Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2024; 15(7):1916-1928. doi:10.7150/jca.91546 This issue Cite
Research Paper
Identification of a lncRNA/circRNA-miRNA-mRNA network in Nasopharyngeal Carcinoma by deep sequencing and bioinformatics analysis
Shilei Liu1,2#, Xiaoxiao Li1,3#, Qingming Xie1,2, Sai Zhang1,2, Xujun Liang1,2, Shisheng Li4, Pengfei Zhang1,2 ![]()
1. NHC Key Laboratory of Cancer Proteomics, Department of Oncology, Xiangya Hospital, Central South University, Changsha, Hunan, P.R. China, 410008.
2. National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, Hunan, P.R. China, 410008.
3. Department of Pathology, Changsha Medical College, Changsha, Hunan, P.R. China, 410219.
4. Department of Otolaryngology Head and Neck Surgery, The Second Xiangya Hospital, Central South University, Changsha, Hunan, P.R. China, 410011.
# These authors contributed equally to this work.
Received 2023-10-25; Accepted 2024-1-12; Published 2024-2-11
Citation:
Liu S, Li X, Xie Q, Zhang S, Liang X, Li S, Zhang P. Identification of a lncRNA/circRNA-miRNA-mRNA network in Nasopharyngeal Carcinoma by deep sequencing and bioinformatics analysis. J Cancer 2024; 15(7):1916-1928. doi:10.7150/jca.91546. https://www.jcancer.org/v15p1916.htm
Other stylesAbstract

Background: Accumulating evidence indicates that non-coding RNAs (ncRNA), including long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs), can function as competitive endogenous RNAs (ceRNAs) by binding to microRNAs (miRNAs) and regulating host gene expression at the transcriptional or post-transcriptional level. Dysregulation in ceRNA network regulation has been implicated in the occurrence and development of cancer. However, the lncRNA/circRNA-miRNA-mRNA regulatory network is still lacking in nasopharyngeal carcinoma (NPC).
Methods: Differentially expressed genes (DEGs) were obtained from our previous sequencing data and Gene Expression Omnibus (GEO). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes pathway (KEGG) were used to explore the biological functions of these common DEGs. Through a series of bioinformatic analyses, the lncRNA/circRNA-miRNA-mRNA network was established. In additional, the external data GSE102349 was used to test the prognostic value of the hub mRNAs through the Kaplan-Meier method.
Results: We successfully constructed a lncRNA/circRNA-miRNA-mRNA network in NPC, consisting of 16 lncRNAs, 6 miRNAs, 3 circRNAs and 10 mRNAs and found that three genes (TOP2A, ZWINT, TTK) were significantly associated with overall survival time (OS) in patients.
Conclusion: The regulatory network revealed in this study may help comprehensively elucidate the ceRNA mechanisms driving NPC, and provide novel candidate biomarkers for evaluating the prognosis of NPC.
Keywords: bioinformatics, competitive endogenous RNA, deep sequencing, nasopharyngeal carcinoma, non-coding RNAs
1. Introduction
Nasopharyngeal carcinoma (NPC) is a malignant tumor that develops from the top and lateral walls of the nasopharynx cavity. It has a distinct geographical distribution and is prevalent in east and southeast Asia, particularly in southern China[1]. In the past decade, studies have shown that its incidence has declined gradually but mortality has been dropped dramatically, which may be attributed to changes in people's lifestyle and diet[2]. It is well recognized that Epstein-Barr virus (EBV) was the first virus identified in humans to have oncogenic potential[3], and numerous studies suggested that over 90% of non-keratinizing nasopharyngeal squamous carcinoma are infected with EBV[4]. Recent advances in the etiology and molecular mechanism of NPC have led to development of new therapeutic approaches, such as programmed death-1 (PD-1) inhibitor[5]. Radiotherapy remains the primary treatment strategy for NPC[6], particularly intensity-modulated radiotherapy (IMRT) for recurrent or distant metastasis NPC. However, advanced NPC with a high risk of both locoregional recurrence and distant metastasis, which are the most common treatment failure patterns [7]. Therefore, further elucidating the molecular mechanism of NPC and finding new potential biomarkers are urgent to improve the prognosis and treatment efficacy.
Non-coding RNAs (ncRNAs), are a major component of the RNA transcripts in the human genome, accounting for more than 90%. The main types of ncRNAs include long non-coding RNAs (lncRNAs), microRNAs (miRNAs) and circular RNAs (circRNAs). Emerging evidence suggests that ncRNAs play a critical role in the development and progression of cancer[8-10]. Despite being one of the hottest topics in biomedical science today, the exact role of ncRNA in tumor is still largely unknown. LncRNAs are typically longer than 200 nucleotides, were originally believed to have no or limited protein-coding ability and are now recognized as a subtype of ncRNAs with various regulatory functions[11]. A growing body of evidence indicates that lncRNA plays an important role in cancer biology mechanisms such as epigenetic regulation or modification, cell cycle and cell differentiation. Dysfunction in lncRNA regulation leads to the occurrence of many diseases, including tumors [12,13]. Some lncRNAs can promote tumor development, while others can inhibit [14]. Lung cancer-related transcript1 (LUCATA) was the first lncRNA to be identified as having an association with smoking-related lung cancer[15]. Researchers also found LUCAT1 is involved in hepatocellular carcinoma, breast cancer and other type of cancers[16,17]. PVT1 expression is upregulated in NPC tissues and associated with poor prognostic outcomes for patients[18]. Therefore, it is thought that the specific lncRNA biomarkers can be used for the prognosis and diagnosis of NPC are of great clinical significance. MiRNAs are a class of small ncRNA molecules, include 21-24 nucleotides[19], which regulate target genes degradation or reduce target products by binding to the 3'-untranslated region (UTR) of target genes at the post-transcriptional level[20]. Circular RNA is a unique type of single-stranded, covalently closed RNA molecules, making it resistant to endonuclease and more structurally stable than other types of non-coding RNAs[21,22]. Numerous studies have shown that circRNAs can be detected in human body fluids like blood and urine[23]. Therefore, researchers are focusing on the potential application of circRNA in early cancer diagnosis and prognostic monitoring, with hopes of developing a new non-invasive testing technique[24-26]. Furthermore, circRNA can also act as sponge for miRNAs, regulating the expression of host target genes[27,28]. Dysregulation of circRNAs has been linked to the carcinogenesis of NPC. For example, Circluar RNA circRNF13 has been found downregulated in NPC tissues and regulated glucose metabolism in cells, thus inhibiting the proliferation and metastasis of NPC cells[29]. Additionally, the upregulated expression of hsa_circ_0006401 in Colorectal Cancer (CRC) tissues is associated with lymph node metastasis, and experimental studies have demonstrated that hsa_circ_0006401 enhanced the growth, migration, and metastasis of CRC and prevented CRC cell apoptosis[30].
In 2011, Salmena proposed the theory of competitive endogenous RNA (ceRNA) regulatory network, which has garnered significant interest among scientific researchers[31]. The central dogma of genetic information states that gene expression involves two processes: transcription and translation. DNA transcripts are transcribed into mRNAs that encode protein products and various ncRNAs. At the post-transcriptional level, miRNAs regulate target gene expression by recognizing the binding sites within the 3 'UTR region of mRNA[32]. In recent years, a growing evidence shows that lncRNAs and circRNAs can also competitively combine with mRNA by absorbing miRNA, forming a wide- range of competitive regulatory network[33-35]. In the context of NPC, the lncRNA-miRNA-mRNA[36]and circRNA-miRNA-mRNA[37] in the ceRNA networks have been reported. However, there is still no research simultaneously including lncRNAs and circRNAs in the ceRNA network of NPC.
In our current study, we used deep sequencing and bioinformatic analysis to establish a lncRNA/circRNA-miRNA-mRNA network in NPC and found three key genes that might could potentially serve as biomarkers for diagnosing NPC and also targets for therapeutic intervention. Our study fills an important knowledge gap in the understanding of the ceRNA network in NPC and provides new insights into the molecular mechanisms underlying NPC pathogenesis.
2. Materials and Methods
2.1. Patients and Tissue Samples
Seven fresh nasopharyngeal carcinoma samples and seven matched healthy samples were obtained from the Second Xiangya Hospital of Central South University for deep sequencing. All specimens used in this experiment were strictly diagnosed and confirmed by the Second Xiangya Hospital of Central South University. The Ethics Committee of Xiangya Hospital, Central South University approved the experiment, and all patients were informed of the experimental purpose and voluntarily signed informed consent. None of the patients included in this study had undergone radiotherapy or chemotherapy before biopsy and specimens were stored in liquid nitrogen(-180℃) immediately after biopsy.
2.2. Total RNA Extraction
The abstraction of total RNA was extraced from the frozen tissues by TRIzol (LifeTechnology,USA) following the manufacturer's guidelines. The tissue samples were placed in 1.5ml RNA enzyme-free EP tubes and ground into tissue fragments in TissueLyser LT (QIAGEN, Dusseldorf, Germany). The purity and quality of the isolated RNA of each sample were assessed by measuring the OD260/280 ratio, with values ranging from 1.8-2.1 being considered acceptable.
2.3. Library Preparation and RNA Sequencing
RNA deep sequencing was carried out by Genergy (Shanghai,China), using a total of 2ug RNA to synthesize the first strand of cDNA, according to Promega GoScript's protocol (Promega,USA). Strand-specific RNA-seq Libraries were accomplished using the TruSeq® RNA LT Sample Prep Kit v2 (Illumina, CA, USA) according to the protocals. The Ribo-Zero Gold kit (Illumina, CA, USA) was used to remove rRNA, and the remaining RNA fragments was used to synthesize double-stranded cDNA. To prepare the cDNA libraries, the double-stranded cDNA products underwent end-repair, A-tailing, and adaptor ligation. The cDNA libraries were then enriched through PCR amplification and the quality of libraries was controlled by Quant-iT™ PicoGreen® dsDNA Assay Kit (Life Technology, MA, USA). Sequencing was conducted using the Illumina Hiseq 2500 instrument (Illumina, CA, USA) with the TruSeq PE Cluster Kit v3-cBot-HS (Illumina, CA, USA).
2.4. Microarry data
Gene Expression Omnibus (GEO) is a valuable public functional genomics repository, which collects microarrays, high-throughput sequencing data. In this study, we utilized GEO database to download GSE61218 dataset, which contains expression profiles of mRNAs in 10 NPC samples and 6 normal samples. Furthermore, we downloaded GSE102349 dataset (containing 113 NPC samples) for validation of prognostic model.
2.5. Data analysis and Identification differentially expressed genes
We analyzed our previous sequencing data and identified 2397 significantly differentially expressed mRNAs (DEmRNAs) using a cutoff criteria of p-value <0.05, false positive rate (FDR) <= 0.05 and the absolute value of log-fold change IlogFCI>=2 and 346 differentially expressed circRNAs (DEcircRNAs) with p-value<0.05, IlogFCI>=2. To screen DEmRNAs between NPC samples and normal samples in GSE61218 dataset, we used the “limma” package in R software (version4.1.3), with p<0.05 and IlogFCI>=1 as the thresholds to identify significantly differentially expressed mRNAs. We visualized the DEGs using the heatmap and volcano plot drawn by the heatmap and ggplot2 packages, respectively. Common differentially expressed genes (commonDEGs) were shown on the Venn diagram.
2.6. Functional enrichment analysis of DEGs
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analysis were performed for the common DEGs obtained above by using the ClusterProfiler package. GO enrichment analysis included biologic process (BP), cellular components (CC), and molecular function (MF) and p-value < 0.05 as the criteria for significant enrichment. The ggplot2 package was used for the visualization of functional enrichment analysis results.
2.7. Protein-protein Interaction analysis (PPI)
The Search Tool for the Retrieval of Interacting Genes database[38] (STRING) is a very powerful online bioinformatics tool that enables the analysis of protein interactions. The minimum interaction score was set to 0.7 as high confidence. The protein-protein interaction networks of common DEmRNAs were visualized using Cytoscape[39] software (version, 3.9.0). To identify key node modules in the network, we employed the MCC algorithm, a Cytoscape plugin. According to the MCC algorithm, the top 10 genes with high scores are screened out as the hub mRNAs for further analysis and research.
2.8. Identification of miRNAs
The miRDB[40], miRWalk[41] and Targetscan[42] databases were used to predict the upstream miRNAs for the hub mRNAs, in order to ensure the accuracy and reliability of the research, we only choose the predicted miRNAs from the aforementioned databases as final common miRNAs. The expression level of miRNA was predicted by OncomiR[43], and p < 0.05 was considered to have statistical significance.
2.9. Identification of lncRNAs and circRNAs
We utilized two databases, LncBase[44] and Starbase[45]to predict the upstream lncRNAs of hub mRNAs. The resulting common lncRNAs were then analyzed and used to construct network. The expression of circRNAs was predicted by circBank[46] and DEcircRNAs were obtained the NPC sequencing data of our research group. Finally, circRNAs from database predictions and sequencing data were selected as the final common circRNAs.
2.10. Verification and prognostic analysis of hub mRNAs
The GEPIA[47] is a robust online website that has been developed based on TCGA and GTEx data that can be used for personalized analysis. We used the GEPIA to validate the hub mRNAs. To further evaluate the prognostic significance of hub mRNAs, we used the GSE102349 as external validation cohort. We performed survival analysis, and considered p < 0.05 as statistically significant.
2.11. Construction of the ceRNA network
Based on the competitive endogenous RNA regulatory network mechanisms, we successfully constructed a lncRNA-circRNA-miRNA-mRNA network, consisting of 16 lncRNAs, 6 miRNAs, 3 circRNAs and 10 mRNAs.
3. Results
3.1. Identification of Differentially Expressed Genes between Nasopharyngeal Carcinoma Tissues and Normal Tissues
Figure 1 shows the overall procedure of the study. We identified 2397 DEmRNAs and 346 DEcircRNAs (Figures 2A, B) from the sequencing data. Additionally, we extracted 1268 DEmRNAs from the GSE61218 dataset (Figures 2C, D). Finally, 313 common DEGs were obtained from between sequencing data and GSE61218 dataset (Figure 2E).
3.2. GO Functions and KEGG Pathway Enrichment Analysis
GO functions and KEGG pathway enrichment analysis was performed on the common DEGs. Figures 3A-D show the enriched GO terms and KEGG pathways for common DEGs. The common DEGs were enriched in nuclear division, DNA recombination, sister chromatid segregation, organelle fission, nuclear chromosome segregation, mitotic sister chromatid segregation in the biologic process category; chromosome, centromeric region, chromosomal region, condensed chromosome, kinetochore in the cellular components; cytoskeletal motor activity, virus receptor activity, ATP-dependent activity, acting on DNA in the molecular functions (Figures 3A, B), the enriched KEGG pathways for common DEGs mainly were involved in Mismatch repair, Homologous recombination, Cell cycle, Metabolism of xenobiotics by cytochrome P450, Ferroptosis, p53 signaling pathway etc (Figures 3C, D).
3.3. Construction of Protein-Protein Interaction Network and Hub Genes Identification
Based on the STRING database and Cytoscape software, the protein-protein interaction network of these common DEGs was built, consisting of 581 edges and 313 nodes (Figure 4A). MCC algorithm, a plug-in for Cytoscape, the top10 genes (Figure 4B) (CCNA2, TTK, CDK1, TOP2A, NDC80, PBK, CCNB2, AURKB, ZWINT, PTTG1) with high scores were selected as the hub mRNAs from the network according its method (Table 1), and hub mRNAs were verified by GEPIA database (Figures 5A-J).
Figure 1
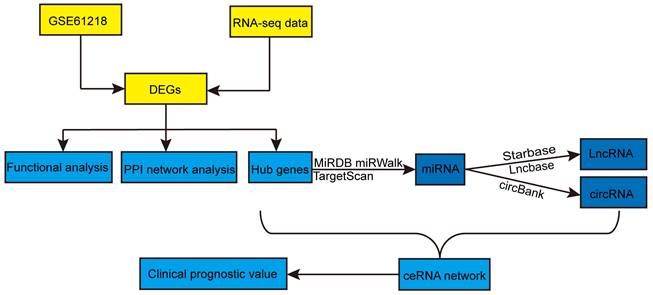
Flowchart of this study. DEGs: differentially expressed genes; PPI: protein-protein interaction; miRNAs: microRNAs; lncRNAs: long non-coding RNAs; circRNAs: circular RNAs; ceRNA: competitive endogenous RNA.
Figure 2
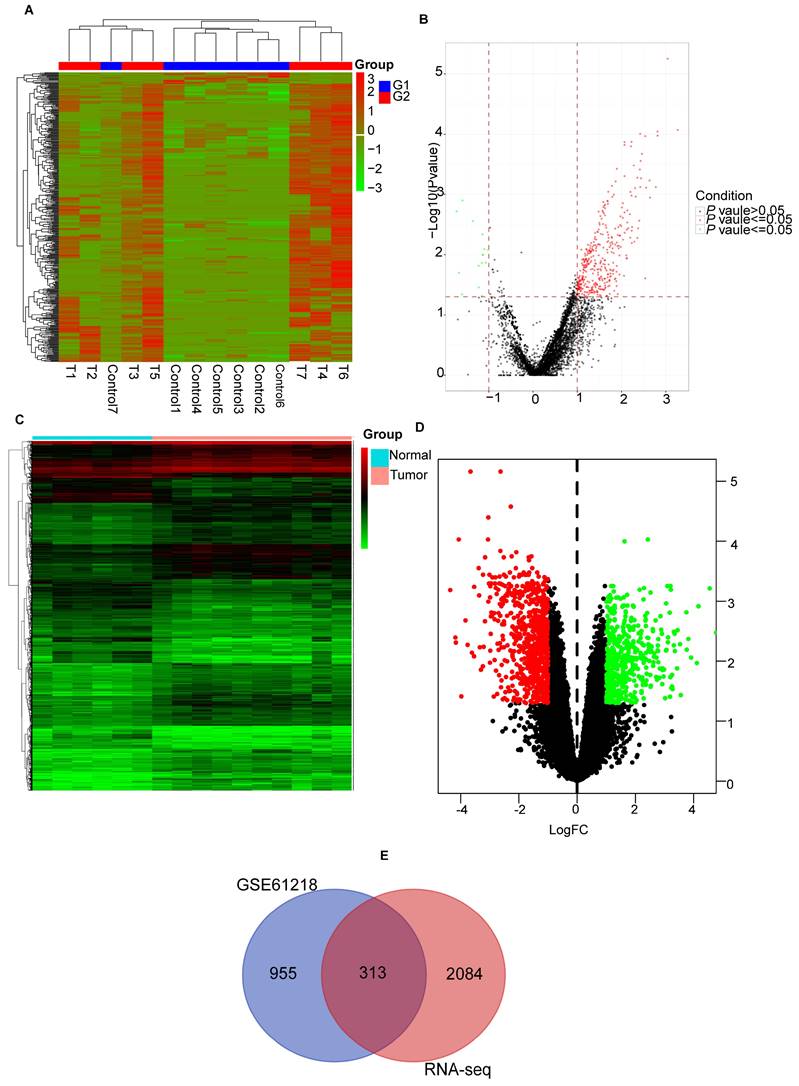
Identification of differentially expressed genes. (A, B) heatmap and volcano plot of Sequencing data. (C, D) heatmap and volcano plot of GSE61218. (E) Venn Diagram of DEGs. DEGs: differentially expressed genes.
Figure 3
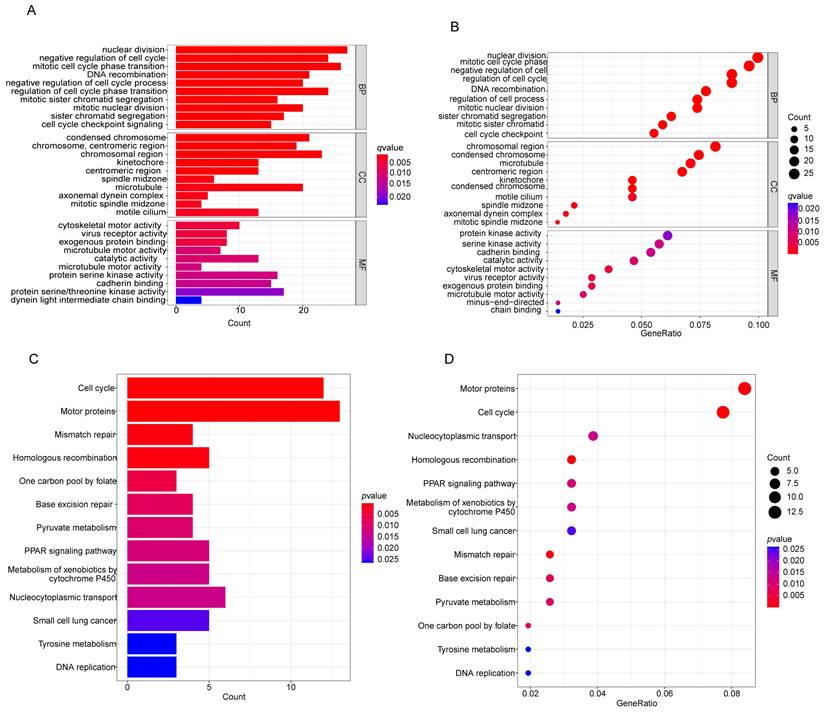
Functional enrichment analysis of common differentially expressed genes. (A, B) The results of GO enrichment analysis of the common DEGs, including BP, CC and MF. (C, D) The results of KEGG pathways analysis of the common DEGs. DEGs: differentially expressed genes, GO: Gene Ontology, KEGG: Kyoto Encyclopedia of Genes and Genomes, BP: biologial process, CC:cellular component, MF: molecular function.
Table 1
The top ten mRNAs ranked as hub mRNAs by the MCC method.
| Rank | Name | Score | |
|---|---|---|---|
| 1 | CCNA2 | 7.55E+14 | |
| 2 | TTK | 7.55E+14 | |
| 3 | CDK1 | 7.55E+14 | |
| 4 | TOP2A | 7.55E+14 | |
| 5 | NDC80 | 7.55E+14 | |
| 6 | PBK | 7.55E+14 | |
| 7 | CCNB2 | 7.55E+14 | |
| 8 | AURKB | 7.55E+14 | |
| 9 | ZWINT | 7.55E+14 | |
| 10 | PTTG1 | 7.54E+14 |
3.4. Identification of lncRNAs, miRNAs and circRNAs, and Construction of a ceRNA network
Overall 26 potential upstream miRNAs (Figure 6B) were found by searching in miRDB, miRWalk, and Targetscan databases, based on the hub mRNAs. Out of these, 6miRNAs were found to be significantly differentially expressed in the OncomiR database (Table 2). Using these miRNAs, a total of 16 lncRNAs were predicted from the Lncbase and Starbase databases (Figure 6A). The circBank identified 481 interactions between circRNAs and miRNAs, we chosen three circRNAs (Figure 6C) that overlapped with DEcircRNAs (Table 3). Finally, the lncRNA-circRNA-miRNA-mRNA regulatory network was successfully constructed, consisting of 16 lncRNAs (LINC01963, EBLN3P, NEAT1, XIST, AC016717.2, PVT1, AC107068.1, AC093297.2, JPX, AC092279.2, AC055713.1, LINC00052, AC092279.1, AC145207.5, AL035071.1, MALAT1), 6 miRNAs (has-miR-410-3p, has-miR-495-3p, has-miR-148a-3p, hsa-miR-655-3p, hsa-miR-152-3p, hsa-miR-524-5p), 3 circRNAs (hsa_circ_0001359, has-circ_0002458, hsa_circ_0008967), 10 mRNAs (CCNA2, TTK, CDK1, TOP2A, NDC80, CCNB2, AURKB, ZWINT, PTTG1) (Figure 6D).
Figure 4
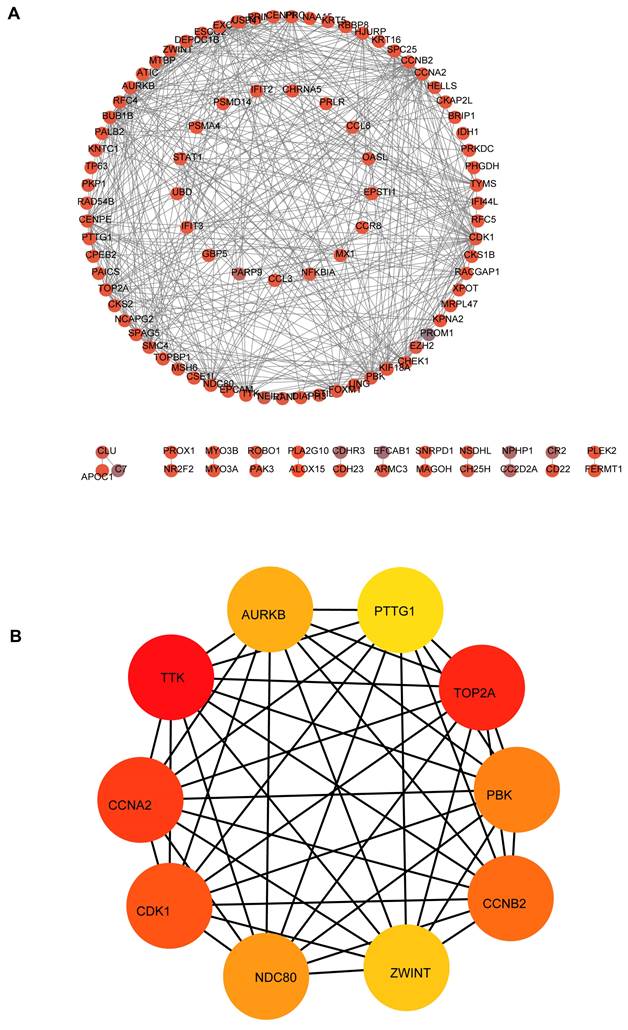
Construction of PPI network and identified hub mRNAs in the network. (A) PPI network of common DEGs. (B) hub module in the network.
Figure 5
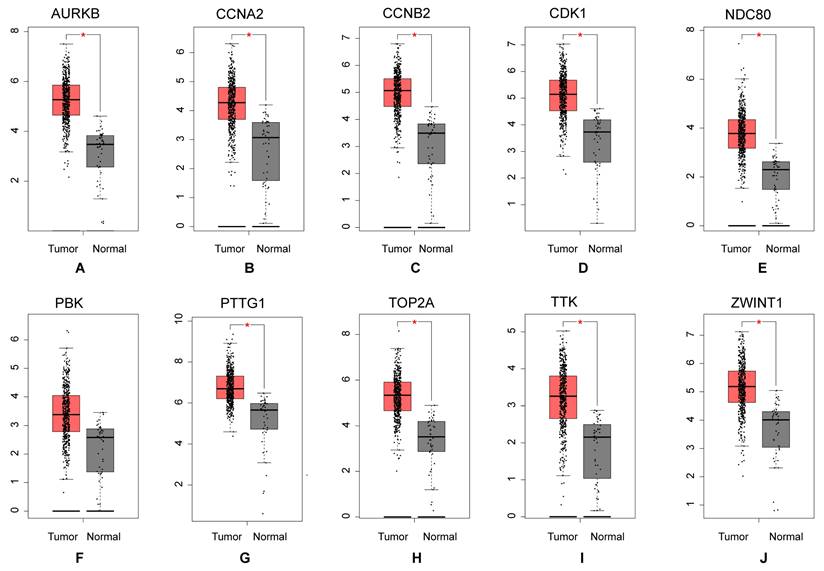
Expression level analysis of hub mRNAs. The red presented tumor, while the gray indicated normal. (A) AURKB: (B) CCNA2: (C) CCNB2: (D) CDK1: (E) NDC80: (F) PBK: (G) PTTG1: (H) TOP2A: (I) TTK: (J) ZWINT.
Table 2
Expression level of the predicted miRNAs in OncomiR.
| Name | t-test-p-value | The expression level of miRNA | |
|---|---|---|---|
| Tumor tissue | Normal tissue | ||
| hsa-miR-410-3p | 6.20E-05 | 3.96 | 3.61 |
| hsa-miR-495-3p | 9.58E-04 | 2.92 | 2.71 |
| hsa-miR-148a-3p | 2.20E-03 | 14.61 | 1.89 |
| hsa-miR-655-3p | 1.24E-02 | 1.66 | 1.57 |
| hsa-miR-152-3p | 2.00E-02 | 8.92 | 8.63 |
| hsa-miR-524-5p | 2.68E-02 | 0.18 | 0.03 |
Table 3
CircRNAs expression levels in tumor tissues and normal tissues.
| CircRNA | Normal tissue | Tumor tissue |
|---|---|---|
| has_circ_0001359 | 0.71 | 2.49 |
| has_circ_0002458 | 0 | 1.03 |
| has_circ_0008967 | 0.86 | 2.55 |
3.5. Prognostic analysis of hub mRNAs
We performed prognostic analysis of these hub mRNAs through external dataset GSE102349 (containing 88 complete NPC patients' survival time) (Figures 7A-J). and found that the high expression of TOP2A (Figure 7I), ZWINT (Figure 7H) and TTK (Figure 7G) were significantly associated with overall survival time (p<0.05).
4. Discussion
In this study, we systematically explored the ceRNA regulatory in various tumors. The ceRNA regulatory network provides a more comprehensive explanation for interactions among diverse RNA types at the gene level. To better understand the role of ceRNA network in NPC, we constructed a ceRNA regulatory network in NPC using RNA sequencing and bioinformatics and explored the prognostic value of core genes. Through RNA-seq and GSE61218 dataset, we identified 313 DEGs. GO and KEGG enrichment analysis suggested that cell cycle progression, energy metabolism and PPAR signaling pathway were involved in the formation of NPC. We have also pinpointed the top 10 hub genes within the PPI network associated with NPC. Additionally, prognostic analysis of the core genes using the external data GSE102349 revealed a significant association between high expression levels of TOP2A, ZWINT, and TTK and poor prognosis in NPC. These findings further underscore the potential clinical relevance of these genes as prognostic indicators in the context of NPC.
The exponential growth and evolution of sequencing technologies have revolutionized our understanding of the genomic landscape, particularly with regard to ncRNAs. The accelerated pace of sequencing advancements, exemplified by techniques such as RNA-seq, has led to a surge in the identification and characterization of diverse classes of ncRNAs. These non-coding transcripts, previously overlooked or underestimated, are now recognized as integral players in the regulation of cellular processes and disease pathogenesis. The most widely studied ncRNA is lncRNA, and numerous studies have shown that lncRNA is not only involved in tumor development, but also promises to be used as a diagnostic marker for tumors. For instance, highly expressed lncRNA RP11-624L4.1 promotes NPC cell cycle progression through interaction with CDK4 and is associated with poor prognosis[48]. Liang et al reported that the immune-associated nine-lncRNAs signature can serve as a promising biomarker of for metastasis prediction through RNA-seq in NPC[49].
Figure 6
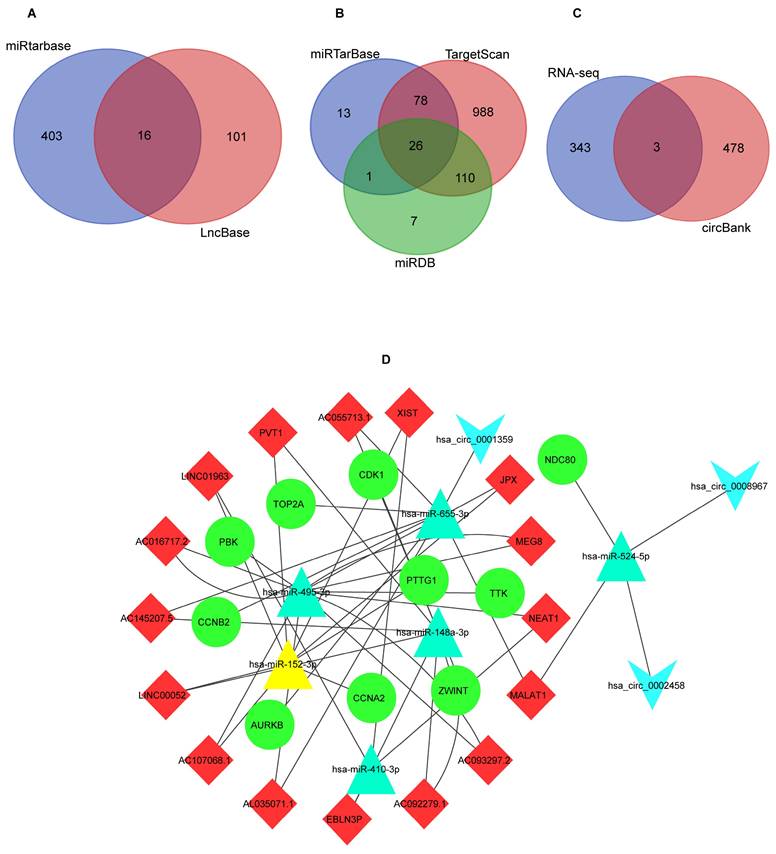
The results of ceRNA network prediction and construction of ceRNA network. (A) Venn diagram of common lncRNAs. (B) Venn diagram of common miRNAs. (C) Venn diagram of common circRNAs. (D) The ceRNA network. As shown in Figure D, diamond represents lncRNAs, college represents mRNAs, triangle represents miRNAs, and inverted triangle represents circRNAs. ceRNA: competitive endogenous RNA; lncRNAs: long non-coding RNAs; miRNA: microRNAs; circRNAs: circular RNAs.
Figure 7
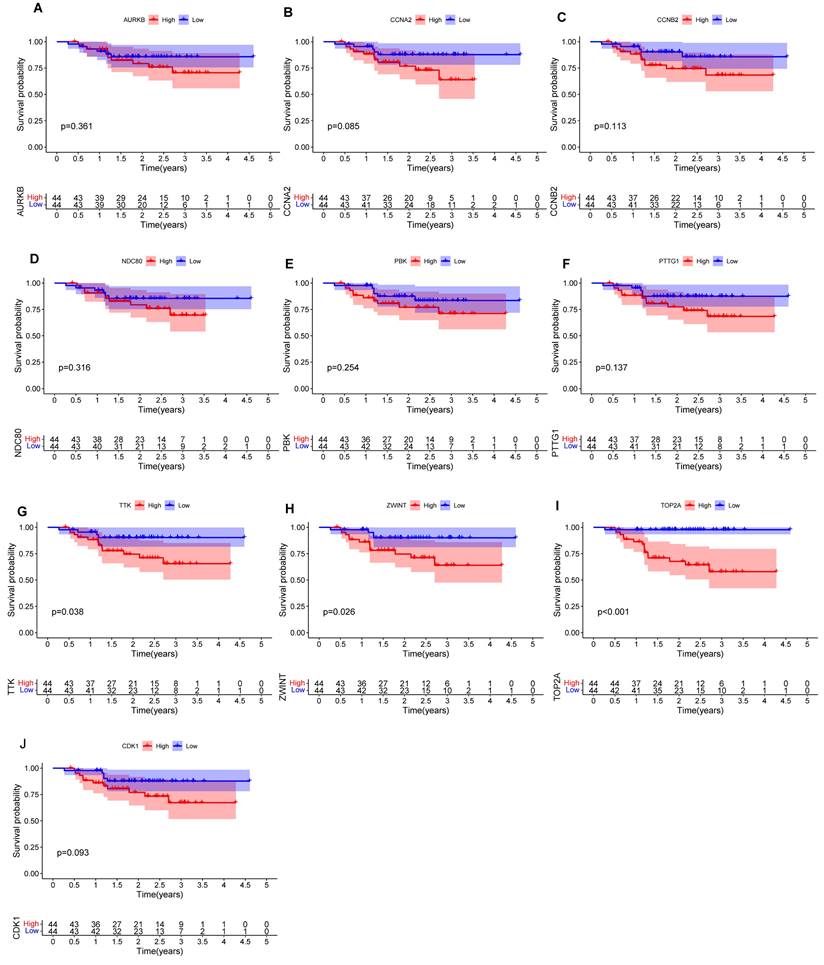
The survival curve of hub mRNAs. (A) AURKB: (B) CCNA2: (C) CCNB2: (D) NDC80: (E) PBK: (F) PTTG1: (G) TTK: (H) ZWINT: (I) TOP2A (J) CDK1.
After the proposal of the ceRNA hypothesis, a growing body of evidence shows that the ceRNA regulatory network is involved in the occurrence and development of many tumors[50]. CircRNAs and lncRNAs, can function as miRNA sponge, which can regulate the expression of parental genes[51,52]. Several studies have delved into and elucidated the significance of ceRNAs in both tumor prognosis and the pathogenesis of diverse cancers. For instance, the lncRNA XIST, as ceRNA, competitively bind to miR-491-5p with NEK5 and promote migration and invasion malignant of NPC[53]. Similarly, MALAT1 promoted proliferation, invasion and EMT of NPC cells through de-repressing Capn4 by sponging miR-124[54]. Furthermore, CircTP63, which binds competitively to miR-873-3p, abolishes the inhibition of miR-873-3p on FOXM1 and then promotes cell proliferation in lung squamous cell carcinoma[55]. CircCCNB1 can act as a miR-106b-5p inhibited GPM6A expression to promote hepatocellular carcinoma progression[56]. However, the ceRNA regulatory networks containing both lncRNAs and cicRNAs are uncommon in NPC. In our study, we conducted transcriptome sequencing on NPC to reveal the role of the ceRNA regulatory network in the tumorgenesis of NPC.
Here, we successfully constructed a regulatory network consisting of 16 lncRNAs, 6 miRNAs, 10 mRNAs and 3 circRNAs. In the ceRNA network, the 10 mRNAs were validated by GSE102349 and found three mRNAs (TOP2A, ZWINT and TTK) could be key factors in poor prognosis of NPC patients. Several studies have demonstrated that the mRNAs identified in this study promote tumor cell proliferation and cell cycle progression[57], participate in substrate ubiquitination[58], contribute to the maintenance of genome stability and organization[59], and play a role in other biological mechanisms of tumor formation. Lan et al. suggested that overexpression of TOP2A was positively associated with the aggressiveness of NPC and could be a key factor in poor prognosis of NPC patients[60]. Interestingly, studies of ZWINT and TTK in NPC have not been reported, while the exact role of the three hub mRNAs with NPC carcinogenesis requires further exploration.
In conclusion, our study successfully built a lncRNA-circRNA-miRNA-mRNA regulatory network in NPC and identified three prognostic mRNAs that could serve as diagnostic biomarkers and targets for early treatment of NPC. The ceRNA network plays a pivotal role in the development and progress of NPC and regulates cancer-related pathways. Our research provides new insights for further exploring the molecular mechanism of NPC tumorigenesis.
Abbreviations
circRNAs: circular RNAs
ceRNAs: competitive endogenous RNAs
CRC: Colorectal Cancer
DEGs: differentially expressed genes
GEO: Gene Expression Omnibus
GO: Gene Ontology
IMRT: intensity-modulated radiotherapy
KEGG: Kyoto Encyclopedia of Genes and Genomes pathway
LUCATA: Lung cancer-related transcript1
lncRNAs: long non-coding RNAs
MCC: Maximal Clique Centrality
miRNAs: microRNAs
NPC: nasopharyngeal carcinoma
ncRNA: non-coding RNAs
PD-1: programmed death-1
PPI: protein-protein interaction
Acknowledgements
Thanks for the help of Zhang group members.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 82073008), Key Research and Development Project of Hunan Province (Nos. 2020SK2071), and Natural Science Foundation of Hunan Province (Nos. 2020JJ4924).
Author contributions
SL and PZ: design and methodology. SL, XXL and XL: software and data procession. LS and SZ: validation. SL, SZ, and XXL: writing-original draft preparation. XL and PZ: writing review, editing, and supervision. All authors contributed to the article and approved the submitted version.
Data availability statement
The information presented in this article can be accessed through the GEO database, which can be found at: https://www.ncbi.nlm.nih.gov/geo. We have uploaded the raw sequencing data to the GEO database with serial number GSE227541 (data are to be held confidential until Dec 31, 2023). You may view our study at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227541.
Ethics Statement
This study involving human participants were reviewed and approved by the ethics committee of Xiangya Hospital of Central South University. Prior to participation, all individuals were informed of the study's purpose and voluntarily signed informed consent.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chang ET, Adami H-O. The Enigmatic Epidemiology of Nasopharyngeal Carcinoma. Cancer Epidemiol Biomarkers Prev. 2006;15:1765-77
2. Chen Y-P, Chan ATC, Le Q-T, Blanchard P, Sun Y, Ma J. Nasopharyngeal carcinoma. The Lancet. 2019;394:64-80
3. Yuan L. et al. EBV infection-induced GPX4 promotes chemoresistance and tumor progression in nasopharyngeal carcinoma. Cell Death Differ [Internet]. 2022 [cited. 2023 Feb 12];29. Available from: https://pubmed.ncbi.nlm.nih.gov/35105963/
4. Al-Anazi AE, Alanazi BS, Alshanbari HM, Masuadi E, Hamed ME, Dandachi I. et al. Increased Prevalence of EBV Infection in Nasopharyngeal Carcinoma Patients: A Six-Year Cross-Sectional Study. Cancers. 2023;15:643
5. Wang F-H, Wei X-L, Feng J, Li Q, Xu N, Hu X-C. et al. Efficacy, Safety, and Correlative Biomarkers of Toripalimab in Previously Treated Recurrent or Metastatic Nasopharyngeal Carcinoma: A Phase II Clinical Trial (POLARIS-02). J Clin Oncol Off J Am Soc Clin Oncol. 2021;39:704-12
6. Guan S, Wei J, Huang L, Wu L. Chemotherapy and chemo-resistance in nasopharyngeal carcinoma. Eur J Med Chem. 2020;207:112758
7. Current Status and Future Perspectives about Molecular Biomarkers of Nasopharyngeal Carcinoma - PubMed [Internet]. [cited 2023 Feb 20]. Available from: https://pubmed.ncbi.nlm.nih.gov/34298701/.
8. Willingham AT, Orth AP, Batalov S, Peters EC, Wen BG, Aza-Blanc P. et al. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science. 2005;309:1570-3
9. Cai L, Ye Y, Jiang Q, Chen Y, Lyu X, Li J. et al. Author Correction: Epstein-Barr virus-encoded microRNA BART1 induces tumour metastasis by regulating PTEN-dependent pathways in nasopharyngeal carcinoma. Nat Commun. 2020;11:3437
10. Mu M, Niu W, Zhang X, Hu S, Niu C. LncRNA BCYRN1 inhibits glioma tumorigenesis by competitively binding with miR-619-5p to regulate CUEDC2 expression and the PTEN/AKT/p21 pathway. Oncogene. 2020;39:6879-92
11. Batista PJ, Chang HY. Long Noncoding RNAs: Cellular Address Codes in Development and Disease. Cell. 2013;152:1298-307
12. Heskett MB, Vouzas AE, Smith LG, Yates PA, Boniface C, Bouhassira EE. et al. Epigenetic control of chromosome-associated lncRNA genes essential for replication and stability. Nat Commun. 2022;13:6301
13. Wang R, Ma Z, Feng L, Yang Y, Tan C, Shi Q. et al. LncRNA MIR31HG targets HIF1A and P21 to facilitate head and neck cancer cell proliferation and tumorigenesis by promoting cell-cycle progression. Mol Cancer. 2018;17:162
14. Bhan A, Soleimani M, Mandal SS. Long Noncoding RNA and Cancer: A New Paradigm. Cancer Res. 2017;77:3965-81
15. Xing C, Sun S, Yue Z-Q, Bai F. Role of lncRNA LUCAT1 in cancer. Biomed Pharmacother. 2021;134:111158
16. Zheng A, Song X, Zhang L, Zhao L, Mao X, Wei M. et al. Long non-coding RNA LUCAT1/miR-5582-3p/TCF7L2 axis regulates breast cancer stemness via Wnt/β-catenin pathway. J Exp Clin Cancer Res. 2019;38:305
17. Jiao Y, Li Y, Ji B, Cai H, Liu Y. Clinical Value of lncRNA LUCAT1 Expression in Liver Cancer and its Potential Pathways. J Gastrointest Liver Dis JGLD. 2019;28:439-47
18. Lu J, Xu F, Lu H. LncRNA PVT1 regulates ferroptosis through miR-214-mediated TFR1 and p53. Life Sci. 2020;260:118305
19. Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597-610
20. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203-22
21. Chen L-L. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol. 2016;17:205-11
22. Arnaiz E, Sole C, Manterola L, Iparraguirre L, Otaegui D, Lawrie CH. CircRNAs and cancer: Biomarkers and master regulators. Semin Cancer Biol. 2019;58:90-9
23. Zhang Z, Yang T, Xiao J. Circular RNAs: Promising Biomarkers for Human Diseases. EBioMedicine. 2018;34:267-74
24. Wang S, Dong Y, Gong A, Kong H, Gao J, Hao X. et al. Exosomal circRNAs as novel cancer biomarkers: Challenges and opportunities. Int J Biol Sci. 2021;17:562-73
25. Roy S, Kanda M, Nomura S, Zhu Z, Toiyama Y, Taketomi A. et al. Diagnostic efficacy of circular RNAs as noninvasive, liquid biopsy biomarkers for early detection of gastric cancer. Mol Cancer. 2022;21:42
26. Peng Z, Hu Q, Fang S, Zhang X, Hong X, Tao L. et al. Circulating circTOLLIP serves as a diagnostic biomarker for liquid biopsy in non-small cell lung cancer. Clin Chim Acta Int J Clin Chem. 2021;523:415-22
27. Chen Q, Wang H, Li Z, Li F, Liang L, Zou Y. et al. Circular RNA ACTN4 promotes intrahepatic cholangiocarcinoma progression by recruiting YBX1 to initiate FZD7 transcription. J Hepatol. 2022;76:135-47
28. Misir S, Wu N, Yang BB. Specific expression and functions of circular RNAs. Cell Death Differ. 2022;29:481-91
29. Mo Y, Wang Y, Zhang S, Xiong F, Yan Q, Jiang X. et al. Circular RNA circRNF13 inhibits proliferation and metastasis of nasopharyngeal carcinoma via SUMO2. Mol Cancer. 2021;20:112
30. Zhang C, Zhou X, Geng X, Zhang Y, Wang J, Wang Y. et al. Circular RNA hsa_circ_0006401 promotes proliferation and metastasis in colorectal carcinoma. Cell Death Dis. 2021;12:443
31. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell. 2011;146:353-8
32. Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol. 2018;141(4):1202-1207
33. Hansen TB, Kjems J, Damgaard CK. Circular RNA and miR-7 in cancer. Cancer Res. 2013;73:5609-12
34. Liu Y, Cheng Z, Pang Y, Cui L, Qian T, Quan L. et al. Role of microRNAs, circRNAs and long noncoding RNAs in acute myeloid leukemia. J Hematol OncolJ Hematol Oncol. 2019;12:51
35. Mirzaei S, Gholami MH, Hushmandi K, Hashemi F, Zabolian A, Canadas I. et al. The long and short non-coding RNAs modulating EZH2 signaling in cancer. J Hematol OncolJ Hematol Oncol. 2022;15:18
36. Zou Z, Liu S, Ha Y, Huang B. Construction and Analysis of lncRNA-Mediated ceRNA Network in Nasopharyngeal Carcinoma Based on Weighted Correlation Network Analysis. BioMed Res Int. 2020;2020:1-16
37. Lin X, Wang S, Lin K, Zong J, Zheng Q, Su Y. et al. Competitive Endogenous RNA Landscape in Epstein-Barr Virus Associated Nasopharyngeal Carcinoma. Front Cell Dev Biol. 2021;9:782473
38. Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S. et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49:D605-12
39. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498-504
40. Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020;48:D127-31
41. Sticht C, De La Torre C, Parveen A, Gretz N. miRWalk: An online resource for prediction of microRNA binding sites. PloS One. 2018;13:e0206239
42. Agarwal V, Bell GW, Nam J-W, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. eLife. 2015;4:e05005
43. Wong NW, Chen Y, Chen S, Wang X. OncomiR: an online resource for exploring pan-cancer microRNA dysregulation. Bioinforma Oxf Engl. 2018;34:713-5
44. Karagkouni D, Paraskevopoulou MD, Tastsoglou S, Skoufos G, Karavangeli A, Pierros V. et al. DIANA-LncBase v3: indexing experimentally supported miRNA targets on non-coding transcripts. Nucleic Acids Res. 2020;48:D101-10
45. starBase v2.0. decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data - PubMed [Internet]. [cited. 2023 Feb 20]. Available from: https://pubmed.ncbi.nlm.nih.gov/24297251/
46. Circbank. a comprehensive database for circRNA with standard nomenclature - PubMed [Internet]. [cited. 2023 Feb 20]. Available from: https://pubmed.ncbi.nlm.nih.gov/31023147/
47. GEPIA2021. integrating multiple deconvolution-based analysis into GEPIA - PubMed [Internet]. [cited. 2023 Feb 20]. Available from: https://pubmed.ncbi.nlm.nih.gov/34050758/
48. Zhou L, Liu R, Liang X, Zhang S, Bi W, Yang M. et al. lncRNA RP11-624L4.1 Is Associated with Unfavorable Prognosis and Promotes Proliferation via the CDK4/6-Cyclin D1-Rb-E2F1 Pathway in NPC. Mol Ther Nucleic Acids. 2020;22:1025-39
49. Liang Y-L, Zhang Y, Tan X-R, Qiao H, Liu S-R, Tang L-L. et al. A lncRNA signature associated with tumor immune heterogeneity predicts distant metastasis in locoregionally advanced nasopharyngeal carcinoma. Nat Commun. 2022;13:2996
50. Karreth FA, Pandolfi PP. ceRNA cross-talk in cancer: when ce-bling rivalries go awry. Cancer Discov. 2013;3:1113-21
51. Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20:675-91
52. Boon RA, Jaé N, Holdt L, Dimmeler S. Long Noncoding RNAs: From Clinical Genetics to Therapeutic Targets? J Am Coll Cardiol. 2016;67:1214-26
53. Zhao C-H, Bai X-F, Hu X-H. Knockdown of lncRNA XIST inhibits hypoxia-induced glycolysis, migration and invasion through regulating miR-381-3p/NEK5 axis in nasopharyngeal carcinoma. Eur Rev Med Pharmacol Sci. 2020;24:2505-17
54. Shi B, Wang Y, Yin F. MALAT1/miR-124/Capn4 axis regulates proliferation, invasion and EMT in nasopharyngeal carcinoma cells. Cancer Biol Ther. 2017;18:792-800
55. Cheng Z, Yu C, Cui S, Wang H, Jin H, Wang C. et al. circTP63 functions as a ceRNA to promote lung squamous cell carcinoma progression by upregulating FOXM1. Nat Commun. 2019;10:3200
56. Liu Y-M, Cao Y, Zhao P-S, Wu L-Y, Lu Y-M, Wang Y-L. et al. CircCCNB1 silencing acting as a miR-106b-5p sponge inhibited GPM6A expression to promote HCC progression by enhancing DYNC1I1 expression and activating the AKT/ERK signaling pathway. Int J Biol Sci. 2022;18:637-51
57. Chen P, He Z, Wang J, Xu J, Jiang X, Chen Y. et al. Hypoxia-Induced ZWINT Mediates Pancreatic Cancer Proliferation by Interacting With p53/p21. Front Cell Dev Biol. 2021;9:682131
58. Chen X, Yu C, Gao J, Zhu H, Cui B, Zhang T. et al. A novel USP9X substrate TTK contributes to tumorigenesis in non-small-cell lung cancer. Theranostics. 2018;8:2348-60
59. Pommier Y, Nussenzweig A, Takeda S, Austin C. Human topoisomerases and their roles in genome stability and organization. Nat Rev Mol Cell Biol. 2022;23:407-27
60. Lan J, Huang H-Y, Lee S-W, Chen T-J, Tai H-C, Hsu H-P. et al. TOP2A overexpression as a poor prognostic factor in patients with nasopharyngeal carcinoma. Tumour Biol J Int Soc Oncodevelopmental Biol Med. 2014;35:179-87
Author contact
![]() Corresponding author: Pengfei Zhang, zhangpf690421edu.cn.
Corresponding author: Pengfei Zhang, zhangpf690421edu.cn.
Citation styles
APA
Liu, S., Li, X., Xie, Q., Zhang, S., Liang, X., Li, S., Zhang, P. (2024). Identification of a lncRNA/circRNA-miRNA-mRNA network in Nasopharyngeal Carcinoma by deep sequencing and bioinformatics analysis. Journal of Cancer, 15(7), 1916-1928. https://doi.org/10.7150/jca.91546.
ACS
Liu, S.; Li, X.; Xie, Q.; Zhang, S.; Liang, X.; Li, S.; Zhang, P. Identification of a lncRNA/circRNA-miRNA-mRNA network in Nasopharyngeal Carcinoma by deep sequencing and bioinformatics analysis. J. Cancer 2024, 15 (7), 1916-1928. DOI: 10.7150/jca.91546.
NLM
Liu S, Li X, Xie Q, Zhang S, Liang X, Li S, Zhang P. Identification of a lncRNA/circRNA-miRNA-mRNA network in Nasopharyngeal Carcinoma by deep sequencing and bioinformatics analysis. J Cancer 2024; 15(7):1916-1928. doi:10.7150/jca.91546. https://www.jcancer.org/v15p1916.htm
CSE
Liu S, Li X, Xie Q, Zhang S, Liang X, Li S, Zhang P. 2024. Identification of a lncRNA/circRNA-miRNA-mRNA network in Nasopharyngeal Carcinoma by deep sequencing and bioinformatics analysis. J Cancer. 15(7):1916-1928.
This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See http://ivyspring.com/terms for full terms and conditions.