Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(16):3798-3809. doi:10.7150/jca.31364 This issue Cite
Review
The Epithelial-Myeloid-Transition (EMyeT) of cancer cells as a wrongly perceived primary inflammatory process eventually progressing to a bone remodeling malignancy: the alternative pathway for Epithelial- Mesenchymal-Transition hypothesis (EMT)?
Henning M. Schramm ![]()
Institute for Integral Cancer Research (IFIK), CH-4144 Arlesheim/Switzerland
Received 2018-11-9; Accepted 2019-5-10; Published 2019-6-9
Abstract
Cancer cells express multiple markers expressed by mesenchymal as well as myeloid cells in common and in addition specific markers of the myeloid lineages, especially those of dendritic cells, macrophages and preosteoclasts. It has also been possible to identify monocyte-macrophage gene clusters in cancer cell specimens as well as in cancer cell lines.
Accordingly, like myeloid cells cancer cells often express pro-inflammatory cytokines, and consequently the carcinoma may be perceived by the organism as a primary inflammatory process comparable to the immune inflammatory reactions in the eye or in the case of arthritis. This would explain why a carcinoma may induce a certain alarm state in the organism by increasing a fatal sympathetic tone in the patient, supplying the carcinomas with nutrients at the cost of other requirements, inducing tolerance against the cancer cells mistaken as myeloid cells, provoking fibrosis and neoangiogenesis, and increasing inflammatory cells at the carcinoma site.
This seemingly inflammatory process of Epithelial-Myeloid-Transition (EMyeT) is superimposed by the progression of part of the myeloid cancer cells to stages comparable to preosteoclasts and osteoclasts, and their development to metastasizing carcinomas often at the site of bone.
This concept of carcinogenesis and malignant progression described here challenges the widely accepted EMT-hypotheses and could deliver the rationale for the various peculiar aspects of cancer and the variety of therapeutic antitumoral measures.
Keywords: Cancer, EMyeT, myeloid lineage cells, cancer as a non-healing wound, carcinoma as an inflammatory process, metastases as a false bone remodeling process
General Introduction
Epithelial-to-mesenchymal transition (EMT) is a process that plays essential roles in embryonic development and wound healing that is characterized by loss of homotypic adhesion and cell polarity and increased invasion and migration. When a carcinoma is progressing in malignancy and starting to metastasize, similar changes in cancer cells are seen. Consequently this is explained by an epithelial-mesenchymal transition (EMT) of cancer cells.
However, during carcinogenesis and malignant progression various phenomena on the molecular level like e.g. the myeloid antigen expression of cancer cells as well as on various clinical aspects like e.g. cancer as a non-healing wound cannot be explained by the widely accepted EMT-hypotheses. Therefore, the findings on which the EMT-hypothesis is based are scrutinized for their validity and we discuss another possible conclusion from them. As cancer cells express besides many common markers with mesenchymal cell specifically myeloid markers and behave like myeloid cells, we hypothesize that they undergo an Epithelial-Myeloid transition (EMyeT).
In the first part of our investigative literature review we point out why a different conclusion i. e. the Epithelial-Myeloid-Transition hypothesis (EMyeT hypothesis) can be drawn from scientific research findings. The EMyeT-hypothesis would allow us to understand the entailing reactions of the organism towards the carcinoma in a more comprehensive way than the EMT hypothesis. In the second part of our review we describe how within the EMyeT concept the myeloid cancer cells progress to pre-, osteoclasts and giant cells and due to their nature often migrate to the bone site. And again, how the reactions of the organism in coping with this bone related tumorous challenge will be discussed in view of the EMyeT hypothesis.
Part 1: The myeloid nature of cancer cells and their perception as an inflammatory process by the organism
Introduction - The difficulties to differentiate between mesenchymal cells and myeloid cells in-vitro
In a former publication we proposed an alternative or additional interpretation of the phenotypical and functional change of cancer cells when progressing in their malignancy, which is usually defined as the epithelial-mesenchymal transition (EMT) of cancer cells. Based on various special features of metastasizing cancer cells we suggested that the change can also be regarded as an epithelial-myeloid transition (EMyeT) [1]. To substantiate this view we will here describe functional, genetic and morphological aspects in addition to those already reported in the former publication. This interpretation may allow us to understand why the organism may perceive the carcinoma as a primary inflammatory process and reacts accordingly which ensures the fatal course of the disease in this context.
According to the EMT hypothesis cancer cells seem to pathologically recapitulate the normal epithelial-mesenchymal transition occurring during mammalian development, and during physiological wound healing [2]. However, the markers of EMT are not specific to mesenchymal cells; they are found also in migrating myeloid cells as well [3, 4]. Even certain myeloid cells may adopt a spindle-like morphology and therefore resemble mesenchymal cells [5-7]. EMT is the physiological process for wound healing and is necessary for the re-epithelialization of the wound. In cancer this does not occur because the cancer process remains at a stage comparable to the proliferation phase of a wound-healing process. This non-healing phase may be explained by the perception of the carcinoma as an uncontrolled primary inflammatory process.
The putative origin of cancer cells
Carcinomas arise in the epithelium, and because of the epithelial markers cancer cells express besides their myeloid or mesenchymal markers, they are thought to be of epithelial origin. As a consequence of this concept the origin of cancer cells is assumed to be purely from immature or mature epithelial cells. But is this conclusion compelling? There are several studies suggesting another origin of cancer cells. In one animal study Houghton et al. demonstrated that cancer in stomach epithelial tissue originated from myeloid bone marrow-derived cells. These cells were recruited to the site of epithelial tissue injury and inflammation, induced by a chronic helicobacter infection. The bone marrow-derived cell adopted a gastric epithelial phenotype. The cells subsequently progressed through metaplasia and dysplasia to intraepithelial cancer [8]. Cancer detected in patients who had received bone marrow transplants demonstrate that hematopoietic cells were incorporated into neoplasia and had adopted the phenotype of epithelial cancer cells [9-11] In an in-vitro study CD34 liver cancer stem cells were initiated by fusion of CD34 hematopoietic precursor-derived myeloid cells with hepatobiliary stem/progenitor cells [12]. As this fusion or exchange occurs via inadequate partners, it may result in aneuploidy and the merged characteristics of myeloid cells with epithelial cells or melanocytes, which are specific features of cancer cells. The findings that cancer cells may derive from fusions of progenitor-epithelial cells with cells committed to the myeloid lineage are substantiated by another animal study from a different viewpoint. MTA transgenic mice are deficient in MHC-II positive cells in the epidermis and therefore lack Langerhans cells or any other myeloid cells in this tissue. Researchers expected MTA transgenic mice to be very prone to skin carcinogenesis due to the lack of Langerhans cells in their epidermis. The contrary was the case. The animals were resistant to squamous cell carcinoma induction in the skin [13]. This study indicates that myeloid cells in the epithelium may be a prerequisite for cancerogenesis.
Based on these results the epithelial myeloid transition hypothesis assumes that cancer originates from epithelial cells which may have fused with committed myeloid cells and thus interprets the origin of cancer cells differently from the EMT.
Some simplistic basics of mesenchymal cells and myeloid cells
Mesenchymal cells and myeloid cells are part of the differentiation of the mesoderm during embryonic development. The mesenchymal precursor cell is their common predecessor. From there on they differentiate in two different cell lineages: the mesenchymal stem cell and the hematopoietic stem cell. The mesenchymal stem cell may differentiate into fibroblasts, myoblasts, osteoblasts, chondroblasts, adipocytes, endothelial cells and cardiomyocytes. Under pathological conditions they may form sarcomas. Myeloid cells on the other hand originate from hematopoietic stem cells and form the cell family of monocytes, macrophages, dendritic cells, osteoclasts, microglia and Kupfer cells. The monocyte cells among the myeloid cells may still preserve their mesodermal plasticity and in-vitro can be reprogrammed to adult stem cells and under conditions known to induce mesenchymal differentiation develop into cells of the mesenchymal lineage [14, 5]. We come back to this aspect, when we discuss the EMT hypotheses as a possible in-vitro artefact in the discussion chapter. Vice-versa, to our knowledge there is no publication showing that mesenchymal cells may transdifferentiated into myeloid cells, although they may enhance their formation.
Common metabolism between cancer and activated myeloid cells: aerobic glycolysis
Tumors generally show a shift in their metabolism from oxidative phosphorylation to glycolysis and thus exhibit increased glycolysis for ATP generation [15]. They obtain as much as 50% of their ATP by metabolizing glucose directly to lactic acid, even in the presence of oxygen [16]. Increased glycolysis induces acidification of the local environment, limiting proliferation and inducing cell death through necrosis and apoptosis [17].
Glycolysis however is not a specific hallmark of cancer cells. Activated monocytes, dendritic cells, macrophages, lymphocytes, astrocytes, retina cells, white muscle cells and auditory hair cells gain their energy by glycolysis even in the presence of sufficient oxygen. An increased speed of energy production, which glycolysis enables, is required for the proliferation or reactivity of these cells. Here we hypothesize that aerobic glycolysis of cancer cells is a hallmark which is based on their myeloid traits and nature.
Common clusters of differentiation between cancer cells and cells of the myeloid lineage
When we compare the surface markers of cancer cells with those of osteoclasts and their myeloid lineage progenitors, we detect multiple correspondences described by us in a former publication [1].
Other common surface markers
Other than the above cited clusters of differentiation, a multiplicity of surface markers are expressed by both cancer and myeloid lineage cells, of which we name here only the following: Trap, TREM2, Cathepsin K, TLRs, RANK, ADAM, DAP12, OSCAR (Osteoclast Associated Receptor), MAC387, DC-STAMP, NK1 receptor, BMP receptor, Protease activated receptor-1, TRAF-6 and calcitonin receptor [18-24]. The calcitonin receptor along with TRAP, OSCAR, NFATc1 and Cathepsin K are specific pre-/osteoclast markers [1, 25, 26]. These markers are expressed neither by epithelial nor mesenchymal cells, and suggest that cancer cells even in their primary site are related to the various stages of myeloid cells. i.e. from stem cells to progenitor cells of monocytes, dendritic cells, macrophages through to preosteoclasts.
Common transcription factors between migratory cancer and myeloid cells
The transcription factors ZEB, Twist1, Slug (Snail), the cytoskeleton Vimentin, the adhesion protein N-cadherin and the matrix substance fibronectin are commonly considered as mesenchymal markers. However, these markers are expressed and activated in migrating leucocytes as well. ZEB2 is upregulated in migratory dendritic cells as well as in monocytes and widely expressed among hematopoietic lineages [27, 28]. ZEB1mediates a diverse array of processes including mesoderm-derived cell differentiation, is activated in migrating dendritic cells and in tumor associated macrophages [29, 27]. N-cadherin plays a role in migrating dendritic cells [27]. Twist1 plays a decisive role in myeloid lineage development and cytokine expression by macrophages [30]. Migrating macrophages express Snail, and downregulation of Snail leads to impaired migration of macrophages [31]. The TGF-beta1-induced migration of activated macrophages is mediated by Snail [31]. The extracellular protein substance fibronectin adheres to integrin alphavbeta3 and alpha1beta3, which are expressed by macrophages/preosteoclasts [32]. Macrophage interaction with fibronectin is recognized as an important aspect of wound repair and activates macrophage migration [33]. The assumed mesenchymal markers of cancer cells could therefore also be myeloid markers, especially of dendritic cells, macrophages and preosteoclasts when they are migrating. The interpretation that cancer cells undergo EMT based on these markers alone could be premature or even a fallacy.
Common gene clusters between cancer cells and cells of the myeloid lineage
In a gene-profiling analysis of mouse mammary EpRas tumor cells and of 38 human breast cancer cell lines gene clusters of monocyte/macrophage were identified after activation by TFG-β1. In human breast cancer cell lines basal B cells and ER- and PR- negative cells displayed the highest activation of myeloid gene clusters [34]. Canine mammary cancer cell lines highly expressed genes of the myeloid lineage [35]. Transcriptome profiling of human prostate cancer cell lines indicated that they harbor a genetic signature of myeloid cells that is associated with the differentiation and maintenance of myeloid cells. However, their myeloid markers were only up-regulated in castration-resistant prostate cancer (CRPC) cells that showed inactivation of the classical AR pathway and were Enzalutamide resistant, i. e. the more malignant cancer cells. These cells upregulated the programmed death ligand 1(PD-L1) inhibiting anticancer immune response [36].
Common role of IGF-1/II in cancer cell proliferation and myeloid cell activation
IGF-1 and 2 (Insulin like growth factor 1 and 2) play a key role in inflammatory processes, in bone remodeling as well as in cancer initiation and progression.
IGF-1 is implicated in the proliferation of many types of cancers such as pancreas, prostate, mammary, and colorectal carcinoma. Mouse models where circulating IGF-1 levels are reduced, while tissue expression of IGF-1 is normal, show lower risk for the development of colon and breast cancer and metastases when compared to control mice [37]. Patients with Laron syndrome (inactive GH receptor resulting in IGF-1 deficiency) seem to be protected from cancer [38, 39]. Upon withdrawal of IGF-1 from the medium the cancer cells revert to an epithelial morphology in in vitro tests. IGF-1 plays an important role in the transition of a sedentary cancer cell to a migratory one [40]. Some cancer cells may secrete IGF-1 or IGF-II acting as an autocrine factor for cancer cell growth [41].
Resident myeloid cells (tissue monocytes and macrophages) can be a prominent source of IGF-1 and IGF-II regulating inflammatory processes and promoting the growth of hematopoietic and other cells [42-44].
Common role of M-CSF (CSF-1)/CSF-1R in the biology of cancer and of myeloid cells
The hematopoietic receptor-ligand pair of macrophage-colony stimulating factor (M-CSF/ CSF-1) and its receptor CSF-1R have been found to be important in the biology of breast cancer, renal cell cancer, pancreas cancer, neuroendocrine pancreas cancer, prostate cancer, ampullary cancer, bladder cancer, ovarian cancer and non-small lung cancer. The increased plasma level of M-CSF can be used in diagnosing breast cancer, especially when discriminating between cancer and non-carcinoma lesions [45]. Invasion and metastasis of breast cancer cells are driven by this cytokine pairing of tumor cells and macrophages. This cytokine pair physiologically regulates the growth and differentiation of hematopoietic progenitor cells into monocytes, dendritic cells and bone-resorbing osteoclasts, and functionally activates mature neutrophils or macrophages [46]. The pair seems to be important for the migratory state of macrophages as well as of cancer cells, thus demonstrating their relationship.
Common activation of the two major stress axes (HPA axis and SNS) in cancer patients and patients with inflammatory diseases
Evidence clearly demonstrate a pivotal role of the sympathetic nervous system (SNS) and its neurotransmitters in regulating inflammation as well as influencing the course of cancer [47-49]. Inflammatory cells as well as cancer cells may secrete inflammatory cytokines like interferons, interleukins and tumor necrosis factor which stimulate the activation of the hypothalamic-pituitary-adrenal axis (HPA axis) [50, 51].
Common activation of NFkappaB, S100A8/S100A9 protein expression, toll-like receptors and Stat3 in cancer and in myeloid lineage cells
In about 40% of the tissue specimens of various cancer sites, constitutive NFkappaB and STAT3 activation has been noted. The transcription factors may be involved in tumor-initiating angiogenesis and invasiveness [52].
Elevated S100A8/S100A9 protein expression and corresponding inflammatory cytokines expression has been detected in various cancer cells and are a marker for activated myeloid cells in an inflammatory response. Toll-like receptors are expressed on immune cell and on cancer cell. When activated both cell types release inflammatory cytokines/chemokines. Immune competence is a decisive trait of cells of the myeloid lineage and is uncharacteristic of matured mesenchymal cells.
MCP-1 secretion by cancer cells and activated monocytes
Besides its production by macrophages, osteoclasts, fibroblasts and endothelial cells, monocyte chemoattractant protein 1 (MCP-1 or CCL2) is secreted by cancer cells as well. MCP-1 plays a critical role in the recruitment and activation of monocytes to sites of inflammation, injury and cancer as well as in bone remodeling [53]. In the later it is involved in the receptor activator of NF-kappa ligand-induced fusion of preosteoclasts, and enhances osteoclastogenesis [54].
In several cancers MCP-1 has been shown to be an important factor for tumor growth and migration [54].
Cancer as a non-healing wound, and adrenergic and cholinergic hyperinnervation
To explain the persistence of glycolysis and malignancy of cancer cells in connection with the carcinoma as a non-healing wound, a further aspect seems to be essential, i.e. the cholinergic and especially the adrenergic hyperinnervation of the tumor site. Cancer cells increase their adrenergic as well as their cholinergic receptors. An increase in beta-adrenergic signaling of cancer cells has been found at many tumor sites, as a decisive factor in the development and progression of malignancy [48, 49]. Moreover, there is an increased secretion of norepinephrine in the circulation of cancer patients. Certain forms of adrenergic signaling stimulate angiogenesis to ensure better energy supply, increasing glucose levels in the blood, and glycolysis, and contributing to immune tolerance of the tumor [55]. Under the persistent adrenergic influence at the tumor site, immigrating macrophages develop into anti-inflammatory M2 macrophages and pro-inflammatory M1 macrophages [56]. Patients with mammary carcinoma have higher norepinephrine levels and a lower heart-rate variability (HRV) indicating that overall sympathetic activity has significantly increased [57]. On the other hand, a relative higher vagal activity is related to increased survival of patients with breast cancer [58].
This process may be comparable to adrenergic hyperinnervation and increases in circulatory norepinephrine levels in the case of a severe injury and wound during the proliferating and remodeling phase. Systemic response to injury is characterized by massive release of norepinephrine into the circulation as a result of global sympathetic activation [59]. The decisive role of the adrenergic activity to the local anti-inflammatory mechanism has been demonstrated for the inflamed eye, with striking similarities in immune reactions to a carcinoma site [47, 60].
Part 2: The seemingly inflammatory process of EMyeT is superimposed by cancer cells progressing into stages of myeloid cells comparable to preosteoclasts and osteoclasts
Do cancer cells hoax osteomimetic properties or are they intrinsically preosteoclastic?
Most known human carcinomas show an increased expression of bone-specific proteins, i.e. osteopontin, osteocalcin and sialoprotein [61]. Osteopontin is produced both by cancer cells and by osteoblasts and osteoclasts. Osteopontin is one of the major noncollagenous bone matrix proteins. It binds to osteoclasts or cancer cells via CD44 or/and integrin alphavbeta3. Osteopontin-integrin alphavbeta3 signaling mediates CD44/MMP-9 complex formation on the cell surface, and enhanced survival in gastrointestinal and other cancer cells [62, 63]. It is involved in MMP-9 secretion and migrating motility. Thus, the expression level of osteopontin correlates with the metastatic potential of several cancer tumors.
When several preosteoclasts fuse into an osteoclast they intracellularly overexpress certain signaling pathways which are likewise overexpressed in cancer cells during their proliferation [64]. STAT3 and NFkappaB are a prerequisite for the maturation of preosteoclasts into osteoclasts, and consequently the inhibition of NFkappa B activity blocks this developmental differentiation. NFkappaB induces the RANKL expression of osteoblasts [65]. The constitutive activation of NFkappaB is also a prerequisite for MMPs secretion and thus for the bone-resolving capacities of osteoclasts [66]. STAT3 activation is required in this context as well [52]. In bone remodeling the RANK-RANKL system belongs as a cytokine system to the tumor-necrosis factor family involved in osteoclastogenesis but as well as in the progression of cancer.
Analysis of surgical biopsy specimens showed the expression of RANKL, RANK and osteoprotegerin (OPG) in primary carcinomas, namely of lung, breast, head and neck, colorectum, kidney, thyroid and liver. The median percentage of RANKL-expressing cells was 60% in primary tumors and metastases, without any statistically significant difference between the two groups [67]. This study highlights the activity and function of the RANKL-RANK system not only in bone metastasis management but also in primary tumors.
Multiple publications describe the osteomimetic properties of cancer cells and relate this to their bone metastasizing properties [68-71]. The biomarkers of cancer cells like RANKL, Runx2, osteocalcin and PTHrP are interpreted as osteomimicry and a hoax by cancer cells to deceive the bone cells in order to metastasize at this site. We interpret these properties not as an osteomimetic feature of mesenchymal cancer cells but as the real nature of migrating cancer cells.
Common migration pattern of cancer cells and myeloid cells: trafficking to lymph nodes and the bone
The migration of cancer cells closely resembles that of cells of the myeloid lineage e.g. dendritic cells, macrophages and preosteoclasts [72, 73]. Both cancer cells and dendritic cells target the regional lymph nodes induced by the same signals [73]. This migration through the lymphatic system is not known in the case of mesenchymal cells. Preosteoclasts show the same extraordinary and unusual ability to migrate and transmigrate through cell layers and tissues in order to reach the skeleton. In cancer cells, as in preosteoclasts, a dynamic actin remodeling appears as one major factor endowing these cells with the migrating property. During transmigration, cancer cells such as osteoclast precursors express integrin alphavbeta3 around actin patches [74]. Matrix metalloproteinases secreted at the podosomes site are required for the degradation of the extracellular matrix and this contributes to the invasiveness of cancer cells. The same function facilitates the migration of preosteoclasts through barriers and to form pits in the bone [75]. Although nearly all cancers may frequently metastasize to the bone, the commonest malignant tumors have a special predilection for the skeleton. Monocytes in the peripheral blood and perhaps in inflamed areas, when becoming committed to the osteoclast lineage, traffic to the bone in the same way [74].
Common role of the chemokine receptor CXCR4 - chemokine SDF-1 (CXCL12) axis in cancer pathogenesis and in osteogenesis
The chemokine receptor-chemokine axis CXCR4/SDF-1 plays multiple roles in tumor pathogenesis [76] as well as in osteogenesis [77]. The tumor cells crosstalk with tumor-associated fibroblasts, as do osteoclasts with osteoblasts via the chemokine axis CXCR4/SDF-1. The trafficking of cancer cells and preosteoclasts to bone involves many common factors, among them chemoattractants such as SDF-1(CXCL-12) /CXCR4 axis, MCP-1, transcription factors such as NFkappaB and STAT3, cytokines like RANK/RANKL and Il-6, hormones such as osteocalcin, estrogen, androgens, further neurotransmitters, alphavbeta3 integrins/osteopontin and calcium signaling pathways [74].
Common neurogenesis and neuronal dependency between bone cells and cancer cells
Bone remodeling, like most other homeostatic functions, but also carcinogenesis and cancer progression, and metastasizing capacities, is subject to sympathetic influence. 70 to 90% of breast, colon, gastric, lung, ovary, nasopharyngeal and prostate cancer tissues express beta2-adrenergic receptors. The migration of breast, prostate, ovary and colon carcinoma cells is enhanced by the stress-related neurotransmitter norepinephrine, and this effect can be inhibited by the beta-blocker propranolol [78].
Osteoblasts as well as osteoclastic cells are equipped with adrenergic receptors and neuropeptide receptors [79]. Sympathetic neurons in the hypothalamus and bone control osteoblast activity in bone formation and osteoclast activity in bone resorption via beta2-adrenergic receptors. Increased sympathetic nervous system activity leads to increased bone resorption through beta2-adrenergic receptors [79].
Osteoblastic and osteoclastic cells on the one hand, and cancer cells on the other, constitutively express diffusible axon-guidance molecules such as netrins and neurotrophins, known to function as chemo-attractants for growing nerve fibers [80, 81].
Estrogens and androgens
Cancer growth and malignancy on the one hand, and bone remodeling on the other, are influenced by hormones e.g. sex steroids such as estrogens and androgens. Therefore, hormonal treatment of cancer often exerts a severe impact on bone remodeling as well. A detailed description of theses interactions, however, would surpass the context of our review and are, therefore, not further discussed.
Common calcium signaling in cancer, related hormones and bone remodeling
The calcium signaling pathways seems to play an important role in cancer like in bone remodeling. Vitamin D, osteocalcin, calcitonin, osteopontin and parathyroid hormone-related peptide are reviewed in this study. Various studies hint at their decisive role not just in bone metastases, but in carcinogenesis and in cancer procession as well.
Calcitonin is a small neuropeptide hormone produced in the thyroid and counteracts the activity of parathormon. Both hormones regulate calcium and phosphate metabolism in the organism. Calcitonin is secreted in response to hypercalcemia. The calcitonin receptor together with TRAP are specific markers of osteoclasts [82]. Calcitonin is not expressed by macrophages [83, 84]. Besides calcitonin's role in bone remodeling by inhibiting the bone resorbing activity of osteoclasts, it is decisively active in cancer, as demonstrated with prostate and breast cancer cells. In animal studies calcitonin inhibited invasion of breast cancer cell and suppressed ERK1/2 phosphorylation [85]. However, in prostate cancer cells it seems to exert an opposite effect. It is produced by prostate cells for autocrine and paracrine stimulation of growth and invasiveness [86].
Parathyroid-hormone-related peptides (PTHrP), among other things, are involved in epithelial-mesenchymal interaction during the formation of the mammary gland. In bone remodeling it acts between osteoclasts and osteoblasts and, under pathological conditions, is an essential growth factor for various cancers. During tumor growth and invasion its expression is modulated by a number of growth and also angiogenic factors, such as interleukins, tumor-derived growth factor beta, platelet-derived growth factor and vascular endothelial-derived growth factor [87]. PTHrPs are the factors responsible for hypercalcemia associated with malignancy [72]. Most malignant human tumors express this hormone abundantly; however this results in hypercalcemia only in a percentage of the tumors [87]. Prostate cancer derived PTHrP acts in the bone marrow to potentiate myeloid cells, which are recruited to tumor tissue where they contribute to tumor angiogenesis and growth [88]. PHTrP might be related to heterotopic ossification associated with malignancies [89].
Osteocalcin is a small, highly conserved peptide hormone, which plays an important role in the remodeling of bone as well as in cancer progression. Osteocalcin has been detected in prostate, breast, lung and pancreas carcinoma as well melanoma. Osteocalcin expressed in pancreatic cancer cell lines enhanced cell growth and invasion through autocrine and paracrine mechanisms [90]. Most studies on osteocalcin`s role in cancer involve prostate and breast cancer. Elevated osteocalcin protein levels in the serum are found in metastatic prostate and breast cancer compared to non-metastatic patients [91].
Coupling between cancer cells and mesenchymal cells is comparable to the coupling between osteoclasts and mesenchymal cells i.e. osteoblasts
The key factors in the coupling of osteoclast with osteoblast and vice-versa are SDF-1, IGF-1 and S1P. All three factors are present at the cancer site and play an important role in the progress of malignancy. Both cancer cells and osteoclasts are coupled with mesenchymal cells and need them for their growth, differentiation, migration activity and survival. Cancer cells are coupled with the mesenchymal tumor-associated fibroblast, as osteoclasts are spliced with mesenchymal osteoblasts.
In the tumor, cancer cells are constantly interacting with tumor-associated stromal cells. The latter are not just bystanders in the tumor region, but contribute to tumor progression, fibrosis and metastasizing capacity [92]. The cancer cell releases factors that enhance the ability of the fibroblast to secrete a variety of tumor-promoting chemokines involving the CXCL12-CXCR4 axis [76], Rankl-Rank, ephrinB2-EphB4, and PTHrP-type 1- PTH receptor. Moreover, preosteoblasts may promote breast cancer migration and seeding to the bone by producing hepatocyte growth factor (HGF) which interacts with the tumor cells [93].
A certain percentage of the stromal cells of the cancer site originate in the bone marrow of the host. Primary tumors are able to encourage the mobilization and recruitment of fibroblast precursors from the bone marrow, indicating that such tumors interact with the bone marrow in order to expedite their own proliferation.
Similar biological functions exist between preosteoclasts and osteoblasts. They also communicate by these chemokines, through cell-to-cell contact, diffusible paracrine factors and cell-bone matrix interaction [94]. The osteoclasts are dependent on this crosstalk for their differentiation [94].
Common sensitivity to antirheumatic, steroids, bisphosphonates, polyphenols and cytostatics
Both cancer cells and osteoclasts are inhibited by antirheumatics, vitamin D [95, 96], bisphosphonates and polyphenols. The antirheumatic and analgetic substance aspirin inhibits the transformation of a benign tumor into malignant one. In vitro studies with various antirheumatics show that they inhibit the growth and proliferation of cancer cells [97].
Bisphosphonates not only inhibit osteoclastogenesis but also show direct anti-tumor effects on cancer cells [98].
Numerous in vitro-, in vivo and epidemiological studies demonstrate that polyphenols, a substance class found in various plants, demonstrate direct antitumoral activity [99] as well as inhibition of osteoclastogenesis [100].
The EMyeT hypotheses could explain specific toxicity of cytostatics. Due to the assumed myeloid nature of cancer cells, cytostatics not only destroy cancer cells but myeloid cells as well, thus causing myeloid depression. On the other hand, bone marrow with its production of myeloid cells may be a source for refilling the tumor site with new cancerous progenitor cells after the termination of cytostatic treatment. A further aspect is the adverse effects of most antitumoral therapeutics on bone remodeling, which again could be explained by the related nature of carcinoma cells and bone cells.
Putative origin of cancer cells by fusion and their migrating ways. Hematopoietic stem cells from adult bone marrow develop into monocytes, macrophages, Langerhans cells and osteoclasts. The progenitor cells of Langerhans cells may fuse with epithelial progenitor cells in hypoxic area in epithelial tissue and may become cancerous. Migrating cancer cells show myeloid traits, target lymph nodes like dendritic cells and metastasize at the bone site like preosteoclasts. The development is fostered by mesenchymal cells, the tumor associated fibroblast and other myeloid cells.
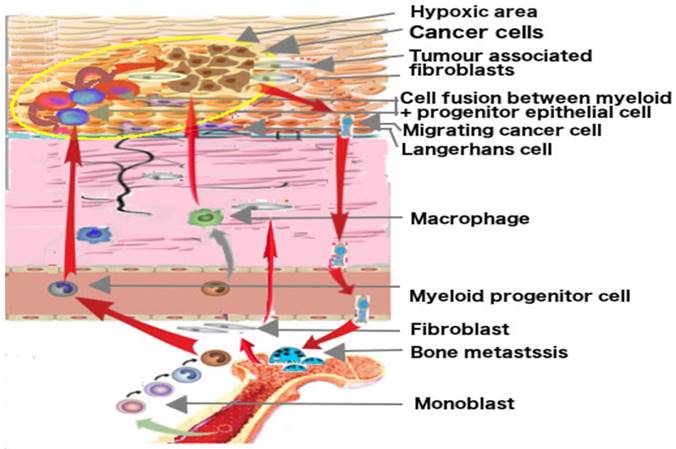
Discussion
EMT is a widely accepted concept for explaining the transition of an epithelial sedentary cancer cell to a migratory one with mesenchymal markers, although some researchers questioned the EMT hypothesis from the outset [101]. Properties of cancer cells not in compliance with these hypotheses are often dismissed as aberrant differentiation or inappropriate gene expression, as mimicking the macrophages phenotype, as hijacking or harnessing the programs of other cell types, as hoaxing osteomimetic properties or behaving like another cell type, for instance dendritic cells, or as an epiphenomenon. In our investigative review, we substantiate the hypothesis that migrating and already sedentary primary cancer cells partly show properties of myeloid cells, and consequently aberrant properties may in fact be their inherent nature. The EMyeT-hypothesis does not require us to postulate a specific immune-escape mechanism or immune evasion of cancer cells. The normal communication of inflammatory cells in a chronic inflammatory process may explain the immune tolerance toward cancer cells. [102, 103] In spite of intensive research by various research groups, no specific immune-escape mechanism of cancer cells has been detected that could be therapeutically exploited, and the checkpoint inhibitors demonstrate that the whole immune homeostasis has to be broken down to affect cancer cells as well as many other functional cells. Besides this, the EMT hypothesis cannot explain the features of a carcinoma as a non-healing wound, the lymphatic access of tumor cells, metastatic spread to bone sites and malignancy of the process. [104] The EMyeT hypothesis can easily explain these characteristics of carcinomas.
A decisive aspect is often overlooked in research on the EMT hypothesis. The mesenchymal markers of cancer cells identified in most studies are not specific to mesenchymal cells but, as we have described in this paper, are expressed by migratory myeloid cells as well. Thus, problematically, there are no specific markers for a mesenchymal cell derived from a cancer cell by which their EMT could unequivocally be determined in the laboratory except by further inducing them to differentiate into adipocytes or osteoblasts. Even the morphology of myeloid cells may adopt that of mesenchymal cells, i.e. a spindle-type morphology. In-vitro peripheral blood monocytes may be reprogrammed to a state of plasticity, where they differentiate into mesenchymal cells when exposed to mesenchymal induction conditions [5, 14]. In view of the hypothesis that cancer cells are myeloid hybrids, the question arises whether the epithelial-myeloid transition of cancer cells may be laboratory artefact.
Based on these results, the epithelial myeloid transition hypothesis assumes that cancer originates from epithelial cells which may have fused with or adopted traits of committed myeloid cells, and thus interprets the origin of cancer cells differently from the EMT-hypothesis [105, 106]. One exception to the EMyeT hypothesis may exist. These are basal cell carcinomas. Their tumor cells may not be initiated in connection with fusion and adoption of myeloid traits. They arise in the basal layer of the skin which is devoid of Langerhans cells resulting in no, or only marginal, contact of cancer cells with myeloid cells. This may explain why no fusion or adoption of myeloid traits is possible, and consequently why this cancer very seldom metastasizes.
But why does the organism react to the carcinoma as if it were an injury, a tissue damage, and a wound? We hypothesize that this reaction is due to the myeloid nature and function of cancer cells. The organism may perceive the cancer cells as proliferating myeloid cells and accordingly interpret their increase and cytokine expressions as a wound process provoked by tissue damage. This reaction provokes a certain alarm state and stimulates further myeloid proliferation, and consequently the progression of the carcinoma. It ensures continuous and increased nutrient supply to the tumor at the cost of other requirements of the organism, immune tolerance, further myeloid attraction, neoangiogenesis; and it increases glycolytic imbalance. As the glycolytic imbalance of carcinoma cells is an intrinsic factor of their malignancy, the inflammatory proliferation phase cannot proceed to the inflammatory resolution and remodeling i.e. the reepithelization phase. In the inflammatory resolution and re-epithelialization phase, the myeloid cells switch their energy supply from glycolysis to oxidative phosphorylation. As this does not occur in cancer cells, final healing is prevented and the carcinoma becomes a non-healing wound.
The question may arise whether the EMT and the EMyeT hypothesis are contradictory, complementary, parallel or overlapping concepts, or whether the EMT hypothesis may be a laboratory artefact and a fallacy? A Canadian and an US-American research group proposed a parallel concept of EMT and EMyeT respectively an Epithelial to Leucocytic Transition in prostate- and colorectal cancer patients [36, 104]. A Swedish research group from Karolinska Institute, Stockholm, hypothesizes that tumor cells having performed the EMT acquire myeloid traits in addition [34]. The majority of studies indicating myeloid traits of cancer cells do not discuss these findings in respect of the EMT hypotheses. Although there are numerous publications on myeloid properties of cancer cells indicating incongruencies of the EMT hypotheses, the number of publications specifically challenging the EMT hypotheses is very scarce.
The EMyeT hypothesis must be seen in connection with three other decisive factors of cancer. Besides glycolysis and the coupling of cancer cells with mesenchymal cells, the third factor is the adrenergic hyperinnervation of the carcinoma and the overall increase of adrenergic activity in the patient [55]. The adrenergic hyperinnervation of the carcinoma may be due to increasing cancer cell numbers with myeloid markers, due to their putative perception and interpretation by the organism as an inflammatory wound process. The adrenergic hyperinnervation and activity causes angiogenesis, a better energy supply and immune tolerance resulting in increased proliferation of the carcinoma [55]. Elevated norepinephrine has been associated with a number of adverse outcomes of breast cancer patients [56]. Thus, the EMyeT hypothesis could explain why carcinomas are non-healing wounds, and why this correlates with the progress of malignancy. On the other hand, EMT is the process central in wound healing, and it is therefore difficult to interpret it as a process preventing wound-healing in the case of cancer. In contrast to physiological wound-healing process, the carcinoma remains at the proliferation phase and does not progress to the remodeling or re-epithelialization phase due to the persistent glycolytic imbalance of cancer cells.
The hypothesis that the organism perceives a carcinoma as a primary inflammatory process due to the myeloid nature of cancer cells, could explain why contrary therapeutic measures may attenuate the disease. An anti-inflammatory treatment may reduce the inflammatory-sustaining reactions of the organism, resulting in less progress of the malignant disease, while immune stimulatory treatment may induce loss of immune homeostasis, disturbing cancer progression.
The decisive question will be whether the EMyeT hypothesis and the interpretation of a carcinoma as a primary inflammatory process has any clinical and therapeutic importance. Research on prostate and breast cancer suggests that myeloid marker expressions on cancer cells correlate with aggressive tumor progression. [107] Furthermore, the myeloid differentiation of prostate cancer cells correlated with an increase in PD-L1 and in circulating PL1/1+ dendritic cells in patients [36].
The myeloid nature of cancer cells contributes to another decisive aspect of cancer. When progressing in disease, cancer cells partly develop to stages comparable to preosteoclasts and osteoclasts. This facilitates their properties of distant migration and their targeting of the bone. Thus, cancer extends beyond the primary tumor site, perceived as a primary inflammatory process, and then may disturb all those processes which are involved in bone remodeling. Solid cancer shows two aspects: a wrongly perceived inflammatory site and a false bone remodeling process, with injurious corresponding dual reactions from the organism.
Conclusions
Conclusion from the EMyeT hypothesis can be drawn on various levels. First, studies on the phenotypic change of cancer cells when progressing in malignancy assumed to be EMT should be done in in-vivo studies and in carcinoma specimens to avoid the artefact of in-vitro studies. To exclude a fallacy from their results specific myeloid markers of cancer cells must be checked prior to the interpretation of the findings. Second, based on the presented EMyeT-hypothesis new interpretations of the mode of action of clinically applied antitumoral therapies will arise and may provoke completely new questions for further research. Third, a new understanding of a disease always entails new concepts of treatment. This may be the case in view of the EMyeT hypotheses as well.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Schramm HM. Should EMT of Cancer Cells Be Understood as Epithelial-Myeloid Transition? J Cancer. 2014;5:125-132
2. Weber CE, Li NY, Wai PY, Kuo PC. Epithelial-mesenchymal transition, TGF-beta, and osteopontin in wound healing and tissue remodeling after injury. J Burn Care Res. 2012;33:311-318
3. Pilz GA, Braun J, Ulrich C. et al. Human mesenchymal stromal cells express CD14 cross-reactive epitopes. Cytometry A. 2011;79:635-645
4. Lv FJ, Tuan RS, Cheung KM, Leung VY. Concise review: the surface markers and identity of human mesenchymal stem cells. Stem Cells. 2014;32:1408-1419
5. Seta N, Kuwana M. Derivation of multipotent progenitors from human circulating CD14+ monocytes. Exp Hematol. 2010;38:557-563
6. Kuwana M, Okazaki Y, Kodama H. et al. Human circulating CD14+ monocytes as a source of progenitors that exhibit mesenchymal cell differentiation. J Leukoc Biol. 2003;74:833-845
7. Kong P, Christia P, Saxena A. et al. Lack of specificity of fibroblast-specific protein 1 in cardiac remodeling and fibrosis. Am J Physiol Heart Circ Physiol. 2013;305:H1363-1372
8. Houghton J, Stoicov C, Nomura S. et al. Gastric cancer originating from bone marrow-derived cells. Science (New York, N.Y.). 2004;306:1568-1571
9. Avital I, Moreira AL, Klimstra DS. et al. Donor-derived human bone marrow cells contribute to solid organ cancers developing after bone marrow transplantation. Stem Cells. 2007;25:2903-2909
10. Cogle CR, Theise ND, Fu D. et al. Bone marrow contributes to epithelial cancers in mice and humans as developmental mimicry. Stem Cells. 2007;25:1881-7
11. Liu C, Chen Z, Chen Z. et al. Multiple tumor types may originate from bone marrow-derived cells. Neoplasia (New York, N.Y.). 2006;8:716-724
12. Zeng C, Zhang Y, Park SC. et al. CD34(+) Liver Cancer Stem Cells Were Formed by Fusion of Hepatobiliary Stem/Progenitor Cells with Hematopoietic Precursor-Derived Myeloid Intermediates. Stem cells and development. 2015;24:2467-2478
13. Modi BG, Neustadter J, Binda E. et al. Langerhans cells facilitate epithelial DNA damage and squamous cell carcinoma. Science (New York, N.Y.). 2012;335:104-108
14. Ungefroren H, Hyder A, Schulze M. et al. Peripheral Blood Monocytes as Adult Stem Cells: Molecular Characterization and Improvements in Culture Conditions to Enhance Stem Cell Features and Proliferative Potential. Stem cells international. 2016;2016:7132751
15. Kim JW, Gao P, Dang CV. Effects of hypoxia on tumor metabolism. Cancer metastasis reviews. 2007;26:291-298
16. Lopez-Lazaro M. The warburg effect: why and how do cancer cells activate glycolysis in the presence of oxygen? Anti-cancer agents in medicinal chemistry. 2008;8:305-312
17. Gillies RJ, Robey I, Gatenby RA. Causes and consequences of increased glucose metabolism of cancers. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 2008;49(Suppl 2):24s-42s
18. Adams LM, Warburton MJ, Hayman AR. Human breast cancer cell lines and tissues express tartrate-resistant acid phosphatase (TRAP). Cell Biol Int. 2007;31:191-195
19. Littlewood-Evans AJ, Bilbe G, Bowler WB. et al. The osteoclast-associated protease cathepsin K is expressed in human breast carcinoma. Cancer Res. 1997;57:5386-5390
20. Sato Y, Goto Y, Narita N, Hoon DS. Cancer Cells Expressing Toll-like Receptors and the Tumor Microenvironment. Cancer Microenviron. 2009;2(Suppl 1):205-214
21. Wang XQ, Tao BB, Li B. et al. Overexpression of TREM2 enhances glioma cell proliferation and invasion: a therapeutic target in human glioma. Oncotarget. 2016;7:2354-2366
22. Shabo I, Svanvik J. Expression of macrophage antigens by tumor cells. Advances in experimental medicine and biology. 2011;714:141-150
23. Warnecke-Eberz U, Metzger R, Holscher AH. et al. Diagnostic marker signature for esophageal cancer from transcriptome analysis. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2016;37:6349-6358
24. Zeng XX, Chu TJ, Yuan JY. et al. Transmembrane 7 superfamily member 4 regulates cell cycle progression in breast cancer cells. European review for medical and pharmacological sciences. 2015;19:4353-4361
25. Kavitha CV, Deep G, Gangar SC. et al. Silibinin inhibits prostate cancer cells- and RANKL-induced osteoclastogenesis by targeting NFATc1, NF-kappaB, and AP-1 activation in RAW264.7 cells. Molecular carcinogenesis. 2014;53:169-180
26. Schramm HM. The cross-over of anticancer agents with osteoclast acitivty. Current Cancer therapy Reviews. 2011:290-302
27. Konradi S, Yasmin N, Haslwanter D. et al. Langerhans cell maturation is accompanied by induction of N-cadherin and the transcriptional regulators of epithelial-mesenchymal transition ZEB1/2. European journal of immunology. 2014;44:553-560
28. Katoh M, Katoh M. Integrative genomic analyses of ZEB2: Transcriptional regulation of ZEB2 based on SMADs, ETS1, HIF1alpha, POU/OCT, and NF-kappaB. International journal of oncology. 2009;34:1737-1742
29. Cortes M, Sanchez-Moral L, de Barrios O. et al. Tumor-associated macrophages (TAMs) depend on ZEB1 for their cancer-promoting roles. The EMBO journal. 2017;36:3336-3355
30. Merindol N, Riquet A, Szablewski V. et al. The emerging role of Twist proteins in hematopoietic cells and hematological malignancies. Blood cancer journal. 2014;4:e206
31. Hotz B, Visekruna A, Buhr HJ, Hotz HG. Beyond epithelial to mesenchymal transition: a novel role for the transcription factor Snail in inflammation and wound healing. Journal of gastrointestinal surgery: official journal of the Society for Surgery of the Alimentary Tract. 2010;14:388-397
32. Hofmann G, Bernabei PA, Crociani O. et al. HERG K+ channels activation during beta(1) integrin-mediated adhesion to fibronectin induces an up-regulation of alpha(v)beta(3) integrin in the preosteoclastic leukemia cell line FLG 29.1. The Journal of biological chemistry. 2001;276:4923-4931
33. Digiacomo G, Tusa I, Bacci M. et al. Fibronectin induces macrophage migration through a SFK-FAK/CSF-1R pathway. Cell adhesion & migration. 2017;11:327-337
34. Johansson J, Tabor V, Wikell A. et al. TGF-beta1-Induced Epithelial-Mesenchymal Transition Promotes Monocyte/Macrophage Properties in Breast Cancer Cells. Frontiers in oncology. 2015;5:3
35. Pawlowski KM, Maciejewski H, Dolka I. et al. Gene expression profiles in canine mammary carcinomas of various grades of malignancy. BMC veterinary research. 2013;9:78
36. Bishop JL, Davies A, Ketola K, Zoubeidi A. Regulation of tumor cell plasticity by the androgen receptor in prostate cancer. Endocrine-related cancer. 2015;22:R165-182
37. Yakar S, Leroith D, Brodt P. The role of the growth hormone/insulin-like growth factor axis in tumor growth and progression: Lessons from animal models. Cytokine & growth factor reviews. 2005;16:407-420
38. Laron Z. The GH-IGF1 axis and longevity. The paradigm of IGF1 deficiency. Hormones (Athens, Greece). 2008;7:24-27
39. Shevah O, Laron Z. Patients with congenital deficiency of IGF-I seem protected from the development of malignancies: a preliminary report. Growth hormone & IGF research: official journal of the Growth Hormone Research Society and the International IGF Research Society. 2007;17:54-57
40. Irie HY, Shrestha Y, Selfors LM. et al. PTK6 regulates IGF-1-induced anchorage-independent survival. PloS one. 2010;5:e11729
41. Quinn KA, Treston AM, Unsworth EJ. et al. Insulin-like growth factor expression in human cancer cell lines. The Journal of biological chemistry. 1996;271:11477-11483
42. Mussar K, Pardike S, Hohl TM. et al. CCR2+ myeloid cell niche required for pancreatic beta cell growth. JCI insight. 2017;2:I
43. Suzuki S, Morimoto S, Fujishiro M. et al. Inhibition of the insulin-like growth factor system is a potential therapy for rheumatoid arthritis. Autoimmunity. 2015;48:251-258
44. McCubrey JA, Steelman LS, Mayo MW. et al. Growth-promoting effects of insulin-like growth factor-1 (IGF-1) on hematopoietic cells: overexpression of introduced IGF-1 receptor abrogates interleukin-3 dependency of murine factor-dependent cells by a ligand-dependent mechanism. Blood. 1991;78:921-929
45. Lawicki S, Bedkowska GE, Wojtukiewicz M, Szmitkowski M, Hematopoietic cytokines as tumor markers in breast malignancies. A multivariate analysis with ROC curve in breast cancer patients. Advances in medical sciences. 2013;58:207-215
46. Cecchini MG, Hofstetter W, Halasy J. et al. Role of CSF-1 in bone and bone marrow development. Molecular reproduction and development. 1997;46:75-83 discussion 83-74
47. Pongratz G, Straub RH. The sympathetic nervous response in inflammation. Arthritis research & therapy. 2014;16:504
48. Cole SW, Sood AK. Molecular pathways: beta-adrenergic signaling in cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:1201-1206
49. Coelho M, Soares-Silva C, Brandao D. et al. Beta-Adrenergic modulation of cancer cell proliferation: available evidence and clinical perspectives. Journal of cancer research and clinical oncology. 2017;143:275-291
50. Nolten WE, Goldstein D, Lindstrom M. et al. Effects of cytokines on the pituitary-adrenal axis in cancer patients. Journal of interferon research. 1993;13:349-357
51. Jia D, Li L, Andrew S. et al. An autocrine inflammatory forward-feedback loop after chemotherapy withdrawal facilitates the repopulation of drug-resistant breast cancer cells. Cell death & disease. 2017;8:e2932
52. Yoshimura A. Signal transduction of inflammatory cytokines and tumor development. Cancer science. 2006;97:439-447
53. Mizutani K, Sud S, McGregor NA. et al. The chemokine CCL2 increases prostate tumor growth and bone metastasis through macrophage and osteoclast recruitment. Neoplasia (New York, N.Y.). 2009;11:1235-1242
54. Kim MS, Day CJ, Morrison NA. MCP-1 is induced by receptor activator of nuclear factor-{kappa}B ligand, promotes human osteoclast fusion, and rescues granulocyte macrophage colony-stimulating factor suppression of osteoclast formation. The Journal of biological chemistry. 2005;280:16163-16169
55. Armaiz-Pena GN, Cole SW, Lutgendorf SK, Sood AK. Neuroendocrine influences on cancer progression. Brain, behavior, and immunity. 2013;30(Suppl):S19-25
56. Qin JF, Jin FJ, Li N. et al. Adrenergic receptor beta2 activation by stress promotes breast cancer progression through macrophages M2 polarization in tumor microenvironment. BMB reports. 2015;48:295-300
57. Fagundes CP, Murray DM, Hwang BS. et al. Sympathetic and parasympathetic activity in cancer-related fatigue: more evidence for a physiological substrate in cancer survivors. Psychoneuroendocrinology. 2011;36:1137-1147
58. Gidron Y, De Couck M, De Greve J. If you have an active vagus nerve, cancer stage may no longer be important. Journal of biological regulators and homeostatic agents. 2014;28:195-201
59. Pan L, Tang J, Liu H, Cheng B. Sympathetic nerves: How do they affect angiogenesis, particularly during wound healing of soft tissues? Clinical hemorheology and microcirculation. 2016;62:181-191
60. Schramm H. Comparison of the impact of pro- and antiinflammatory immune processes at the two immune-deviated sites, eye and solid tumor and possible consequences for the antitumoral therapy with fever inducers. Forschende Komplementarmedizin und klassische Naturheilkunde = Research in complementary and natural classical medicine. 2005;12:37-46
61. Bellahcene A, Castronovo V, Ogbureke KU. et al. Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): multifunctional proteins in cancer. Nature reviews. Cancer. 2008;8:212-226
62. Lee JL, Wang MJ, Sudhir PR. et al. Osteopontin promotes integrin activation through outside-in and inside-out mechanisms: OPN-CD44V interaction enhances survival in gastrointestinal cancer cells. Cancer Res. 2007;67:2089-2097
63. Samanna V, Wei H, Ego-Osuala D, Chellaiah MA. Alpha-V-dependent outside-in signaling is required for the regulation of CD44 surface expression, MMP-2 secretion, and cell migration by osteopontin in human melanoma cells. Experimental cell research. 2006;312:2214-2230
64. Schramm H. Extraterritorial osteoclast traits of primary cancer cells. Journal of Solid Tumors. 2011:65-79
65. Idris AI, Krishnan M, Simic P. et al. Small molecule inhibitors of IkappaB kinase signaling inhibit osteoclast formation in vitro and prevent ovariectomy-induced bone loss in vivo. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2010;24:4545-4555
66. Duan L, de Vos P, Fan M, Ren Y. IkappaBalpha regulates Hes1 in osteoclast differentiation and resorption. Frontiers in bioscience (Elite edition). 2010;2:1065-1072
67. Santini D, Perrone G, Roato I. et al. Expression pattern of receptor activator of NFkappaB (RANK) in a series of primary solid tumors and related bone metastases. Journal of cellular physiology. 2011;226:780-784
68. Koeneman KS, Yeung F, Chung LW. Osteomimetic properties of prostate cancer cells: a hypothesis supporting the predilection of prostate cancer metastasis and growth in the bone environment. The Prostate. 1999;39:246-261
69. Detry C, Waltregny D, Quatresooz P. et al. Detection of bone sialoprotein in human (pre)neoplastic lesions of the uterine cervix. Calcified tissue international. 2003;73:9-14
70. Chaplet M, De Leval L, Waltregny D. et al. Dentin matrix protein 1 is expressed in human lung cancer. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2003;18:1506-1512
71. Rucci N, Teti A. Osteomimicry: how tumor cells try to deceive the bone. Frontiers in bioscience (Scholar edition). 2010;2:907-915
72. Fiaschi-Taesch NM, Stewart AF. Minireview: parathyroid hormone-related protein as an intracrine factor-trafficking mechanisms and functional consequences. Endocrinology. 2003;144:407-411
73. Pang MF, Georgoudaki AM, Lambut L. et al. TGF-beta1-induced EMT promotes targeted migration of breast cancer cells through the lymphatic system by the activation of CCR7/CCL21-mediated chemotaxis. Oncogene. 2016;35:748-760
74. Saltel F, Chabadel A, Bonnelye E, Jurdic P. Actin cytoskeletal organisation in osteoclasts: a model to decipher transmigration and matrix degradation. European journal of cell biology. 2008;87:459-468
75. Yu X, Huang Y, Collin-Osdoby P, Osdoby P. Stromal cell-derived factor-1 (SDF-1) recruits osteoclast precursors by inducing chemotaxis, matrix metalloproteinase-9 (MMP-9) activity, and collagen transmigration. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2003;18:1404-1418
76. Egeblad M, Littlepage LE, Werb Z. The fibroblastic coconspirator in cancer progression. Cold Spring Harbor symposia on quantitative biology. 2005;70:383-388
77. Hwang HD, Lee JT, Koh JT. et al. Sequential Treatment with SDF-1 and BMP-2 Potentiates Bone Formation in Calvarial Defects. Tissue engineering. Part A. 2015;21:2125-2135
78. Powe DG, Voss MJ, Zanker KS. et al. Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget. 2010;1:628-638
79. Togari A, Arai M, Kondo A. The role of the sympathetic nervous system in controlling bone metabolism. Expert opinion on therapeutic targets. 2005;9:931-940
80. Varner JA. Stem cells and neurogenesis in tumors. Progress in experimental tumor research. 2007;39:122-129
81. Togari A, Mogi M, Arai M. et al. Expression of mRNA for axon guidance molecules, such as semaphorin-III, netrins and neurotrophins, in human osteoblasts and osteoclasts. Brain research. 2000;878:204-209
82. Fujikawa Y, Sabokbar A, Neale S, Athanasou NA. Human osteoclast formation and bone resorption by monocytes and synovial macrophages in rheumatoid arthritis. Annals of the rheumatic diseases. 1996;55:816-822
83. Yamamoto Y, Yamamoto Y, Udagawa N. et al. Effects of calcitonin on the function of human osteoclast-like cells formed from CD14-positive monocytes. Cellular and molecular biology (Noisy-le-Grand, France). 2006;52:25-31
84. Shen Z, Crotti TN, McHugh KP. et al. The role played by cell-substrate interactions in the pathogenesis of osteoclast-mediated peri-implant osteolysis. Arthritis research & therapy. 2006;8:R70
85. Han B, Nakamura M, Zhou G. et al. Calcitonin inhibits invasion of breast cancer cells: involvement of urokinase-type plasminogen activator (uPA) and uPA receptor. International journal of oncology. 2006;28:807-814
86. Shah GV, Thomas S, Muralidharan A. et al. Calcitonin promotes in vivo metastasis of prostate cancer cells by altering cell signaling, adhesion, and inflammatory pathways. Endocrine-related cancer. 2008;15:953-964
87. Massfelder T, Lang H, Schordan E. et al. Parathyroid hormone-related protein is an essential growth factor for human clear cell renal carcinoma and a target for the von Hippel-Lindau tumor suppressor gene. Cancer Res. 2004;64:180-188
88. Park SI, Lee C, Sadler WD. et al. Parathyroid hormone-related protein drives a CD11b+Gr1+ cell-mediated positive feedback loop to support prostate cancer growth. Cancer Res. 2013;73:6574-6583
89. Yamamura-Idei Y, Kitazawa S, Kitazawa R. et al. Parathyroid hormone-related protein in gastric cancers with heterotopic ossification. Cancer. 1993;72:1849-1852
90. Kayed H, Bekasi S, Keleg S. et al. BGLAP is expressed in pancreatic cancer cells and increases their growth and invasion. Molecular cancer. 2007;6:83
91. Salem AM, Zohny SF, Abd El-Wahab MM, Hamdy R. Predictive value of osteocalcin and beta-CrossLaps in metastatic breast cancer. Clinical biochemistry. 2007;40:1201-1208
92. Mishra P, Banerjee D, Ben-Baruch A. Chemokines at the crossroads of tumor-fibroblast interactions that promote malignancy. J Leukoc Biol. 2011;89:31-39
93. Vallet S, Bashari MH, Fan FJ. et al. Pre-Osteoblasts Stimulate Migration of Breast Cancer Cells via the HGF/MET Pathway. PloS one. 2016;11:e0150507
94. Sims NA, Gooi JH. Bone remodeling: Multiple cellular interactions required for coupling of bone formation and resorption. Seminars in cell & developmental biology. 2008;19:444-451
95. Fedirko V, Bostick RM, Goodman M. et al. Blood 25-hydroxyvitamin D3 concentrations and incident sporadic colorectal adenoma risk: a pooled case-control study. American journal of epidemiology. 2010;172:489-500
96. Sakai S, Takaishi H, Matsuzaki K. et al. 1-Alpha, 25-dihydroxy vitamin D3 inhibits osteoclastogenesis through IFN-beta-dependent NFATc1 suppression. Journal of bone and mineral metabolism. 2009;27:643-652
97. Axelsson H, Lonnroth C, Andersson M, Lundholm K. Mechanisms behind COX-1 and COX-2 inhibition of tumor growth in vivo. International journal of oncology. 2010;37:1143-1152
98. Neville-Webbe HL, Gnant M, Coleman RE. Potential anticancer properties of bisphosphonates. Seminars in oncology. 2010;37(Suppl 1):S53-65
99. Fresco P, Borges F, Diniz C, Marques MP. New insights on the anticancer properties of dietary polyphenols. Medicinal research reviews. 2006;26:747-766
100. Wattel A, Kamel S, Prouillet C. et al. Flavonoid quercetin decreases osteoclastic differentiation induced by RANKL via a mechanism involving NF kappa B and AP-1. Journal of cellular biochemistry. 2004;92:285-295
101. Tarin D, Thompson EW, Newgreen DF. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res. 2005;65:5996-6000 discussion 6000-5991
102. Lin D, Wang X, Choi SYC. et al. Immune phenotypes of prostate cancer cells: Evidence of epithelial immune cell-like transition? Asian journal of urology. 2016;3:195-202
103. Choi SY, Gout PW, Collins CC, Wang Y. Epithelial immune cell-like transition (EIT): a proposed transdifferentiation process underlying immune-suppressive activity of epithelial cancers. Differentiation; research in biological diversity. 2012;83:293-298
104. Luddy KA, Robertson-Tessi M, Tafreshi NK. et al. The role of toll-like receptors in colorectal cancer progression: evidence for epithelial to leucocytic transition. Frontiers in immunology. 2014;5:429
105. Pawelek JM. Tumour-cell fusion as a source of myeloid traits in cancer. The Lancet. Oncology. 2005;6:988-993
106. Chakraborty AK, Pawelek J, Ikeda Y. et al. Fusion hybrids with macrophage and melanoma cells up-regulate N-acetylglucosaminyltransferase V, beta1-6 branching, and metastasis. Cell growth & differentiation: the molecular biology journal of the American Association for Cancer Research. 2001;12:623-630
107. Garvin S, Oda H, Arnesson LG. et al. Tumor cell expression of CD163 is associated to postoperative radiotherapy and poor prognosis in patients with breast cancer treated with breast-conserving surgery. Journal of cancer research and clinical oncology. 2018;144:1253-1263
Author contact
![]() Corresponding author: e-mail: h.schrammch
Corresponding author: e-mail: h.schrammch