Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2023; 14(18):3387-3396. doi:10.7150/jca.88561 This issue Cite
Research Paper
Potentially Functional Genetic Variants in the NRF2 Signaling Pathway Genes are Associated With HBV-related Hepatocellular Carcinoma Survival
Rongbin Gong1,2,#, Moqin Qiu3,#, Ji Cao4, Zihan Zhou4, Yuying Wei1, Qiuping Wen1, Qiuling Lin5, Xiaoxia Wei5, Xiumei Liang6, Yanji Jiang7, Peiqin Chen1, Junjie Wei2, Shicheng Zhan2, Yingchun Liu1,8, ![]() , Hongping Yu1,8,9,
, Hongping Yu1,8,9, ![]()
1. Department of Experimental Research, Guangxi Medical University Cancer Hospital, Nanning 530000, China.
2. Department of Epidemiology and Health Statistics, School of Public Health, Guangxi Medical University, Nanning 530000, China.
3. Department of Respiratory Oncology, Guangxi Medical University Cancer Hospital, Nanning 530000, China.
4. Department of Cancer Prevention and Control, Guangxi Medical University Cancer Hospital, Nanning 530000, China.
5. Department of Clinical Research, Guangxi Medical University Cancer Hospital, Nanning 530000, China.
6. Department of Disease Process Management, Guangxi Medical University Cancer Hospital, Nanning 530000, China.
7. Department of Scientific Research Dept, Guangxi Medical University Cancer Hospital, Nanning 530000, China.
8. Key Cultivated Laboratory of Cancer Molecular Medicine of Guangxi Health Commission, Guangxi Medical University Cancer Hospital, Nanning 530000, China.
9. Key Laboratory of Early Prevention and Treatment for Regional High Frequency Tumor (Guangxi Medical University), Ministry of Education, Nanning 530000, China.
# These authors contributed equally to this work
Received 2023-7-27; Accepted 2023-9-17; Published 2023-10-16
Citation:
Gong R, Qiu M, Cao J, Zhou Z, Wei Y, Wen Q, Lin Q, Wei X, Liang X, Jiang Y, Chen P, Wei J, Zhan S, Liu Y, Yu H. Potentially Functional Genetic Variants in the NRF2 Signaling Pathway Genes are Associated With HBV-related Hepatocellular Carcinoma Survival. J Cancer 2023; 14(18):3387-3396. doi:10.7150/jca.88561. https://www.jcancer.org/v14p3387.htm
Other stylesAbstract

The nuclear factor E2-related factor 2 (NRF2) signaling pathway is one of the most important cell defense pathways. However, it is unclear whether genetic variants in NRF2 signaling pathway genes are associated with the survival of hepatitis B virus (HBV)-related hepatocellular carcinoma (HCC). In the present study, we utilized a new hypothesis-driven approach based on biological pathways to investigate the associations between 17919 single nucleotide polymorphisms (SNPs) in 137 NRF2 signaling pathway genes and the overall survival (OS) of 866 patients with HBV-related HCC. As a result, two independent SNPs with potential biological function were identified to be significantly associated with HBV-related HCC OS: [SLC2A9 rs28643326 T>C: hazard ratio (HR) = 0.74, 95% confidence interval (95% CI) = 0.62-0.89, P < 0.001 and SLC5A10 rs2472711 G>T: HR = 0.81, 95% CI = 0.71-0.93, P = 0.003, respectively]. The expression quantitative trait loci (eQTL) analysis further revealed that the rs28643326 C allele was significantly associated with increased levels of SLC2A9 mRNA expression (P < 0.001), and higher mRNA expression levels of SLC2A9 in adjacent normal liver tissues were associated with better survival. Although the association between the rs2472711 T allele and the mRNA expression of SLC5A10 was not statistically significant (P = 0.200), the fact that rs2472711 is located at the DNase I hypersensitivity site and is a marker for promoter and enhancer histones also suggests that it may have the function of regulating its corresponding gene expression. In conclusion, genetic variants of NRF2 signaling pathway genes may serve as potential prognostic biomarkers for HBV-related HCC and also provide a solid basis for further mechanistic exploration.
Keywords: NRF2, HBV-related HCC, SNPs, OS, eQTL
Introduction
Primary liver cancer is one of the most common malignancies worldwide and has become the second leading cause of cancer deaths in China, with approximately 410,000 new cases and 391,000 deaths in 2020 [1]. Hepatocellular carcinoma (HCC) is the most common primary liver cancer, accounting for approximately 80% of all cases, and hepatitis B virus (HBV) is the major risk factor, with nearly 84% of HCC patients infected in China [2, 3]. Although the level of treatment for HCC has been greatly improved in recent years, the 5-year survival rate for patients with middle-advanced HCC is still pessimistic with only 14.1% [4]. Currently, the major clinicopathological variables that are utilized to predict the prognosis of HCC include age, sex, alpha-fetoprotein (AFP) levels, cirrhosis, and tumor stage. Notably, due to individual heterogeneity, even the HCC patients with the same clinical characteristics may have different outcomes, suggesting that genetic susceptibility may have an impact on the prognosis of HCC patients [5]. Therefore, it is important to explore proper prognostic biomarkers for HCC.
As the most common type of genetic variant, single nucleotide polymorphisms (SNPs) can regulate gene expression by altering transcriptional activity. To date, the genome-wide association analysis (GWAS) possesses the ability to efficiently identify the SNPs associated with the development and progression of tumors, and a growing number of SNPs have been successively confirmed to be associated with the risk or survival of HCC [6, 7]. However, a large number of the SNPs with significant regulatory functions have been overlooked because of the failure to meet the stringent statistical P values. With the advent of the post-GWAS era, a new hypothesis-driven approach based on biological pathways has emerged for better investigating the impact of biological pathways and is now widely utilized to identify more SNPs of the pathway genes that have potential function on the HCC survival [8].
The nuclear factor E2-related factor 2 (NRF2) signaling pathway is one of the most important cell defense pathways [9], primarily by regulating gene expression to resist oxidative stress, maintain cellular homeostasis, and play an important protective role in tumorigenesis and development [10]. It has been shown that over-activation of the NRF2 signaling pathway enhances cellular resistance to the oxidative stress and electromagnetic waves involved in multiple stages of tumor development by regulating cell cycle and cell motility [11]. NRF2, a master transcription factor with functions in regulating inflammatory and antiviral responses, has also been shown to act as a tumor suppressor that protects cells from endogenous and exogenous stresses and inhibits carcinogenesis [12, 13]. NRF2 has been found to play an important protective role in various liver diseases, including liver inflammation, liver fibrosis, and hepatocarcinogenesis, through the inhibition of reactive oxygen species (ROS) [14]. Additionally, it has been suggested that NRF2 may regulate several important genes to detoxify and combat oxidative stress in the prevention of carcinogenesis [15]. In recent years, a number of SNPs in NRF2 signaling pathway genes have been identified to be associated with the risk and prognosis of pancreatic cancer [16], breast cancer [17], and cholangiocarcinoma [18], further supporting the vital role of the NRF2 pathway playing in cancer susceptibility and prognosis. Unfortunately, little is known about the effect of genetic variants in NRF2 signaling pathway genes on the survival of the HBV-related HCC. Considering the importance of the NRF2 pathway in HCC, based on genotyping data from the Guangxi population in China, we aimed to investigate the associations between SNPs with potential functions in NRF2 signaling pathway genes and the survival of the HBV-related HCC.
Material and Methods
Study populations
From June 2007 to December 2017, 866 HCC patients who underwent hepatectomy and were seropositive for hepatitis B surface antigen (HBsAg) were recruited at Guangxi Medical University Cancer Hospital and randomized into discovery and validation groups in a 1:1 ratio. The inclusion and exclusion criteria were as follows: 1) All participants must be pathologically diagnosed HCC patients; 2) All patients must have positive serum HBsAg test results; 3) Patients with surface-positive anti-hepatitis C virus were excluded; 4) Patients with cardiovascular, renal or pulmonary diseases were excluded; 5) Patients who had developed pathological distant metastases, and those who had received preoperative radiotherapy or chemotherapy for hepatectomy were excluded; 6) Patients lacking complete clinical and follow-up information were further excluded. Detailed information has been described in a previous study [19]. We gathered demographic and clinical information on all individuals, including age, sex, smoking status, drinking status, AFP level, cirrhosis, embolus, and Barcelona Clinic Liver Cancer (BCLC) stage. In addition, 5 ml of peripheral blood samples were collected from all individuals for genotyping with the Illumina GSA genotyping chip (Infnium Global Screening Array-24 v1.0), and Minimac3 was utilized for imputation based on 1000 Genomes Project (Phase 3 v5) reference population information (https://imputationserver.sph.umich.edu/index.htm). We used overall survival (OS) as the primary endpoint and acquired it through a rigorous follow-up that ended in March 2020. Informed consent was obtained from all patients, and the study was approved by the Ethics Committee of Guangxi Medical University Cancer Hospital (Approval Number: LW2023105).
Genes and SNPs selection
NRF2 signaling pathway genes were identified in the Molecular Signatures Database (http://software.broadinstitute.org/gsea/msig-db/index.jsp) using “NRF2” as a keyword. After excluding four genes on the X chromosome, 137 genes remained as candidate genes (Table S1). All SNPs within candidate genes and their flanking regions of ± 2 kb were extracted according to the following criteria: genotyping rate ≥ 95%, minimum allele frequency (MAF) ≥ 0.05, and Hardy-Weinberg equilibrium (HWE) P value ≥ 1×10-6.
RNA sequencing
After obtaining consent, we collected 100 pairs HCC tissues and adjacent normal tissues from Guangxi Medical University Cancer Hospital for RNA sequencing to detect differential expression of genes in HCC.
Statistical analyses
By using the GenABEL package in R software [20], the multivariate Cox proportional hazards regression analysis adjusted for age, sex, smoking status, drinking status, AFP level, cirrhosis, embolus, and BCLC stage was used to calculate the hazard ratio (HR) and 95% confidence interval (95% CI) for all the SNPs in an additive model to assess the associations between SNPs in 137 candidate genes of the NRF2 signaling pathway and the HBV-related HCC OS. Next, to reduce the possibility of false positive results, the Bayesian false discovery probability (BFDP) approach was utilized for multiple testing correction. Only SNPs with a BFDP value ≤ 0.8 were chosen for validation. Subsequently, we included the validated SNPs in a multivariate stepwise Cox model with adjustment for clinical variables to identify independent SNPs and employed additive and dominant genetic models to assess the associations between different genotypes of independent SNPs and the HBV-related HCC OS. In addition, the combination of protective genotypes was conducted to estimate the joint effects of identified SNPs, and Kaplan-Meier survival curves with log-rank tests were used to observe the influence of the joint effect on OS. Then, we further utilized stratified analysis to assess whether the combined effect of protective genotypes on OS was influenced by clinical covariates. Finally, Haploview v4.2 was used to generate Manhattan plots [21], and LocusZoom [22] was used to construct regional association plots.
All statistical analyses were performed using the R software (versions 3.1.3 and 4.2.2), and P < 0.05 was considered statistically significant.
Function prediction
In order to predict the potential function of the identified SNPs, we utilized three available online bioinformatics tools: RegulomeDB (https://www.regulomedb.org/regulome-search/) [23], HaploReg v4.2 (https://pubs.broadinstitute.org/mammals/haploreg/haploreg.php) [24], and SNPinfo (https://manticore.niehs.nih.gov/snpinfo/snpfunc.html) [25]. The expression quantitative trait locus (eQTL) analysis was performed to assess the correlation between SNPs and the corresponding genes mRNA expression levels by using the RNA sequencing data of lymphoblastoid cell lines (n = 76) generated from Chinese descendants in the 1000 Genomes Project Han Chinese in Beijing and the data of whole blood (n = 369) and normal liver tissues (n = 220) from the Genotype-Tissue Expression Project (GTEx) database (http://www.gtexportal.org/home/) [26]. We further compared the variation of gene mRNA expression levels between HCC tissues and adjacent normal tissues by using the Cancer Genome Atlas (TCGA) database [27] and in-house RNA sequencing data. In addition, the Kaplan-Meier Plotter (https://kmplot.com/analysis/), an online survival analysis software, was used to illustrate the correlation between the corresponding gene mRNA expression levels and the survival in patients with liver cancer. At last, the public database of the cBioPortal for Cancer Genomics was utilized to evaluate the somatic mutational status of genes in HCC.
Results
Associations between SNPs in NRF2 pathway genes and HBV-related HCC OS
The workflow of this study is presented in Figure 1. As described in the previous study, the demographic and clinical characteristics of 866 patients with the HBV-related HCC were shown in Table S2. We extracted 17919 SNPs (1109 genotyped and 16810 imputed) in the 137 NRF2 signaling pathway genes with 2 kb flanking regions from the discovery dataset. In the single locus analysis of the discovery dataset, 1207 SNPs were significantly associated with the OS of the HBV-related HCC in the additive genetic model (P < 0.05, BFDP < 0.80), of which rs28643326 in solute carrier family 2, member 9 (SLC2A9), and rs2472711 in solute carrier family 5, member 10 (SLC5A10) remained statistically significant after further replication in the validation dataset (Table 1). These results were also depicted by Manhattan plots in Figure S1.
Independent SNPs in NRF2 pathway genes were associated with HBV-related HCC OS
The multivariable stepwise Cox regression model was performed with adjustment for clinical variables in the combined dataset to observe whether the identified SNPs were independent predictors for the HCC OS. As a result, two SNPs remained statistically significant (Table 2). Consequently, the results of genetic model analysis in the combined dataset showed that rs28643326 TC/CC and rs2472711 GT/TT were presented to be associated with better prognosis in the HBV-related HCC patients (HR = 0.71, 95% CI = 0.57-0.87, P < 0.001 and HR = 0.74, 95% CI = 0.60-0.90, P = 0.003, respectively) (Table 3). For illustrative purposes, two SNPs in their corresponding genes with 50kb flanking region are shown in regional association plots (Figure S2).
Figure 1
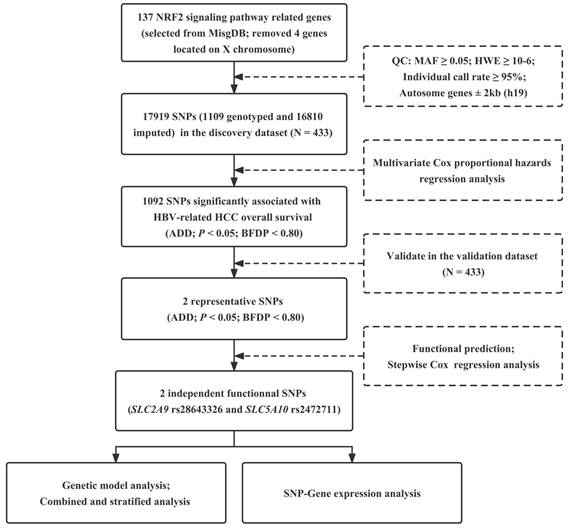
Workflow of the study process. NRF2: nuclear factor E2-related factor 2; MsigDB: Molecular Signatures Database; QC: quality control; MAF: minor allele frequency; HWE: Hardy-Weinberg equilibrium; SNP: single nucleotide polymorphism; HBV: hepatitis B virus; HCC: hepatocellular carcinoma; ADD: additive model; BFDP: Bayesian false-discovery probability.
Table 1
Associations of two validated significant SNPs with HBV-related HCC OS in the discovery, validation, and combined dataset.
| SNP | Chr | Gene | Allelea | Discovery dataset (N = 433) | Validation dataset (N = 433) | Combined dataset (N = 866) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EAF | HR (95% CI) | Pb | BFDP | EAF | HR (95% CI) | Pb | BFDP | EAF | HR (95% CI) | Pb | BFDP | ||||
| rs28643326 | 4 | SLC2A9 | T>C | 0.22 | 0.73 (0.57-0.94) | 0.014 | 0.66 | 0.20 | 0.75 (0.57-0.97) | 0.029 | 0.76 | 0.21 | 0.74 (0.62-0.89) | 0.001 | 0.23 |
| rs2472711 | 17 | SLC5A10 | G>T | 0.45 | 0.80 (0.66-0.97) | 0.022 | 0.75 | 0.49 | 0.81 (0.67-0.98) | 0.032 | 0.79 | 0.47 | 0.81 (0.71-0.93) | 0.003 | 0.38 |
Abbreviations: SNPs: single nucleotide polymorphisms; HBV: hepatitis B virus; HCC: hepatocellular carcinoma; OS: overall survival; EAF: effect allele frequency; HR: hazards ratio; 95% CI: 95% confidence interval; BFDP: Bayesian false-discovery probability.
a Referring allele/effect allele.
b Multivariate Cox proportional hazards regression analysis was adjusted for age, sex, smoking status, drinking status, AFP level, cirrhosis, embolus, and BCLC stage.
Table 2
Two independent predictors of OS obtained from stepwise Cox regression analysis in the combine dataset
| Characteristics | Category | Frequency | HR (95% CI) | Pa |
|---|---|---|---|---|
| Age (year) | ≤47 | 434 | 1.00 | |
| >47 | 432 | 0.80 (0.66-0.97) | 0.026 | |
| Sex | Female | 106 | 1.00 | |
| Male | 760 | 1.28 (0.93-1.77) | 0.135 | |
| AFP level (ng/ml) | ≤400 | 522 | 1.00 | |
| >400 | 344 | 1.30 (1.06-1.60) | 0.011 | |
| Embolus | No | 636 | 1.00 | |
| Yes | 230 | 1.72 (1.36-2.18) | <0.001 | |
| BCLC stage | 0/A | 427 | 1.00 | |
| B/C | 439 | 2.02 (1.59-2.57) | <0.001 | |
| SLC2A9 rs28643326 T>C | TT/TC/CC | 534/295/37 | 0.73 (0.61-0.88) | <0.001 |
| SLC5A10 rs2472711 G>T | GG/GT/TT | 261/397/208 | 0.81 (0.71-0.92) | 0.002 |
Abbreviations: OS: overall survival; HR: hazards ratio; 95% CI: 95% confidence interval; AFP: alpha-fetoprotein; BCLC: Barcelona Clinic Liver Cancer.
a Stepwise Cox regression analysis was adjusted for age, sex, smoking status, drinking status, AFP level, cirrhosis, embolus, and BCLC stage.
Figure 2
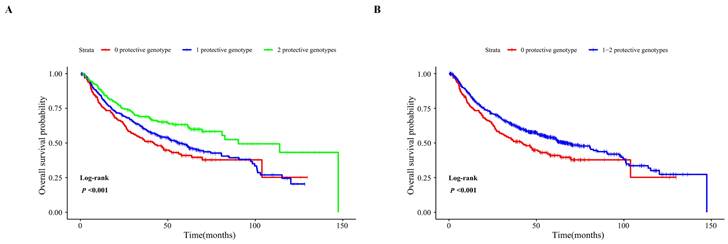
Kaplan-Meier analysis for patients with HBV-related HCC by the combined protective genotypes. Kaplan-Meier analysis for patients with HBV-related HCC by 0, 1, and 2 protective genotypes (A), by 0 and 1-2 protective genotypes (B).
Combined and stratified analysis of the two independent SNPs
To assess the joint effect of the two independent SNPs on the OS of HBV-related HCC, we combined protective genotypes (SLC2A9 rs28643326 TC/CC and SLC5A10 rs2472711 GT/TT) into a genetic score based on the number of protective genotypes (NPGs). All HCC patients were divided into three groups with genetic scores of 0, 1, and 2, respectively. The results indicated that patients with a genetic score of 1 or 2 had a more favorable OS than that with a score of 0 (P = 0.026 and < 0.001, respectively), and a higher genetic score was associated with a progressively better survival in the combined dataset (Ptrend < 0.001). Next, all HCC patients were further dichotomized into a low-protection group (score 0) and a high-protection group (score 1-2). We found that the patients in the high-protection group had better survival compared to those in the low-protection group in the combined dataset (HR = 0.67, 95% CI = 0.53-0.85, P < 0.001) (Table 4), and Kaplan-Meier survival curves were employed to describe these results (Figure 2). The stratified analysis results showed that the HCC patients in the high-protection group had a favorable prognosis in all the subgroups (P < 0.05) except age > 47, female, drinking, AFP ≤ 400ng/ml, and patients with cancer embolus. However, no significant interaction between NPG and clinical variables was found (Table 5).
In silico functional validation
As shown in Table S3, biological function annotations showed that SLC2A9 rs28643326 and SLC5A10 rs2472711 were both located in the intron region with RegulomeDB scores of 5 and 3a, respectively, which might change multiple motifs. Furthermore, SLC2A9 rs28643326 was located at a transcription factor binding site (TFBS), and SLC5A10 rs2472711 was located at 7 DNase I hypersensitive sites and may be markers of promoter or enhancer histones. The eQTL analysis was performed to further explore the potential functions of the two identified SNPs. We found that the rs28643326 C allele was significantly associated with elevated mRNA expression levels of SLC2A9 in normal liver tissues from the GTEx project (P < 0.001, Figure 3A), as well as in lymphoblastoid cells from the 1000 Genomes Project Han Chinese in Beijing (P = 0.093 for the additive genetic model, Figure 3C, and P = 0.031 for dominant genetic model, Figure 3D, respectively). However, genotyping data for SLC5A10 rs2472711 in normal liver tissues were not available, and there was no significant correlation between the rs2472711 T allele and mRNA expression levels of SLC5A10 in whole blood (P = 0.200, Figure 3B) from the GTEx project. Additionally, gene expression data from the TCGA and in-house RNA-Seq dataset illustrated that the mRNA expression of SLC2A9 was significantly lower in HCC tissues compared to adjacent normal tissues (Figure 4A and Figure 4B). In contrast, in comparison with adjacent normal tissues, HCC tissues had a higher mRNA expression level of SLC5A10 (Figure 4D and Figure 4E). Finally, K-M survival curves obtained based on the Kaplan-Meier Plotter website showed that higher expression of SLC2A9 was significantly associated with improved liver cancer survival (P = 0.004, Figure 4C), whereas higher expression levels of SLC5A10 were was associated with a worse survival of liver cancer (P = 0.210, Figure 4F).
Mutation analysis
The public database of the cBioPortal for Cancer Genomics was utilized to explore the mutation status of SLC2A9 and SLC5A10 in HCC. As shown in Figure S3, both genes had very low somatic mutation rates in different HCC datasets (SLC2A9: 0.82%, 2/243 in the INSERM; 0.27%, 1/366 in the TCGA PanCancer Atlas study; 0.27%, 1/373 in the TCGA Firehose Legacy study; and 0.43%, 1/231 in the AMC; SLC5A10: 0.41%, 1/243 in the INSERM; 0.55%, 2/366 in the TCGA PanCancer Atlas study; 0.54%, 2/373 in the TCGA Firehose Legacy study; and 0.43%, 1/231 in the AMC). Thus, it was evident that the mutations in SLC5A10 and SLC2A9 had negligible effects on their expression levels, supporting functional genetic variants as the key influencing factor leading to the imbalance of SLC2A9 and SLC5A10 expression in HCC.
Figure 3
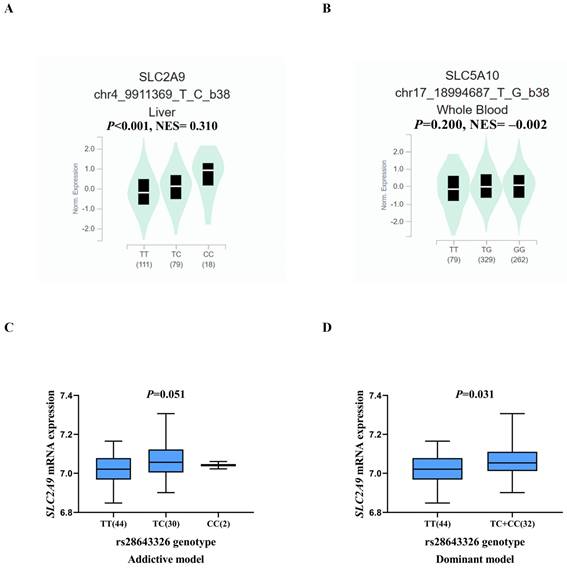
Correlation of the SNPs with mRNA expression. eQTL analysis of SLC2A9 rs28643326 in liver tissue from the GTEx database (n = 208, P < 0.001) (A), eQTL analysis of SLC5A10 rs2472711 in whole blood from the GTEx database (n = 670, P = 0.200) (B), correlation of rs28643326 with SLC2A9 mRNA expression in lymphoblastoid cells from the 1000 Genomes Project Han Chinese in Beijing in the additive (n = 76, P = 0.051) (C) and dominant (n = 76, P = 0.031) (D) models. SNPs: single nucleotide polymorphisms; eQTL: expression quantitative trait loci; GTEx: Genotype-Tissue Expression project; NES: normalized effect size; Norm: normalized.
Figure 4
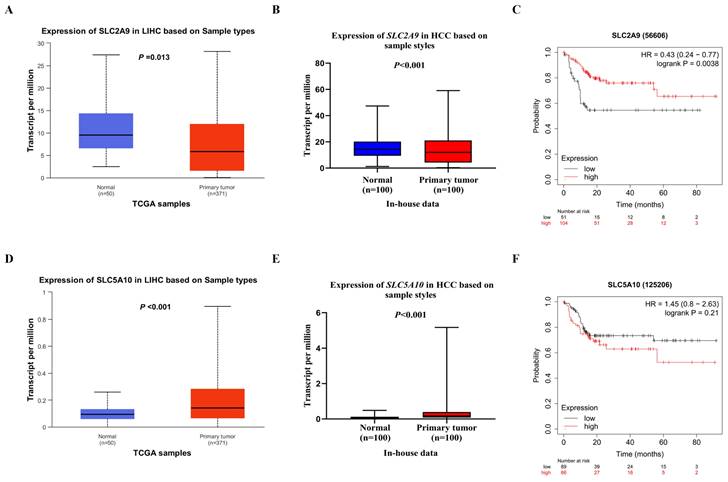
Differential mRNA expression analysis and survival of HCC. The relative expression levels of the SLC2A9 mRNA in the TCGA database (A) and in-house RNA-Seq data (B), the relative expression levels of the SLC5A10 mRNA in the TCGA database (D) and in-house RNA-seq data (E), liver cancer patients with high mRNA expression levels of SLC2A9 had a better OS (C), liver cancer patients with high mRNA expression levels of SLC5A10 had a worse OS (F). TCGA: the Cancer Genome Atlas; HCC: hepatocellular carcinoma; HR: hazard ratio; LIHC: liver hepatocellular carcinoma; RNA-seq: RNA sequencing; OS: overall survival.
Table 3
Associations between two independent SNPs and OS of HBV-related HCC in different genetic models.
| Genotype | Discovery dataset (N = 433) | Validation dataset (N = 433) | Combined dataset (N = 866) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All | Death (%) | HR (95% CI) | Pa | All | Death (%) | HR (95% CI) | Pa | All | Death (%) | HR (95% CI) | Pa | |
| SLC2A9 rs28643326 T>C | ||||||||||||
| TT | 263 | 134 (51.0) | 1.00 | 271 | 148 (54.6) | 1.00 | 534 | 282 (52.8) | 1.00 | |||
| TC | 147 | 60 (40.8) | 0.79 (0.58-1.08) | 0.140 | 148 | 64 (43.2) | 0.66 (0.48-0.89) | 0.007 | 295 | 124 (42.0) | 0.72 (0.58-0.89) | 0.002 |
| CC | 23 | 6 (26.1) | 0.41 (0.18-0.93) | 0.032 | 14 | 7 (50.0) | 1.00 (0.47-2.15) | 0.992 | 37 | 13 (35.1) | 0.63 (0.36-1.10) | 0.101 |
| Trend test | 0.014 | 0.029 | 0.001 | |||||||||
| TT | 263 | 134 (51.0) | 1.00 | 271 | 148 (54.6) | 1.00 | 534 | 282 (52.8) | 1.00 | |||
| TC+CC | 170 | 66 (38.8) | 0.73 (0.54-0.98) | 0.036 | 162 | 71 (43,8) | 0.68 (0.51-0.91) | 0.010 | 332 | 137 (41.3) | 0.71 (0.57-0.87) | <0.001 |
| SLC5A10 rs2472711 G>T | ||||||||||||
| GG | 141 | 75 (53.2) | 1.00 | 120 | 65 (54.2) | 1.00 | 261 | 140 (53.6) | 1.00 | |||
| GT | 191 | 85 (44.5) | 0.75 (0.54-1.03) | 0.071 | 206 | 105 (51.0) | 0.76 (0.56-1.04) | 0.092 | 397 | 190 (47.9) | 0.77 (0.62-0.96) | 0.022 |
| TT | 101 | 40 (39.6) | 0.65 (0.44-0.96) | 0.030 | 107 | 49 (45.8) | 0.67 (0.46-0.97) | 0.035 | 208 | 89 (42.8) | 0.67 (0.51-0.88) | 0.003 |
| Trend test | 0.022 | 0.032 | 0.003 | |||||||||
| GG | 141 | 75 (53.2) | 1.00 | 120 | 65 (54.2) | 1.00 | 261 | 140 (53.6) | 1.00 | |||
| GT+TT | 292 | 125 (42.8) | 0.71 (0.53-0.95) | 0.022 | 313 | 154 (49.2) | 0.73 (0.54-0.98) | 0.035 | 605 | 279 (46.1) | 0.74 (0.60-0.90) | 0.003 |
Abbreviations: SNPs: single nucleotide polymorphisms; OS: overall survival; HBV: hepatitis B virus; HCC: hepatocellular carcinoma; HR: hazards ratio; 95% CI: 95% confidence interval.
a Multivariate Cox proportional hazards regression analysis was adjusted for age, sex, smoking status, drinking status, AFP level, cirrhosis, embolus, and BCLC stage.
Discussion
In the present study, we evaluated the associations between 17919 SNPs in 137 NRF2 signaling pathway genes and HBV-related HCC OS. Two independent and potentially functional SNPs (SLC2A9 rs28643326 T>C and SLC5A10 rs2472711 G>T) have been demonstrated to reduce the risk of death in the patients with HBV-related HCC after hepatectomy. Importantly, an increased number of protective genotypes of these two independent SNPs were significantly associated with better HCC OS. Further eQTL analysis results indicated that the rs28643326 C allele predicted an increased mRNA expression of SLC2A9. Moreover, we found that higher mRNA expression levels of SLC2A9 in adjacent normal liver tissues were associated with better survival, whereas higher mRNA expression levels of SLC5A10 in HCC tissues were associated with poorer survival. These evidences suggested that these two SNPs in the NRF2 signaling pathway genes may serve as the prognostic biomarkers for HBV-related HCC, especially the SLC2A9 rs28643326.
SLC2A9 (also known as GLUT9), located on chromosome 4p16.1, is a member of the glucose transporter family, which plays a key role in cellular metabolism mainly by transporting glucose and fructose [28, 29]. A study revealed that SLC2A9 mRNA expression levels were significantly lowered in a variety of cancer tissues, including kidney, prostate, and testicular cancers. Meantime, lower SLC2A9 mRNA levels were significantly associated with poorer survival in the patients with gastric cancer [30]. In addition, it was reported that SLC2A9 might be a novel tumor suppressor gene that induced apoptosis in HCC cells by inhibiting the expression of caspase 3, and high expression of SLC2A9 could effectively inhibit the proliferation of HCC cells [31], suggesting that SLC2A9 may have an important biological role in HCC.
Table 4
Combined protective genotypes of the two independent SNPs and OS of HBV-related HCC in the combined dataset
| NPGsa | Combined dataset (N = 866) | Multivariable analysis | ||
|---|---|---|---|---|
| All | Death (%) | HR (95% CI) | Pb | |
| 0 | 161 | 91 (56.5) | 1.00 | |
| 1 | 473 | 240 (50.7) | 0.76 (0.59-0.97) | 0.026 |
| 2 | 232 | 88 (37.9) | 0.51 (0.38-0.69) | <0.001 |
| Trend test | <0.001 | |||
| 0 | 161 | 91 (56.5) | 1.00 | |
| 1-2 | 705 | 328 (46.5) | 0.67 (0.53-0.85) | <0.001 |
Abbreviations: SNPs: single nucleotide polymorphisms; OS: overall survival; HBV: hepatitis B virus; HCC: hepatocellular carcinoma; HR: hazards ratio; 95% CI: 95% confidence interval; NPGs: number of protective genotypes.
a Protective genotypes were SLC2A9 rs28643326 TC/CC and SLC5A10 rs2472711 GT/TT.
b Multivariate Cox proportional hazards regression analysis was adjusted for age, sex, smoking status, drinking status, AFP level, cirrhosis, embolus, and BCLC stage.
Table 5
Stratified analysis of combined protective genotypes with OS of HBV-related HCC
| Characteristics | NPG 0 | NPGs 1-2 | Multivariable analysis | ||||
|---|---|---|---|---|---|---|---|
| All | Death (%) | All | Death (%) | HR (95% CI) | Pa | Pinterb | |
| Age (year) | 0.061 | ||||||
| ≤47 | 81 | 55 (67.9) | 353 | 178 (50.4) | 0.57 (0.42-0.77) | <0.001 | |
| >47 | 80 | 36 (45.0) | 352 | 150 (42.6) | 0.85 (0.58-1.22) | 0.373 | |
| Sex | 0.299 | ||||||
| Female | 20 | 12 (60.0) | 86 | 30 (34.9) | 0.53 (0.26-1.09) | 0.083 | |
| Male | 141 | 79 (56.0) | 619 | 298 (48.1) | 0.67 (0.52-0.87) | 0.002 | |
| Smoking status | 0.724 | ||||||
| No | 109 | 64 (58.7) | 436 | 204 (46.8) | 0.72 (0.54-0.96) | 0.025 | |
| Yes | 52 | 27 (51.9) | 269 | 124 (46.1) | 0.57 (0.37-0.88) | 0.011 | |
| Drinking status | 0.499 | ||||||
| No | 125 | 71 (56.8) | 489 | 221 (45.2) | 0.66 (0.50-0.86) | 0.003 | |
| Yes | 36 | 20 (55.6) | 216 | 107 (49.5) | 0.68 (0.41-1.11) | 0.122 | |
| AFP level (ng/ml) | 0.060 | ||||||
| ≤400 | 94 | 45 (47.9) | 428 | 187 (43.7) | 0.76 (0.55-1.06) | 0.100 | |
| >400 | 67 | 46 (68.7) | 277 | 141 (50.9) | 0.58 (0.41-0.81) | 0.002 | |
| Cirrhosis | 0.424 | ||||||
| No | 72 | 38 (52.8) | 318 | 146 (45.9) | 0.66 (0.45-0.95) | 0.026 | |
| Yes | 89 | 53 (59.6) | 387 | 182 (47.0) | 0.70 (0.51-0.95) | 0.023 | |
| Embolus | 0.831 | ||||||
| No | 124 | 62 (50.0) | 512 | 198 (38.7) | 0.67 (0.50-0.90) | 0.008 | |
| Yes | 37 | 29 (78.4) | 193 | 130 (67.4) | 0.68 (0.44-1.04) | 0.076 | |
| BCLC stage | 0.658 | ||||||
| 0/A | 82 | 33 (40.2) | 345 | 113 (32.8) | 0.66 (0.45-0.98) | 0.042 | |
| B/C | 79 | 58 (73.4) | 360 | 215 (59.7) | 0.67 (0.50-0.90) | 0.008 | |
Abbreviations: OS: overall survival; HBV: hepatitis B virus; HCC: hepatocellular carcinoma; NPGs: number of protective genotypes; HR: hazards ratio; 95% CI: 95% confidence interval; Pinter: P-value for interaction.
a Multivariate Cox proportional hazards regression analysis was adjusted for age, sex, smoking status, drinking status, AFP level, cirrhosis, embolus, and BCLC stage.
b P-value for multiplicative interaction analysis between variables and NPG.
Consistent with previous reports, in this study, we have observed that the rs28643326 C allele was associated with an increased mRNA expression level of SLC2A9, and the SLC2A9 mRNA expression levels were significantly higher in adjacent normal tissues than in HCC tissues, and their high expression levels were associated with a more favorable HCC OS. Remarkably, rs28643326 is located at the transcription factor binding site that may affect SLC2A9 expression by enhancing transcriptional activity, which further supports the biological plausibility of the findings. Thus, we further validated that SLC2A9 played a carcinogenic role in HCC biology and that SLC2A9 rs28643326 might be an important factor affecting the prognosis of HCC.
SLC5A10 is located on chromosome 17p11.2 and encodes a protein that may function as a sodium-dependent mannose and fructose co-transporter. Previous research reported that SLC5A10 rs2257609 C>T affected the prognosis of early-stage non-small-cell lung cancer by influencing the expression of DRG2 [32]. Unfortunately, few studies have revealed the role of SLC5A10 in HCC. In the present study, we have found that the rs2472711 T allele is capable of reducing the risk of death in patients with HBV-related HCC, and rs2472711 is located at DNase I hypersensitive sites, which also suggests that it may have the function of regulating its corresponding gene expression. However, due to the lack of data supporting the correlation between the rs2472711 T allele and SLC5A10 mRNA expression levels, more investigations are needed to prove whether the molecular mechanisms of the observed associations are due to biological processes other than affecting the expression of the corresponding gene at the transcriptional level.
Noticeably, some limitations of this study cannot be ignored. Firstly, all subjects in this study were recruited from Southern China, and thus our results may not be generalizable to other ethnic populations due to differences in genetic variation across ethnic groups. Secondly, the sample size of this study is relatively small, and all patients were enrolled at Guangxi Medical University Cancer Hospital, and thus more studies with a larger sample size from multi-centers are urgently needed to validate our results. Finally, further biological and functional experiments for these two independent SNPs should be performed to fully understand their exact molecular mechanisms in HCC.
Conclusions
In conclusion, we found that two independent functional SNPs (SLC2A9 rs28643326 T>C and SLC5A10 rs2472711 G>T) were significantly associated with HBV-associated HCC OS, suggesting that these two SNPs may serve as potential biomarkers for predicting HCC prognosis. It is likely that the SLC2A9 rs28643326 C allele elevates the expression of the corresponding gene by affecting transcriptional activity. Our results also provide a solid foundation for further functional studies to identify the molecular mechanisms underlying the observed significant association between genetic variants of NRF2 signaling pathway genes and HCC prognosis.
Abbreviations
NRF2: nuclear factor E2-related factor 2; HBV: hepatitis B virus; HCC: hepatocellular carcinoma; SNPs: single nucleotide polymorphisms; OS: overall survival; eQTL: expression quantitative trait loci; AFP: alpha-fetoprotein; GWAS: genome-wide association analysis; ROS: reactive oxygen species; HBsAg: hepatitis B surface antigen; BCLC: Barcelona Clinic Liver Cancer; MAF: minimum allele frequency; HWE: Hardy-Weinberg equilibrium; HR: hazard ratio; 95% CI: 95% confidence interval; BFDP: Bayesian false discovery probability; GTEx: Genotype-Tissue Expression Project; TCGA: the Cancer Genome Atlas; NPGs: number of protective genotypes; TFBS: transcription factor binding site.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We would like to thank all study participants, researchers and clinicians who contributed to this study.
Funding
This study was supported by grants from the Natural Science Foundation of Guangxi Province (2023GXNSFBA026201); Youth Science Foundation of Guangxi Medical University (GXMUYSF202312); Youth Program of Scientific Research Foundation of Guangxi Medical University Cancer Hospital (YQJ2022-7); Natural Science Foundation of Guangxi Province 2023GXNSFBA026091; Youth Program of Scientific Research Foundation of Guangxi Medical University Cancer Hospital (2021-10); Key Laboratory of Early Prevention and Treatment for Regional High Frequency Tumor, Ministry of Education (GKE-ZZ202104); Key Cultivated Laboratory of Cancer Molecular Medicine of Guangxi Health Commission, Guangxi Medical University Cancer Hospital (ZPTJ2020001).
Author contributions
Y.L. and H.Y. designed the study; Z.Z., P.C., J.W., S.Z., R.G., Y.W., Q.W. and M.Q. collected the clinical data; R.G. and M.Q. analyzed the clinical data and wrote the manuscript; Q.L., J.C., X.W., Y.J., A.T. and X.L. provided clinical care to the patients; Y.L. and H.Y. provided comments; and all the authors approved the final version of the manuscript.
Data availability
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Ethics approval
Approval of the research protocol by an Institutional Reviewer Board: The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by Ethics Committee of the Guangxi Medical University Cancer Hospital (LW2023105).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cao W, Chen H-D, Yu Y-W, Li N, Chen W-Q. Changing profiles of cancer burden worldwide and in China: a secondary analysis of the global cancer statistics 2020. Chinese Medical Journal. 2021;134:783-91
2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: A Cancer Journal for Clinicians. 2021;71:209-49
3. Lin J, Zhang H, Yu H, Bi X, Zhang W, Yin J. et al. Epidemiological Characteristics of Primary Liver Cancer in Mainland China From 2003 to 2020: A Representative Multicenter Study. Frontiers in Oncology. 2022;12:906778
4. Zeng H, Chen W, Zheng R, Zhang S, Ji JS, Zou X. et al. Changing cancer survival in China during 2003-15: a pooled analysis of 17 population-based cancer registries. The Lancet Global Health. 2018;6:e555-e67
5. Zhu X, Wang Z, Qiu X, Wang W, Bei C, Tan C. et al. Rs2303428 ofMSH2Is Associated with Hepatocellular Carcinoma Prognosis in a Chinese Population. DNA and Cell Biology. 2018;37:634-41
6. Li Y, Zhai Y, Song Q, Zhang H, Cao P, Ping J. et al. Genome-Wide Association Study Identifies a New Locus at 7q21.13 Associated with Hepatitis B Virus-Related Hepatocellular Carcinoma. Clinical Cancer Research. 2018;24:906-15
7. Wei J, Sheng Y, Li J, Gao X, Ren N, Dong Q. et al. Genome-Wide Association Study Identifies a Genetic Prediction Model for Postoperative Survival in Patients with Hepatocellular Carcinoma. Medical Science Monitor. 2019;25:2452-78
8. Lin Q, Qiu M, Wei X, xiang Z, Zhou Z, Ji I. et al. Genetic variants of SOS2, MAP2K1 and RASGRF2 in the RAS pathway genes predict survival of HBV-related hepatocellular carcinoma patients. Archives of Toxicology. 2023;97:1599-611
9. Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes & Development. 2013;27:2179-91
10. Ma Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annual Review of Pharmacology and Toxicology. 2013;53:401-26
11. Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the Hallmarks of Cancer. Cancer Cell. 2018;34:21-43
12. Zhang L, Wang J, Wang Z, Li Y, Wang H, Liu H. Upregulation of nuclear factor E2-related factor 2 (Nrf2) represses the replication of herpes simplex virus type 1. Virology Journal. 2022;19:23
13. Sánchez-Ortega M, Carrera AC, Garrido A. Role of NRF2 in Lung Cancer. Cells. 2021;10:1879
14. Shin SM, Yang JH, Ki SH. Role of the Nrf2-ARE Pathway in Liver Diseases. Oxidative Medicine and Cellular Longevity. 2013;2013:1-9
15. Iida K, Itoh K, Maher JM, Kumagai Y, Oyasu R, Mori Y. et al. Nrf2 and p53 cooperatively protect against BBN-induced urinary bladder carcinogenesis. Carcinogenesis. 2007;28:2398-403
16. Yang W, Liu H, Duan B, Xu X, Carmody D, Luo S. et al. Three novel genetic variants in NRF2 signaling pathway genes are associated with pancreatic cancer risk. Cancer Science. 2019;110:2022-32
17. Hartikainen JM, Tengström M, Winqvist R, Jukkola-Vuorinen A, Pylkäs K, Kosma V-M. et al. KEAP1 Genetic Polymorphisms Associate with Breast Cancer Risk and Survival Outcomes. Clinical Cancer Research. 2015;21:1591-601
18. Khunluck T, Kukongviriyapan V, Puapairoj A, Khuntikeo N, Senggunprai L, Zeekpudsa P. et al. Association of NRF2 Polymorphism with Cholangiocarcinoma Prognosis in Thai Patients. Asian Pacific Journal of Cancer Prevention. 2014;15:299-304
19. Huang Q, Liu Y, Qiu M, Lin Q, Wei X, Zhou Z. et al. Potentially functional variants of MAP3K14 in the NF-κB signaling pathway genes predict survival of HBV-related hepatocellular carcinoma patients. Frontiers in Oncology. 2022;12:990160
20. Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:1294-6
21. Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263-5
22. Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336-7
23. Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M. et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Research. 2012;22:1790-7
24. Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Research. 2011;40:D930-D4
25. Xu Z, Taylor JA. SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Research. 2009;37:W600-W5
26. Consortium G. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648-60
27. Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK. et al. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia. 2017;19:649-58
28. Le MT, Shafiu M, Mu W, Johnson RJ. SLC2A9-a fructose transporter identified as a novel uric acid transporter. Nephrology Dialysis Transplantation. 2008;23:2746-9
29. Long W, Panigrahi R, Panwar P, Wong K, O′Neill D, Chen X-Z. et al. Identification of Key Residues for Urate Specific Transport in Human Glucose Transporter 9 (hSLC2A9). Scientific Reports. 2017;7:41167
30. Itahana Y, Han R, Barbier S, Lei Z, Rozen S, Itahana K. The uric acid transporter SLC2A9 is a direct target gene of the tumor suppressor p53 contributing to antioxidant defense. Oncogene. 2014;34:1799-810
31. Han X, Yang J, Li D, Guo Z. Overexpression of Uric Acid Transporter SLC2A9 Inhibits Proliferation of Hepatocellular Carcinoma Cells. Oncology Research Featuring Preclinical and Clinical Cancer Therapeutics. 2019;27:533-40
32. Hong MJ, Yoo SS, Choi JE, Kang HG, Do SK, Lee JH. et al. Functional intronic variant of SLC5A10 affects DRG2 expression and survival outcomes of early-stage non-small-cell lung cancer. Cancer Science. 2018;109:3902-9
Author contact
![]() Corresponding authors: Hongping Yu: yuhongpinggxmu.edu.cn; Yingchun Liu: liuyingchungxmu.edu.cn.
Corresponding authors: Hongping Yu: yuhongpinggxmu.edu.cn; Yingchun Liu: liuyingchungxmu.edu.cn.
Citation styles
APA
Gong, R., Qiu, M., Cao, J., Zhou, Z., Wei, Y., Wen, Q., Lin, Q., Wei, X., Liang, X., Jiang, Y., Chen, P., Wei, J., Zhan, S., Liu, Y., Yu, H. (2023). Potentially Functional Genetic Variants in the NRF2 Signaling Pathway Genes are Associated With HBV-related Hepatocellular Carcinoma Survival. Journal of Cancer, 14(18), 3387-3396. https://doi.org/10.7150/jca.88561.
ACS
Gong, R.; Qiu, M.; Cao, J.; Zhou, Z.; Wei, Y.; Wen, Q.; Lin, Q.; Wei, X.; Liang, X.; Jiang, Y.; Chen, P.; Wei, J.; Zhan, S.; Liu, Y.; Yu, H. Potentially Functional Genetic Variants in the NRF2 Signaling Pathway Genes are Associated With HBV-related Hepatocellular Carcinoma Survival. J. Cancer 2023, 14 (18), 3387-3396. DOI: 10.7150/jca.88561.
NLM
Gong R, Qiu M, Cao J, Zhou Z, Wei Y, Wen Q, Lin Q, Wei X, Liang X, Jiang Y, Chen P, Wei J, Zhan S, Liu Y, Yu H. Potentially Functional Genetic Variants in the NRF2 Signaling Pathway Genes are Associated With HBV-related Hepatocellular Carcinoma Survival. J Cancer 2023; 14(18):3387-3396. doi:10.7150/jca.88561. https://www.jcancer.org/v14p3387.htm
CSE
Gong R, Qiu M, Cao J, Zhou Z, Wei Y, Wen Q, Lin Q, Wei X, Liang X, Jiang Y, Chen P, Wei J, Zhan S, Liu Y, Yu H. 2023. Potentially Functional Genetic Variants in the NRF2 Signaling Pathway Genes are Associated With HBV-related Hepatocellular Carcinoma Survival. J Cancer. 14(18):3387-3396.
This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See http://ivyspring.com/terms for full terms and conditions.