Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(18):4264-4269. doi:10.7150/jca.32646 This issue Cite
Research Paper
SphK1 functions downstream of IGF-1 to modulate IGF-1-induced EMT, migration and paclitaxel resistance of A549 cells: A preliminary in vitro study
Xingping Wu1,2,3,*, Qibiao Wu2,*, ![]() , Xiqiao Zhou4,*, Jianan Huang1
, Xiqiao Zhou4,*, Jianan Huang1 ![]()
1. Department of Respiratory Medicine, the First Affiliated Hospital of Soochow University, Jiangsu, P.R. China
2. State Key Laboratory of Quality Research in Chinese Medicines, Macau University of Science and Technology, Macau, P.R. China
3. Department of Respirology, the First People's Hospital of Lianyungang, Jiangsu, P.R. China
4. Department of Gastroenterology, the First Affiliated Hospital of Nanjing Medical University, Jiangsu, P.R. China
*Equal contributors and co-first authors.
Received 2018-12-28; Accepted 2019-5-25; Published 2019-7-10
Abstract
Insulin-like growth factor-1 (IGF-1) -induced epithelial-mesenchymal transition (EMT) plays a key role in the metastasis and drug resistance of non-small cell lung cancer (NSCLC). Sphingosine kinase-1 (SphK1) is also involved in EMT of NSCLC. However, the interaction between SphK1 and IGF-1 in the EMT of NSCLC is largely unknown. To clarify this issue, we examined the involvement of SphK1 in IGF-1-induced EMT using human lung cancer cell line A549, and its paclitaxel-resistant subline. Cell viability was evaluated by cell counting kit-8 assay; Migratory ability was examined using scratch wound healing test; Protein expression levels of SphK1, vimentin, fibronectin, N-cadherin and E-cadherin were detected by western blot analysis, respectively. The results showed that, IGF-1 treatment of A549 cells stimulated the expression of SphK1, the activation of ERK and AKT, the cell migration, and the expression of EMT hallmark proteins, while inhibition of SphK1 by its specific inhibitor SKI-II suppressed all the above changes and increased the sensitivity of A549 cells to paclitaxel. Our data demonstrate that SphK1 acts as a downstream effector of IGF-1 and plays a critical role in IGF-1-induced EMT, cell migration and paclitaxel resistance of A549 cells, suggesting that SphK1 might be a potential therapeutic target for NSCLC.
Keywords: SphK1, IGF-1, EMT, NSCLC, signaling pathway, therapeutic target.
1. Introduction
Worldwide, lung cancer remains the leading cause of cancer-related mortality, and approximately 85% of lung cancers are Non-small cell lung cancer (NSCLC) [1,2]. The major causes of mortality in NSCLC patients are metastasis and drug resistance, which are closely associated with Epithelial-mesenchymal transition (EMT) [3-6]. EMT is a process during which the epithelial cells lose their phenotype and acquire the characteristics of mesenchymal cells [5]. Accumulating evidence shows that insulin-like growth factor (IGF) -1 signaling pathway is involved in EMT and IGF-1-induced EMT plays an important role in the development and progression of many types of solid tumors, including NSCLC [7-11].
Recently, sphingosine kinase (SphK) -1, an oncogenic kinase, has attracted increasing attention because of its important functions in many processes of cancer cells [12-14]. A few studies showed that SphK1 might mediate the EMT of A549 cells, but the mechanisms remain ambiguous [15-16]. Both IGF-1 and SphK1 have been shown to be involved in the EMT process of NSCLC. And a previous study provided the first illustration of SphK1 role as an crucial regulator of neuroblastoma cells' death by mediating IGF-1 [17]. These findings subsequently lead to a hypothesis that, there is probably a similar functional interaction between SphK1 and IGF-1 in the EMT process of NSCLC. However, little is known about this issue. In this study, we explored the role of SphK1 in IGF-1-induced EMT, migration and paclitaxel resistance of the human lung cancer cell line A549.
Material and Methods
2.1 Cells and antibodies
A549 cell line was purchased from the American Type Culture Collection (Manassas, VA) and cultured in F-12K medium (Gibco) supplemented with 10% fetal bovine serum (FBS, Gibico), 100U/ml penicillin, and 100μg/ml streptomycin. Paclitaxel resistant-A549 was generated by culturing A549 cells in complete F-12K medium containing 2 μM paclitaxel. When indicated, cells were treated with IGF-1 (Peprotech) or SphK1 inhibitor SKI-II (Sigma Cat# S5696).
Antibodies used for immunoblot included anti-SphK1 (Cell Signaling Technology Cat# 3297), anti-phospho SphK1(Ser225) (ECM Biosciences Cat# SP1641), anti-Fibronection (Abcam Cat# ab32419), anti-N-Cadherin (Abcam Cat# ab76011), anti-E-Cadherin (Abcam Cat# ab40772), anti-Vimentin (Abcam Cat# 92547), anti-p-ERK1/2 (Cell Signaling Technology Cat# 4377), anti-ERK (Cell Signaling Technology Cat# 4697), anti-p-AKT(S473) (Cell Signaling Technology Cat# 9271 ), anti-AKT (Abcam Cat# AB8805), anti-GAPDH (Beyotime Cat# AG019-1), and horseradish peroxidase-conjugated secondary antibodies (Nanjing SunShine Biotechnology Co., LTD.).
2.2 Cell Proliferation Assay
A549 or paclitaxel-resistant A549 cells were seeded at 3000 cells/well in 96-well plates and cultured overnight. SphK1 specific inhibitor SKI-II, Cisplatin or paclitaxel were then added to the wells at varying concentrations. The cells were further cultured for additional 3 days. After that, the cell viability was evaluated by using CCK8 kit (Dojindo, Cat# CK04) according to the manufacturer's instructions.
2.3 Cell Migration Assay
Cell migration assay was performed using scratch / wound healing test. A549 cells were cultured to 90% confluence in 6-well plates. Scratches were made using a 200μl pipette tip. The cells were then cultured overnight under serum-starved conditions before being supplemented with IGF-1 or SKI-II. Culturing lasted for up to 96 hrs. Images of the cells were taken at selected time points to evaluate the wound-healing condition.
3. Results
3.1 IGF-1 stimulates SphK1 expression and activation in A549
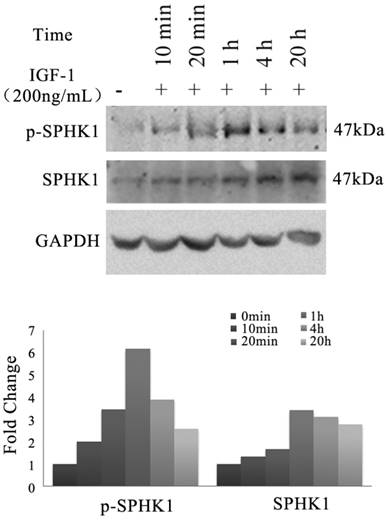
We investigated whether IGF-1 signaling pathway has an impact on the activity of SphK1 in NSCLC cells. A549 cells were treated with 200ng/mL IGF-1, the expression and phosphorylation of SphK1 were examined by Western blot (Figure 1). The results revealed that the expression of SphK1 was increased gradually as early as 20 minutes after IGF-1 stimulation. Meanwhile, phosphorylation of SphK1 at Ser225 was upregulated, which has been shown to increase the catalytic activity and translocation of SphK1 to the plasma membrane, and is crucial for oncogenic signaling [18].
Stimulation of A549 cancer cells with IGF-1 induces overexpression and activation of SphK1. A549 cells were stimulated with 200ng/ml IGF-1 for indicated period of time. Cell lysates were then electrophoresed in 8% PAGE gel and immunoblotted using anti-SphK1 and anti-phospho-SphK1 (Ser225), respectively.
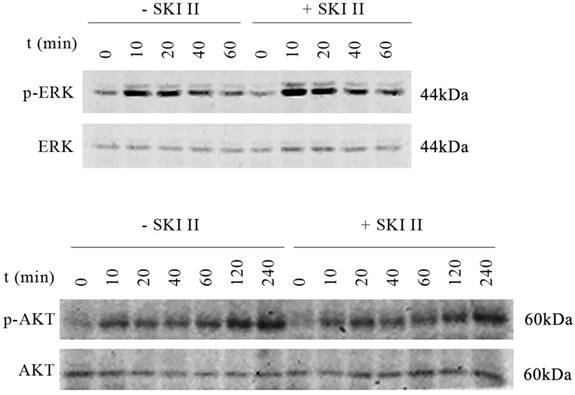
On the other hand, inhibition of SphK1 activity by its specific inhibitor SKI-II [19] led to minor changes in the activation of either ERK or AKT. Upon IGF-1 treatment, extended activation of phosphorylation of ERK and AKT was indicated (Figure 2). The above results suggest that SphK1 functions downstream of IGF-1 in IGF-1 signaling pathway.
IGF-1 activates both AKT and ERK, treatment of cells with SKI-II has no impact on IGF-1 induced ERK or AKT activation. A549 cells were stimulated with 200ng/ml IGF-1 in the presence or absence of 10μM of SKI-II at various time points. IGF-1 activates both AKT and ERK as indicated by phosphorylation of the proteins. On the other hand, treatment of cells with SKI-II has no impact on the activation of either AKT or ERK.
3.2 SphK1 activation induces IGF-1-mediated EMT of A549 cells
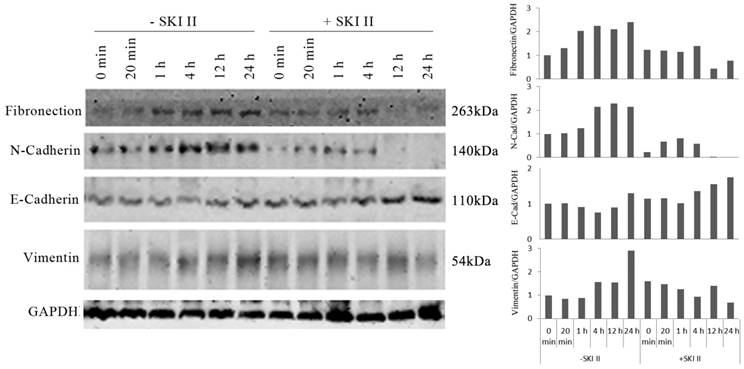
We further investigated whether inhibition of SphK1 by its specific inhibitor SKI-II changes the ability of IGF-1 to induce EMT in A549 cells. IGF-1 treatment of A549 cells resulted in changes of EMT biomarkers, including the elevated expression of vimentin, fibronectin, N-Cadherin, and loss of E-cadherin expression. These changes were inhibited by SKI-II (Figure 3). These results suggest that SphK1 activity is critical for IGF-1-induced EMT in A549 cells.
IGF-1-induced EMT in A549 cells was dependent on SphK1 activity. IGF-1 treatment led to decreased E-cadherin expression and increased expression of N-Cadherin, Vimentin and Fibronectin, which are the markers of EMT. Pretreatment of the cells with 10μM of SKI-II reversed the changes of these EMT markers.
3.3 Inhibition of SphK1 alleviates IGF-1-induced cell migration of A549
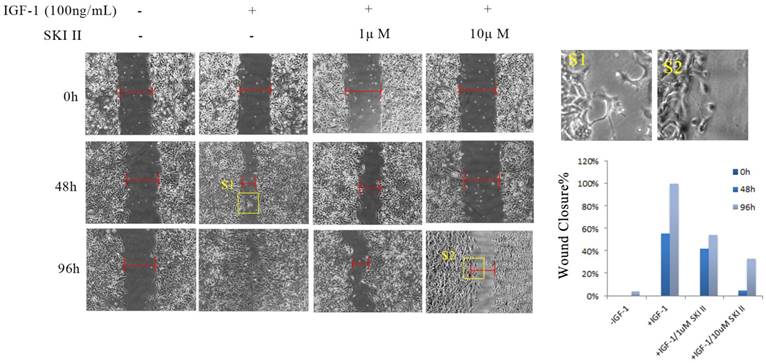
A549 cells were cultured in serum-starved medium, and treatment with IGF-1 at a concentration of 100ng/mL induced apparent cell migration. In a time-course scratch wound healing assay, treating cells with SKI-II at dosages that have no obvious impact on cell proliferation significantly inhibited IGF-1-induced cell migration (Figure 4).
Inhibition of SphK1 suppressed IGF-1-induced cell migration. IGF-1 exhibited potent migratory stimulating activity on A549 cells in a scratch wound healing assay. 96 hours post addition of 100ng/ml IGF-1, the scratch mark almost completed the seal, while little change was seen by 1μM or 10μM SKI-II.
3.4 Inhibition of SphK1 increases the sensitivity of A549 cells to paclitaxel
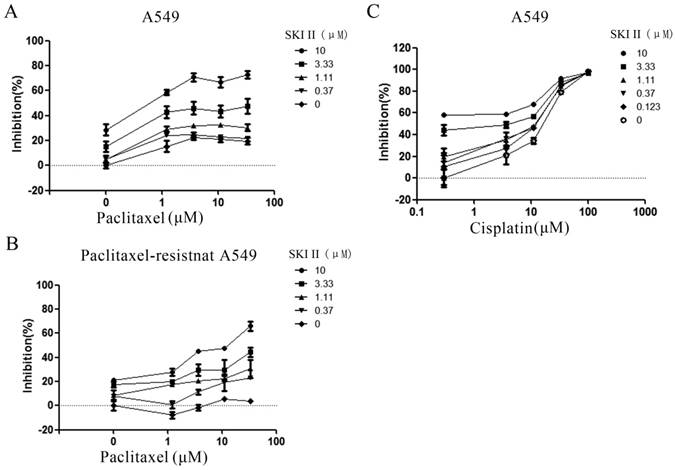
In order to investigate whether SphK1 is involved in the sensitivity of A549 cells to paclitaxel, we evaluated the anti-proliferation effect of paclitaxel in the presence of different concentrations of SKI-II. The results showed that paclitaxel alone only demonstrated very weak cytotoxic effect on A549 cells. Addition of SKI-II enhanced paclitaxel's cytotoxicity in a dose dependent manner (Figure 5A). This synergistic effect was even more evident in paclitaxel-resistant A549 cell line. Treatment of cells with paclitaxel (30μM) showed little cytotoxic effect, while addition of SKI-II significantly restored the sensitivity of the cells to paclitaxel (Figure 5B).
Synergistic inhibition on A549 proliferation by SKI-II and paclitaxel. (A) A549 cells were treated with different combination of SKI-II and paclitaxel as indicated in the figure. Both agents showed dose-dependent inhibitory effects on cell proliferation and synergistic effect was observed in the tested range of concentrations. (B) Paclitaxel alone showed little effect on the proliferation of paclitaxel-resistant A549 cells at as high as 30μM. Addition of SKI-II sensitized the cells to paclitaxel in a dose-dependent manner. (C) No obvious synergistic effect was observed when A549 cell was treated by SKI-II and Cisplatin.
4. Discussion
Most of newly diagnosed patients with NSCLC are already at advanced stage, and the prognosis of them remains extremely poor. Therefore, there is a pressing need for optimal therapies [20,21]. Mounting evidence has demonstrated involvement of IGF-1-induced EMT in the metastasis and drug resistance, thus contributing to the gloomy prognosis of NSCLC patients [3-6]. This scenario promotes the efforts to develop the potential antitumor agents for NSCLC that target IGF-1 pathways.
Our study found that, stimulation of A549 cancer cells with IGF-1 induced overexpression and activation of SphK1, decreased E-cadherin expression and increased expression of N-Cadherin, vimentin and fibronectin, which were dependent on SphK1 activity, while SKI-II, the specific inhibitor of SphK1, could suppress the above effects. These results are consistent with the findings of a previous study, suggesting that SphK1 modulates EMT in colorectal cancer cells [22].
The activation of IGF-I/AKT and IGF-I//ERK pathways contributes to the cell proliferation, migration, invasion, and drug resistance in lung cancer [23-26]. Our study showed that IGF-1 activated both AKT and ERK, but, treatment of cells with SKI-II had no impact on IGF-1 induced ERK or AKT activation.
Drug resistance is one of the biggest obstacles to successful treatment and remains a major concern about cancer chemotherapy [27-29]. Paclitaxel is a widely used chemotherapy agent for NSCLC treatment. We investigated whether SphK1 activity plays a role in paclitaxel resistance. Cell viability assay showed that enhanced SphK1 activity was involved in paclitaxel resistance of lung cancer cells. Addition of SKI-II enhanced paclitaxel's cytotoxicity in a dose dependent manner, and the synergistic effect was even more evident in paclitaxel-resistant A549 cell line. Interestingly, this mechanism appeared to be drug-related as no synergistic effect was observed when SKI-II worked with Cisplatin (Figure 5C). It is necessary to further investigate what leads to such difference.
A previous study reported that the change of SphK1 expression could affect cell migration, invasiveness and the expression of EMT-related hallmark proteins in A549 cells [15]. (Ni, et al., 2015) Our study confirmed the results of this study, and strengthened the evidence base for the role of SphK1 in EMT of A549 cells; Besides, our study uncovered the functional interaction between SphK1 and IGF-1 in the EMT process, and provided new evidence to SphK1 role as a downstream effector of IGF-1.
5. Conclusions
In summary, our study demonstrates that, SphK1 acts as a downstream effector of IGF-1 and plays a critical role in IGF-1-induced EMT, migration and paclitaxel resistance of A549 cells, suggesting that SphK1 might be a promising target for the development of a more effective lung cancer therapy, and for the prevention of tumor metastases and drug resistance. Based on this preliminary in vitro study, more in-depth studies are warranted to decipher IGF-1/SphK1 signaling pathway networks and develop potential target specific anticancer agents for NSCLC.
Abbreviations
AKT, protein kinase B; EMT, epithelial-mesenchymal transition; ERK, extracellular signal-regulated kinase; IGF, insulin-like growth factor (IGF)-1; NSCLC, non-small cell lung cancer; SphKs, sphingosine kinases; SKI-II, SphK inhibitor 2-(p-hydroxyanilino)-4-(p-chlorophenyl) thiazole.
Acknowledgements
This work was supported by grants from The Clinical Medicine Center of Suzhou (No.Szzx201502), Suzhou Key Laboratory for Respiratory Medicine (No.SZS201617), the Societal and Developmental Project of Suzhou (No.SS201630), the National Natural Science Foundation of China (No.81570522), the Science and Technology Development Fund, Macau SAR (130/2017/A3, 0099/2018/A3).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I. et al. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2018;68:394-424 doi: 10.3322/caac.21492. Epub 2018 Sep 12
2. Siegel RL, Miller KD, Jemal A. et al. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7-34 doi: 10.3322/caac.21551
3. Cevenini A, Orrù S, Mancini A. et al. Molecular Signatures of the Insulin-like Growth Factor 1-mediated Epithelial-Mesenchymal Transition in Breast. Lung and Gastric Cancers. Int J Mol Sci. 2018:19 doi: 10.3390/ijms19082411
4. Marcucci F, Stassi G, De Maria R. Epithelial-mesenchymal transition: a new target in anticancer drug discovery. Nat Rev Drug Discov. 2016;15(5):311-25 doi: 10.1038/nrd.2015.13
5. Thiery JP, Acloque H, Huang RY. et al. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871-890 doi:10.1016/j.cell.2009.11.007
6. Zhou J, Wang JJ, Zeng YY. et al. Implication of epithelial-mesenchymal transition in IGF1R-induced resistance to EGFR-TKIs in advanced non-small cell lung cancer. Oncotarget. 2015;6:44332-44345 doi:10.18632/oncotarget.6293
7. Chu PC, Lin PC, Wu HY. et al. Mutant KRAS promotes liver metastasis of colorectal cancer, in part, by upregulating the MEK-Sp1-DNMT1-miR-137-YB-1-IGF-IR signaling pathway. Oncogene. 2018;37:3440-3455 doi: 10.1038/s41388-018-0222-3
8. Lyons A, Coleman M, Riis S. et al. SphK1 promotes tumor cell migration and invasion in colorectal cancer. Tumour Biol. Insulin-like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells. J Biol Chem. 2017;292:16983-16998 doi: 10.1074/jbc.M117.792838
9. Numata K, Oshima T, Sakamaki K. et al. Clinical significance of IGF1R gene expression in patients with stage ii/iii gastric cancer who receive curative surgery and adjuvant chemotherapy with s-1. J. Cancer Res. Clin. Oncol. 2016;142:415-22 doi:10.1007/s00432-015-2039-6
10. Su C, Wang W, Wang C. IGF-1-induced MMP-11 expression promotes the proliferation and invasion of gastric cancer cells through the JAK1/STAT3 signaling pathway. Oncol Lett. 2018;15:7000-7006 doi: 10.3892/ol.2018.8234
11. Werner H, Bruchim I. IGF-1 and BRCA1 signalling pathways in familial cancer. Lancet Oncol. 2012;13(12):e537-44 doi:10.1016/S1470-2045(12)70362-5
12. Liu SQ, Xu CY, Wu WH. et al. Sphingosine kinase 1 promotes the metastasis of colorectal cancer by inducing the epithelial-mesenchymal transition mediated by the FAK/AKT/MMPs axis. Int J Oncol. 2019;54:41-52 doi: 10.3892/ijo.2018.4607
13. Long J, Xie Y, Yin J. et al. SphK1 promotes tumor cell migration and invasion in colorectal cancer. Tumour Biol. 2016;37:6831-6 doi: 10.1007/s13277-015-4542-4
14. Maceyka M, Harikumar KB, Milstien S. et al. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012;22:50-60 doi:10.1016/j.tcb.2011.09.003
15. Ni M, Shi XL, Qu ZG. et al. Epithelial mesenchymal transition of non-small-cell lung cancer cells A549 induced by SphK1. Asian Pac J Trop Med. 2015;8:142-146 doi:10.1016/S1995-7645(14)60305-9
16. Fan Z, Jiang H, Wang Z, Qu J. Atorvastatin partially inhibits the epithelial-mesenchymal transition in A549 cells induced by TGF-β1 by attenuating the upregulation of SphK1. Oncol Rep. 2016;36:1016-22 doi: 10.3892/or.2016.4897
17. Gomez-Brouchet A, Pchejetski D, Brizuela L. et al. Critical role for sphingosine kinase-1 in regulating survival of neuroblastoma cells exposed to amyloid-beta peptide. Mol Pharmacol. 2007;72:341-9 doi:10.1124/mol.106.033738
18. Pitson SM, Xia P, Leclercq TM. et al. Phosphorylation-dependent translocation of sphingosine kinase to the plasma membrane drives its oncogenic signalling. J Exp Med. 2005;201:49-54 doi:10.1084/jem.20040559
19. French K.J, Schrecengost RS, Lee BD. et al. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003;63:5962-9
20. Wang J, Li G, Yu L. et al. Aidi injection plus platinum-based chemotherapy for stage IIIB/IV non-small cell lung cancer: A meta-analysis of 42 RCTs following the PRISMA guidelines. J Ethnopharmacol. 2018;221:137-150 doi: 10.1016/j.jep.2018.04.013
21. Tiwana MS, Lee HN Saini S. et al. Outcomes of patients with unresected stage III and stage IV non-small cell lung cancer: A single institution experience. Lung India : Official Organ of Indian Chest Society. 2013;30:187-192 doi: 10.4103/0970-2113.116250
22. Xu CY, Liu SQ, Qin MB. et al. SphK1 modulates cell migration and EMT-related marker expression by regulating the expression of p-FAK in colorectal cancer cells. Int J Mol Med. 2017;39:1277-1284 doi: 10.3892/ijmm.2017.2921
23. Cortés-Sempere M, de Miguel MP, Pernía O. et al. IGFBP-3 methylation-derived deficiency mediates the resistance to cisplatin through the activation of the IGFIR/Akt pathway in non-small cell lung cancer. Oncogene. 2013;32:1274-83 doi: 10.1038/onc.2012.146
24. Fritz JM, Dwyer-Nield LD, Malkinson AM. Stimulation of neoplastic mouse lung cell proliferation by alveolar macrophage-derived, insulin-like growth factor-1 can be blocked by inhibiting MEK and PI3K activation. Mol Cancer. 2011;10:76. doi: 10.1186/1476-4598-10-76
25. Sun L, Liu X, Fu H. et al. 2-Deoxyglucose Suppresses ERK Phosphorylation in LKB1 and Ras Wild-Type Non-Small Cell Lung Cancer Cells. PLoS One. 2016;11:e0168793. doi: 10.1371/journal.pone.0168793. eCollection 2016
26. Zhou Y, Li S, Li J. et al. Effect of microRNA-135a on Cell Proliferation, Migration, Invasion, Apoptosis and Tumor Angiogenesis Through the IGF-1/PI3K/Akt Signaling Pathway in Non-Small Cell Lung Cancer. Cell Physiol Biochem. 2017;42:1431-1446 doi: 10.1159/000479207
27. Leung EL, Wu QB. Concurrent use of herbal products with prescription drugs is a double-edged sword and evidence-based medicine contributes to reshaping the practice. Pharmacol Res. 2019;141:609-610 doi: 10.1016/j.phrs.2019.01.033
28. Wang XW, Liu ZT, Sui XB. et al. 2018. Elemene injection as adjunctive treatment to platinum-based chemotherapy in patients with stage III/IV non-small cell lung cancer: a meta-analysis following the PRISMA guidelines. Phytomedicine. 2018;59:152787. doi: 10.1016/j.phymed.2018.12.010. [Epub ahead of print]
29. Xu J, Wang Q, Leung ELH. et al. Compound C620-0696, a new potent inhibitor targeting BPTF, the chromatin-remodeling factor in non-small-cell lung cancer. Front Med. 2019 doi: 10.1007/s11684-019-0694-8. [Epub ahead of print]
Author contact
![]() Corresponding authors: huang_jian_ancom (J. Huang); qbwuedu.mo (Q. Wu).
Corresponding authors: huang_jian_ancom (J. Huang); qbwuedu.mo (Q. Wu).