Proc Natl Acad Sci USA. 1997;94(11):5820-5825
109. Cortez D, Wang Y, Qin J. et al. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science. 1999;286(5442):1162-1166
110. Tibbetts RS, Cortez D, Brumbaugh KM. et al. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev. 2000;14(23):2989-3002
111. Lee JS, Collins KM, Brown AL. et al. hCds1-mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature. 2000;404(6774):201-204
112. Chen J. Ataxia telangiectasia-related protein is involved in the phosphorylation of BRCA1 following deoxyribonucleic acid damage. Cancer Res. 2000;60(18):5037-5039
113. Aprelikova ON, Fang BS, Meissner EG. et al. BRCA1-associated growth arrest is RB-dependent. Proc Natl Acad Sci USA. 1999;96(21):11866-11871
114. MacLachlan TK, Somasundaram K, Sgagias M. et al. BRCA1 effects on the cell cycle and the DNA damage response are linked to altered gene expression. J Biol Chem. 2000;275(4):2777-2785
115. Deng CX. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006;34(5):1416-1426
116. Yarden RI, Pardo-Reoyo S, Sgagias M. et al. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat Genet. 2002;30(3):285-289
117. Stewart GS, Wang B, Bignell CR. et al. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421(6926):961-966
118. Wang RH, Yu H, Deng CX. A requirement for breast-cancer-associated gene 1 (BRCA1) in the spindle checkpoint. Proc Natl Acad Sci USA. 2004;101(49):17108-17113
119. Goto H, Tomono Y, Ajiro K. et al. Identification of a novel phosphorylation site on histone H3 coupled with mitotic chromosome condensation. J Biol Chem. 1999;274(36):25543-25549
120. Marmorstein LY, Kinev AV, Chan GK. et al. A human BRCA2 complex containing a structural DNA binding component influences cell cycle progression. Cell. 2001;104(2):247-257
121. Rocca CJ, Soares DG, Bouzid H. et al. BRCA2 is needed for both repair and cell cycle arrest in mammalian cells exposed to S23906, an anticancer monofunctional DNA binder. Cell Cycle. 2015;14(13):2080-2090
122. Sumpter R Jr, Sirasanagandla S, Fernández ÁF. et al. Fanconi Anemia Proteins Function in Mitophagy and Immunity. Cell. 2016;165(4):867-881
123. Chen Q. The Functions of BRCA1 in Mitochondria and Mitophagy. J Microbiol Pathol. 2017;1(1):e102
124. Arun B, Akar U, Gutierrez-Barrera AM. et al. The PARP inhibitor AZD2281 (Olaparib) induces autophagy/mitophagy in BRCA1 and BRCA2 mutant breast cancer cells. Int J Oncol. 2015;47(1):262-268
125. Tang MK, Kwong A, Tam KF. et al. BRCA1 deficiency induces protective autophagy to mitigate stress and provides a mechanism for BRCA1 haploinsufficiency in tumorigenesis. Cancer Lett. 2014;346(1):139-147
126. Tu Z, Aird KM, Zhang R. Chromatin remodeling, BRCA1, SAHF and cellular senescence. Cell Cycle. 2013;12(11):1653-1654
127. Zhu Q, Pao GM, Huynh AM. et al. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature. 2011;477(7363):179-184
128. Bochar DA, Wang L, Beniya H. et al. BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell. 2000;102(2):257-265
129. Ye Q, Hu YF, Zhong H. et al. BRCA1-induced large-scale chromatin unfolding and allele-specific effects of cancer-predisposing mutations. J Cell Biol. 2001;155(6):911-921
130. Stefansson OA, Esteller M. BRCA1 as a tumor suppressor linked to the regulation of epigenetic states: keeping oncomiRs under control. Breast Cancer Res. 2012;14(2):304
131. Chang S, Wang RH, Akagi K. et al. Tumor suppressor BRCA1 epigenetically controls oncogenic microRNA-155. Nat Med. 2011;17(10):1275-1282
132. Tommasi S, Pinto R1, Danza K. et al. miR-151-5p, targeting chromatin remodeler SMARCA5, as a marker for the BRCAness phenotype. Oncotarget. 2016;7(49):80363-80372
133. Li D, Bi FF, Chen NN. et al. Epigenetic repression of phosphatidylethanolamine N-methyltransferase (PEMT) in BRCA1-mutated breast cancer. Oncotarget. 2014;5(5):1315-1325
134. Li D, Bi FF, Chen NN. et al. A novel crosstalk between BRCA1 and sirtuin 1 in ovarian cancer. Sci Rep. 2014;4:6666
135. Rahman S, Islam R. Mammalian Sirt1: insights on its biological functions. Cell Commun Signal. 2011:9 (11). doi: 10.1186/1478-811X-9-11
136. Wang L, Huang H. EZH2 takes the stage when BRCA1 loses. Cell Cycle. 2013;12(23):3575-3576
137. Wang L, Zeng X, Chen S. et al. BRCA1 is a negative modulator of the PRC2 complex. EMBO J. 2013;32(11):1584-1597
138. Ouchi T, Monteiro AN, August A. et al. BRCA1 regulates p53-dependent gene expression. Proc Natl Acad Sci USA. 1998;95(5):2302-2306
139. Ueki T, Park JH, Nishidate T. et al. Ubiquitination and downregulation of BRCA1 by ubiquitin-conjugating enzyme E2T overexpression in human breast cancer cells. Cancer Res. 2009;69(22):8752-8760
140. Kim S, Jin H, Seo HR. et al. Regulating BRCA1 protein stability by cathepsin S-mediated ubiquitin degradation. Cell Death Differ. 2019;26(5):812-825
141. Wu W, Sato K, Koike A. et al. HERC2 is an E3 ligase that targets BRCA1 for degradation. Cancer Res. 2010;70(15):6384-6392
142. Lu Y, Li J, Cheng D. et al. The F-box protein FBXO44 mediates BRCA1 ubiquitination and degradation. J Biol Chem. 2012;287(49):41014-41022
143. Choudhury AD, Xu H, Baer R. Ubiquitination and proteasomal degradation of the BRCA1 tumor suppressor is regulated during cell cycle progression. J Biol Chem. 2004;279(32):33909-33918
144. Nelson AC, Lyons TR, Young CD. et al. AKT regulates BRCA1 stability in response to hormone signalling. Mol Cell Endocrinol. 2010;319(1-2):129-142
145. Kim JL, Ha GH, Campo L. et al. The role of Rak in the regulation of stability and function of BRCA1. Oncotarget. 2017;8(49):86799-86815
146. Mogilyansky E, Clark P, Quann K. et al. Post-transcriptional Regulation of BRCA2 through Interactions with miR-19a and miR-19b. Front Genet. 2016;7:143
147. Podszywalow-Bartnicka P, Wolczyk M, Kusio-Kobialka M. et al. Downregulation of BRCA1 protein in BCR-ABL1 leukemia cells depends on stress-triggered TIAR-mediated suppression of translation. Cell Cycle. 2014;13(23):3727-3741
148. Jin W, Chen L, Chen Y. et al. UHRF1 is associated with epigenetic silencing of BRCA1 in sporadic breast cancer. Breast Cancer Res Treat. 2010;123(2):359-373
149. Lee EY, Abbondante S. Tissue-specific tumor suppression by BRCA1. Proc Natl Acad Sci USA. 2014;111(12):4353-4354
150. Welcsh PL, King MC. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum Mol Genet. 2001;10(7):705-713
151. Jeffy BD, Hockings JK, Kemp MQ. et al. An estrogen receptor-alpha/p300 complex activates the BRCA-1 promoter at an AP-1 site that binds Jun/Fos transcription factors: repressive effects of p53 on BRCA-1 transcription. Neoplasia. 2005;7(9):873-882
152. Gorrini C, Gang BP, Bassi C. et al. Estrogen controls the survival of BRCA1-deficient cells via a PI3K-NRF2-regulated pathway. Proc Natl Acad Sci USA. 2014;111(12):4472-4477
153. Gorrini C, Baniasadi PS, Harris IS. et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J Exp Med. 2013;210(8):1529-1544
154. Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401-426
155. Fan S, Ma YX, Wang C. et al. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene. 2001;20(1):77-87
156. Zheng L, Annab LA, Afshari C. et al. BRCA1 mediates ligand-independent transcriptional repression of the estrogen receptor. Proc Natl Acad Sci USA. 2001;98(17):9587-9592
157. Hosey AM, Gorski JJ, Murray MM. et al. Molecular basis for estrogen receptor alpha deficiency in BRCA1-linked breast cancer. J Natl Cancer Inst. 2007;99(22):1683-1694
158. Ma Y, Katiyar P, Jones LP. et al. The breast cancer susceptibility gene BRCA1 regulates progesterone receptor signaling in mammary epithelial cells. Mol Endocrinol. 2006;20(1):14-34
159. Wang S, Li Y, Hsu PH. et al. Progesterone receptor A stability is mediated by glycogen synthase kinase-3β in the Brca1-deficient mammary gland. J Biol Chem. 2013;288(36):26265-26274
160. Poole AJ, Li Y, Kim Y. et al. Prevention of Brca1-mediated mammary tumorigenesis in mice by a progesterone antagonist. Science. 2006;314(5804):1467-1470
161. Xu X, Wagner KU, Larson D. et al. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet. 1999;22(1):37-43
162. Widschwendter M, Rosenthal AN, Philpott S. et al. The sex hormone system in carriers of BRCA1/2 mutations: a case-control study. Lancet Oncol. 2013;14(12):1226-1232
163. Kim J, Oktay K. Baseline E2 Levels Are Higher in BRCA2 Mutation Carriers: A Potential Target for Prevention? Cancer Causes Control. 2013;24(3):421-426
164. Foulkes WD, Metcalfe K, Sun P. et al. Estrogen receptor status in BRCA1- and BRCA2-related breast cancer: the influence of age, grade, and histological type. Clin Cancer Res. 2004;10(6):2029-2034
165. Mote PA, Leary JA, Avery KA. et al. Germ-line mutations in BRCA1 or BRCA2 in the normal breast are associated with altered expression of estrogen-responsive proteins and the predominance of progesterone receptor A. Genes Chromosomes Cancer. 2004;39(3):236-248
166. Bramley M, Clarke RB, Howell A. et al. Effects of oestrogens and anti-oestrogens on normal breast tissue from women bearing BRCA1 and BRCA2 mutations. Br J Cancer. 2006;94(7):1021-1028
167. Malone JL, Nelson AC, Lieberman R. et al. Oestrogen-mediated phosphorylation and stabilization of BRCA2 protein in breast. J Pathol. 2009;217(3):380-388
168. Hajdu SI. A note from history: the first tumor pathologist. Ann Clin Lab Sci. 2004;34(3):355-356
169. Heppner GH, Dexter DL, DeNucci T. et al. Heterogeneity in drug sensitivity among tumor cell subpopulations of a single mammary tumor. Cancer Res. 1978;38(11):3758-3763
170. Pui CH, Behm FG, Singh B. et al. Heterogeneity of presenting features and their relation to treatment outcome in 120 children with T-cell acute lymphoblastic leukemia. Blood. 1190; 75(1): 174-179.
171. Gray JM, Pierce GB. Relationship between growth rate and differentiation of melanoma in vivo. J Natl Cancer Inst. 1964;32:1201-1210
172. Danielson KG, Anderson LW, Hosick HL. Selection and characterization in culture of mammary tumor cells with distinctive growth properties in vivo. Cancer Res. 1980;40(6):1812-1819
173. Ryu D, Joung JG, Kim NK. et al. Deciphering intratumor heterogeneity using cancer genome analysis. Hum Genet. 2016;135(6):635-642
174. Wikstrand CJ, Bigner SH, Bigner DD. Demonstration of complex antigenic heterogeneity in a human glioma cell line and eight derived clones by specific monoclonal antibodies. Cancer Res. 1983;43(7):3327-3334
175. Heppner GH. Tumor heterogeneity. Cancer Res. 1984;44(6):2259-2265
176. Gyanchandani R, Lin Y, Lin HM. et al. Intratumor Heterogeneity Affects Gene Expression Profile Test Prognostic Risk Stratification in Early Breast Cancer. Clin Cancer Res. 2016;22(21):5362-5369
177. Lapidot T, Sirard C, Vormoor J. et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645-648
178. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730-737
179. Al-Hajj M, Wicha MS, Benito-Hernandez A. et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100(7):3983-3988
180. Galli R, Binda E, Orfanelli U. et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64(19):7011-7021
181. Ricci-Vitiani L, Lombardi DG, Pilozzi E. et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445(7123):111-115
182. Taylor RA, Risbridger GP. The path toward identifying prostatic stem cells. Differ Res Biol Divers. 2008;76(6):671-681
183. Skvortsov S, Skvortsova II, Tang DG. et al. Concise Review: Prostate Cancer Stem Cells: Current Understanding. Stem Cells. 2018;36(10):1457-1474
184. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501(7467):328-337
185. Rycaj K, Tang DG. Cell-of-Origin of Cancer versus Cancer Stem Cells: Assays and Interpretations. Cancer Res. 2015;75(19):4003-4011
186. Kuhlmann JD, Hein L, Kurth I. et al. Targeting Cancer Stem Cells: Promises and Challenges. Anticancer Agents Med Chem. 2016;16(1):38-58
187. Yu H, Lee H, Herrmann A. et al. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14(11):736-746
188. Gao B, Shen X, Kunos G. et al. Constitutive activation of JAK-STAT3 signaling by BRCA1 in human prostate cancer cells. FEBS Lett. 2001;488(3):179-184
189. Welcsh PL, Lee MK, Gonzalez-Hernandez RM. et al. BRCA1 transcriptionally regulates genes involved in breast tumorigenesis. Proc Natl Acad Sci USA. 2002;99(11):7560-7565
190. Buckley NE, Hosey AM, Gorski JJ. et al. BRCA1 regulates IFN-gamma signaling through a mechanism involving the type I IFNs. Mol Cancer Res. 2007;5(3):261-270
191. Takebe N, Miele L, Harris PJ. et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015;12(8):445-464
192. Hanna A, Shevde LA. Hedgehog signaling: modulation of cancer properies and tumor mircroenvironment. Mol Cancer. 2016;15(24):35
193. De Luca P, Moiola CP, Zalazar F. et al. BRCA1 and p53 regulate critical prostate cancer pathways. Prostate Cancer Prostatic Dis. 2013;16(3):233-238
194. Buckley NE, Nic An tSaoir CB, Blayney JK. et al. BRCA1 is a key regulator of breast differentiation through activation of Notch signalling with implications for anti-endocrine treatment of breast cancers. Nucleic Acids Res. 2013;41(18):8601-8614
195. Gorski JJ, James CR, Quinn JE. et al. BRCA1 transcriptionally regulates genes associated with the basal-like phenotype in breast cancer. Breast Cancer Res Treat. 2010;122(3):721-731
196. Korkaya H, Paulson A, Charafe-Jauffret E. et al. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signalling. PLoS Biol. 2009;7(6):e1000121
197. Yi YW, Kang HJ, Kim HJ. et al. Inhibition of constitutively activated phosphoinositide 3-kinase/AKT pathway enhances antitumor activity of chemotherapeutic agents in breast cancer susceptibility gene 1-defective breast cancer cells. Mol Carcinog. 2013;52(9):667-675
198. Zhao J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol Ther. 2016;160:145-158
199. Cojoc M, Mäbert K, Muders MH. et al. A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms. Semin Cancer Biol. 2015;31:16-27
200. Peitzsch C, Kurth I, Kunz-Schughart L. et al. Discovery of the cancer stem cell related determinants of radioresistance. Radiother Oncol. 2013;108(3):378-387
201. Zhang M, Behbod F, Atkinson RL. et al. Identification of tumor-initiating cells in a p53-null mouse model of breast cancer. Cancer Res. 2008;68(12):4674-4682
202. Desai A, Webb B, Gerson SL. CD133+ cells contribute to radioresistance via altered regulation of DNA repair genes in human lung cancer cells. Radiother Oncol. 2014;110(3):538-545
203. Mathews LA, Cabarcas SM, Hurt EM. et al. Increased expression of DNA repair genes in invasive human pancreatic cancer cells. Pancreas. 2011;40(5):730-739
204. Ferlay J, Soerjomataram I, Dikshit R. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359-386
205. Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007;25(11):1329-1333
206. Domchek SM, Eisen A, Calzone K. et al. Application of breast cancer risk prediction models in clinical practice. J Clin Oncol. 2003;21:593-601
207. Ginestier C, Hur MH, Charafe-Jauffret E. et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1(5):555-567
208. Liu S, Ginestier C, Charafe-Jauffret E. et al. BRCA1 regulates human mammary stem/progenitor cell fate. Proc Natl Acad Sci USA. 2008;105(5):1680-1685
209. Heerma van Voss MR, van der Groep P, Bart J. et al. Expression of the stem cell marker ALDH1 in the normal breast of BRCA1 mutation carriers. Breast Cancer Res Treat. 2010;123(2):611-612
210. Heerma van Voss MR, van der Groep P, Bart J. et al. Expression of the stem cell marker ALDH1 in BRCA1 related breast cancer. Cell Oncol Dordr. 2011;34(1):3-10
211. Kubista M, Rosner M, Kubista E. et al. Brca1 regulates in vitro differentiation of mammary epithelial cells. Oncogene. 2002;21(31):4747-4756
212. Liu Y, Burness ML, Martin-Trevino R. et al. RAD51 Mediates Resistance of Cancer Stem Cells to PARP Inhibition in Triple-Negative Breast Cancer. Clin Cancer Res. 2017;23(2):514-522
213. Hunn J, Rodriguez GC. Ovarian cancer: etiology, risk factors, and epidemiology. Clin Obstet Gynecol. 2012;55(1):3-23
214. Bapat SA, Mali AM, Koppikar CB. et al. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005;65(8):3025-3029
215. Latifi A, Luwor RB, Bilandzic M. et al. Isolation and characterization of tumor cells from the ascites of ovarian cancer patients: molecular phenotype of chemoresistant ovarian tumors. PloS One. 2012;7(10):e46858
216. Ho CM, Chang SF, Hsiao CC. et al. Isolation and characterization of stromal progenitor cells from ascites of patients with epithelial ovarian adenocarcinoma. J Biomed Sci. 2012;19:23
217. Meng E, Mitra A, Tripathi K. et al. ALDH1A1 maintains ovarian cancer stem cell-like properties by altered regulation of cell cycle checkpoint and DNA repair network signalling. PloS One. 2014;9(9):e107142
218. Lim JJ, Yang K, Taylor-Harding B. et al. VEGFR3 inhibition chemosensitizes ovarian cancer stemlike cells through down-regulation of BRCA1 and BRCA2. Neoplasia. 2014;16(4):343-353
219. van Asperen CJ, Brohet RM, Meijers-Heijboer EJ. et al. Cancer risks in BRCA2 families: estimates for sites other than breast and ovary. J Med Genet. 2005;42(9):711-719
220. Liu X, Chen X, Rycaj K. et al. Systematic dissection of phenotypic, functional, and tumorigenic heterogeneity of human prostate cancer cells. Oncotarget. 2015;6(27):23959-23986
221. Mateo J, Boysen G, Barbieri CE. et al. DNA Repair in Prostate Cancer: Biology and Clinical Implications. Eur Urol. 2017;71(3):417-425
222. Francis JC, McCarthy A, Thomsen MK. et al. Brca2 and Trp53 Deficiency Cooperate in the Progression of Mouse Prostate Tumourigenesis. PLoS Genet. 2010;6(6):e1000995
223. Moro L, Arbini AA, Yao JL. et al. Loss of BRCA2 promotes prostate cancer cell invasion through up-regulation of matrix metalloproteinase-9. Cancer Sci. 2008;99(3):553-563
224. Alter BP. Fanconi's anemia and malignancies. Am J Hematol. 1996;53(2):99-110
225. Garcia-Higuera I, Taniguchi T, Ganesan S. et al. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7(2):249-262
226. Hussain S, Wilson JB, Medhurst AL. et al. Direct interaction of FANCD2 with BRCA2 in DNA damage response pathways. Hum Mol Genet. 2004;13(12):1241-1248
227. Eaves CJ. Hematopoietic stem cells: concepts, definitions, and the new reality. Blood. 2015;125(17):2605-2613
228. Vasanthakumar A, Arnovitz S, Marquez R. et al. Brca1 deficiency causes bone marrow failure and spontaneous hematologic malignancies in mice. Blood. 2016;127(3):310-313
229. Mgbemena VE, Signer RAJ, Wijayatunge R. et al. Distinct Brca1 Mutations Differentially Reduce Hematopoietic Stem Cell Function. Cell Rep. 2017;18(4):947-960
230. Antoniou A, Pharoah PD, Narod S. et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72(5):1117-1130
231. Kauff ND, Satagopan JM, Robson ME. et al. Risk-reducing salpingo-oophorectomy in women with a BRCA1 or BRCA2 mutation. N Engl J Med. 2002;346(21):1609-1615
232. Rebbeck TR, Friebel T, Lynch HT. et al. Bilateral prophylactic mastectomy reduces breast cancer risk in BRCA1 and BRCA2 mutation carriers: the PROSE Study Group. J Clin Oncol. 2004;22(6):1055-1062
233. Aysola K, Desai A, Welch C. et al. Triple Negative Breast Cancer - An Overview. Hered Genet. 2013;2013(Suppl 2):001
234. Jiang T, Shi W, Natowicz R. et al. Statistical measures of transcriptional diversity capture genomic heterogeneity of cancer. BMC Genomics. 2014;15:876
235. Kennedy RD, Quinn JE, Mullan PB. et al. The role of BRCA1 in the cellular response to chemotherapy J Natl Cancer Inst. 2004; 96(22): 1659-1668.
236. Pan Z, Xie X1. BRCA mutations in the manifestation and treatment of ovarian cancer. Oncotarget. 2017;8(57):97657-97670
237. Jiang T, Shi W1, Wali VB. et al. Predictors of Chemosensitivity in Triple Negative Breast Cancer: An Integrated Genomic Analysis. PLoS Med. 2016;13(12):e1002193
238. Kriege M, Jager A, Hooning MJ. et al. The efficacy of taxane chemotherapy for metastatic breast cancer in BRCA1 and BRCA2 mutation carriers. Cancer. 2012;118(4):899-907
239. Arun B, Bayraktar S, Liu DD. et al. Response to neoadjuvant systemic therapy for breast cancer in BRCA mutation carriers and noncarriers: a single-institution experience. J Clin Oncol. 2011;29(28):3739-3746
240. Bignon L, Fricker JP, Nogues C. et al. Efficacy of anthracycline/taxane-based neo-adjuvant chemotherapy on triple-negative breast cancer in BRCA1/BRCA2 mutation carriers. Breast J. 2018;24(3):269-277
241. Nientiedt C, Heller M, Endris V. et al. Mutations in BRCA2 and taxane resistance in prostate cancer. Sci Rep. 2017;7(1):4574
242. Mateo J, Cheng HH, Beltran H. et al. Clinical Outcome of Prostate Cancer Patients with Germline DNA Repair Mutations: Retrospective Analysis from an International Study. Eur Urol. 2018;73(5):687-693
243. Byrski T, Gronwald J, Huzarski T. et al. Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J Clin Oncol. 2010;28(3):375-379
244. Byrski T, Dent R, Blecharz P. et al. Results of a phase II open-label, non-randomized trial of cisplatin chemotherapy in patients with BRCA1-positive metastatic breast cancer. Breast Cancer Res. 2012;14(4):110
245. Isakoff SJ, Mayer EL, He L. et al. TBCRC009: A Multicenter Phase II Clinical Trial of Platinum Monotherapy With Biomarker Assessment in Metastatic Triple-Negative Breast Cancer. J Clin Oncol. 2015;33(17):1902-1909
246. Vencken PM, Kriege M, Hoogwerf D. et al. Chemosensitivity and outcome of BRCA1- and BRCA2-associated ovarian cancer patients after first-line chemotherapy compared with sporadic ovarian cancer patients. Ann Oncol. 2011;22(6):1346-1352
247. Pennington KP, Walsh T, Harrell MI. et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20(3):764-775
248. Afghahi A, Timms KM, Vinayak S. et al. Tumor BRCA1 Reversion Mutation Arising during Neoadjuvant Platinum-Based Chemotherapy in Triple-Negative Breast Cancer Is Associated with Therapy Resistance. Clin Cancer Res. 2017;23(13):3365-3370
249. Sakai W, Swisher EM, Jacquemont C. et al. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009;69(16):6381-6386
250. Edwards SL, Brough R, Lord CJ. et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451(7182):1111-1115
251. Herceg Z, Wang ZQ. Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat Res. 2001;477(1-2):97-110
252. Dziadkowiec KN, Gąsiorowska E, Nowak-Markwitz E. et al. PARP inhibitors: review of mechanisms of action and BRCA1/2 mutation targeting. Prz Menopauzalny. 2016;15(4):215-219
253. Farmer H, McCabe N, Lord CJ. et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917-921
254. Fong PC, Boss DS, Yap TA. et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123-134
255. Litton JK, Rugo HS, Ettl J. et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N Engl J Med. 2018;379(8):753-763
256. Ison G, Howie LJ, Amiri-Kordestani L. et al. FDA Approval Summary: Niraparib for the Maintenance Treatment of Patients with Recurrent Ovarian Cancer in Response to Platinum-Based Chemotherapy. Clin Cancer Res. 2018;24(17):4066-4071
257. Konecny GE, Kristeleit RS. PARP inhibitors for BRCA1/2-mutated and sporadic ovarian cancer: current practice and future directions. Br J Cancer. 2016;115(10):1157-1173
258. Zimmer AS, Gillard M, Lipkowitz S. et al. Update on PARP Inhibitors in Breast Cancer. Curr Treat Options Oncol. 2018;19(5):21
259. Atun R, Jaffray DA, Barton MB. et al. Expanding global access to radiotherapy. Lancet Oncol. 2015;16(10):1153-1186
260. Lomax ME, Folkes LK, O'Neill P. Biological consequences of radiation-induced DNA damage: relevance to radiotherapy. Clin Oncol R Coll Radiol. 2013;25(10):578-585
261. Domina EA, Philchenkov A, Dubrovska A. Individual Response to Ionizing Radiation and Personalized Radiotherapy. Crit Rev Oncog. 2018;23(1-2):69-92
262. Kan C, Zhang J. BRCA1 Mutation: A Predictive Marker for Radiation Therapy? Int J Radiat Oncol Biol Phys. 2015;93(2):281-293
263. Bernier J, Poortmans P. Clinical relevance of normal and tumour cell radiosensitivity in BRCA1/BRCA2 mutation carriers: a review. Breast. 2015;24(2):100-106
264. Peñuelas S, Anido J, Prieto-Sánchez RM. et al. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell. 2009;15(4):315-327
265. Ouchi T, Lee SW, Ouchi M. et al. Collaboration of signal transducer and activator of transcription 1 (STAT1) and BRCA1 in differential regulation of IFN-gamma target genes. Proc Natl Acad Sci USA. 2000;97(10):5208-5213
Author contact
![]() Corresponding authors: Ielizaveta Gorodetska, email: Liza.Gorodetskade or Anna Dubrovska, email: Anna.Dubrovskade
Corresponding authors: Ielizaveta Gorodetska, email: Liza.Gorodetskade or Anna Dubrovska, email: Anna.Dubrovskade