Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2024; 15(9):2538-2548. doi:10.7150/jca.90822 This issue Cite
Research Paper
PHGDH is Key to a Prognostic Multigene Signature and a Potential Therapeutic Target in Acute Myeloid Leukemia
Jiagui Zhong1,2,3*, Kezhi Huang1,2,4*, Shaofan Xie1,2, Ailian Tan1,2, Jiaqin Peng1,2, Danian Nie1,2, Liping Ma1,2 ![]() , Yiqing Li1,2
, Yiqing Li1,2 ![]()
1. Department of Hematology, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou 510120, China.
2. Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou 510120, China.
3. Department of Hematology, The Affiliated Kashi Hospital, Sun Yat-sen University, Kashi 844099, China.
4. Internal Medicine Ward I, JieXi People's Hospital (Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University-JieXi Medical Center), JieYang 515499, China
*JZ and KH contributed equally to this work.
Received 2023-10-6; Accepted 2024-2-4; Published 2024-3-11
Citation:
Zhong J, Huang K, Xie S, Tan A, Peng J, Nie D, Ma L, Li Y. PHGDH is Key to a Prognostic Multigene Signature and a Potential Therapeutic Target in Acute Myeloid Leukemia. J Cancer 2024; 15(9):2538-2548. doi:10.7150/jca.90822. https://www.jcancer.org/v15p2538.htm
Other stylesAbstract

As a rate-limiting enzyme for the serine biosynthesis pathway (SSP) in the initial step, phosphoglycerate dehydrogenase (PHGDH) is overexpressed in many different tumors, and pharmacological or genetic inhibition of PHGDH promotes antitumor effects. In the present research, by analyzing several acute myeloid leukemia (AML) datasets in the Gene Expression Omnibus (GEO), we identified prognosis-related genes and constructed a multigene signature by univariate, multivariate Cox regression and LASSO regression. Subsequently, the multigene signature was confirmed through Cox, Kaplan-Meier, and ROC analyses in the validation cohort. Moreover, PHGDH acted as a risk factor and was correlated with inferior overall survival. We further analysed other datasets and found that PHGDH was overexpressed in AML. Importantly, the expression of PHGDH was higher in drug-resistant AML compared to drug-sensitive ones. In vitro experiments showed that inhibition of PHGDH induced apoptosis and reduced proliferation in AML cells, and these antitumor effects could be related to the Bcl-2/Bax signaling pathway by the noncanonical or nonmetabolic functions of PHGDH. In summary, we constructed a twenty-gene signature that could predicate prognosis of AML patients and found that PHGDH may be a potential target for AML treatment.
Keywords: Gene signature, Acute myeloid leukemia, Overall survival, PHGDH, Therapeutic target
Introduction
Acute myeloid leukemia (AML) is a heterogeneous haematopoietic malignancy arising from the malignant clonal expansion of undifferentiated myeloid precursors with complex molecular pathogenesis, which includes a series of cytogenetic or chromosomal aberrations [1]. Its pathological features are mainly manifested as the aberrant accumulation of undifferentiated myeloid progenitor cells in bone marrow with uncontrolled cell proliferation, obstruction of cell apoptosis, and arrested development. In addition, there is high heterogeneity both at the clinical level and prognosis: some patients might achieve long-term remission and survival, but others could become refractory or relapsed AML patients. These clinical heterogeneities of AML are related to several factors, of which age and cytogenetic and molecular alterations in tumour cells are particularly crucial [2]. While such potential mutations may contribute to leukemic development and progression, they may be potential therapeutic targets that provide new insights for drug development [3].
Indeed, chromosomal aberrations and genetic mutations, including an increasing number of recurrent mutations, are considered drivers of AML and may significantly affect the prognosis of AML patients [4, 5]. It has been suggested that, based on increasingly advanced sequencing technology, transcriptomic analysis can even provide predictive models with more prognostic value than traditional clinical parameters or genomic biomarkers [6]. In recent years, many different studies have established survival-related multigene signatures [7-11]. These gene signatures usually contain a cluster of genes, which may be of great significance for further study to discover critical genes from them.
In this study, we constructed a multigene signature containing twenty genes based on public databases, which was highly consistent with the survival of AML patients. Further analysis found phosphoglycerate dehydrogenase (PHGDH) was overexpressed in AML and correlated with inferior overall survival of AML patients. In vitro experiments confirmed that inhibition of PHGDH could effectively induce apoptosis and inhibit proliferation of AML cells, indicating that PHGDH may be a potential therapeutic target for AML treatment.
Materials and methods
Dataset acquisition
All original gene expression profile datasets (GSE37642, GSE9476, GSE106291) were downloaded from the Gene Expression Omnibus (GEO) database. The GSE37642 dataset includes GPL570, GPL96, and GPL97 platforms, of which the dataset based on the GPL570 platform contains 140 AML adult patients, and the other two platforms contain the same 422 AML adult patients. Here, we used only the data of the GPL570 and GPL96 platforms for analysis, with the data from the GPL96 platform as the training cohort and the GPL570 data as the validation cohort.
Data screening and identification and validation of the prognostic gene signature
We filtered all data from the two platforms in the GSE37642 dataset, excluding the following data: 1. no survival status information; 2. overall survival (OS) less than 30 days; 3. uncertain diagnosis or not AML; 4. FAB classification (French-American-British system) unknown or M3 AML; 5. severe comorbidity; 6. presence of other malignancy; 7. prior anti-leukemic treatment. The inclusion criteria were:1. age starting from 18 years with no upper age limit; 2. newly diagnosed AML. First, in the training cohort, univariate Cox regression analysis was used to discover potential genes (p < 0.05) associated with survival. Then these genes were inputted into the LASSO Cox regression model analysis and the best penalty parameter lambda was tested using a 10-fold cross-validation [12, 13]. With the optimal lambda value, the most important survival-related genes in newly diagnosed AML were detected. Next, prognosis-related risk profiles were established by stepwise multivariate Cox regression analysis. Each patient's risk score was calculated which was determined from the formula for the combination of the Cox coefficient and gene expression in this model [14]. By using a cut-off point, the median risk score, patients in both cohorts were split into two groups: low- and high-risk. Kaplan‒Meier survival curves and logarithmic rank tests were used to assess survival differences between two groups. Then, we used the "survivalROC" package to compute the area under the receiver operating characteristic (ROC) curve (AUC). The "pheatmap" package was utilized to make a risk map in R. The same formula was also applied to verify the prognostic value of the multigene signature in the validation cohort. Finally, we performed separate survival analyses of these genes in the model in two cohorts to identify genes that had prognostic implications in both cohorts.
Cell culture and patient samples
Human HL60, THP1, MV4‒11, and MOLM13 cells (all these four cell lines were purchased from ATCC) were cultured in RPMI 1640 medium (GIBCO, USA), with 10% foetal bovine serum and 100 units/ml penicillin and streptomycin and in a moistened environment of 5% CO2 at 37 °C. The PHGDH inhibitor NCT503 (S8619) was purchased from Selleck.
In this study, three healthy donors and three blood samples were acquired from newly diagnosed AML patients from our hospital in 2023. Before participation, written informed consent was obtained from all patients involved in this study. The research protocol was approved by the local ethics committee and conducted according to the ethical guidelines outlined by the World Medical Association Declaration of Helsinki.
Cell viability assay
Cells were inoculated on 96-well tissue culture plates in triplicate at a density of 10,000 cells/wells and treated with different concentrations of NCT503 for 72 h. Each well was then added with 10 μL of Cell Counting Kit-8 solution assay reagent (CCK-8, APExBIO, USA), and the cultures were incubated at 37 °C for 2 to 3 h. Then a microplate reader (Biotek Synergy H1, USA) was used to measure the absorbance at 450 nm.
Measurement of cell apoptosis
The Annexin V-FITC/PI Apoptosis Kit (E-CK-A211, Elabscience, China) was used to detect apoptotic cells by flow cytometry. After treatments, cells were collected and centrifuged for 5 min at 1,000 rpm and 4 °C. The supernatant was discarded and cells were resuspended in 150 μL mixture of 5 μL of annexin V-FITC and 145 μL 1× binding buffer, and incubated for 15 min at 37 ° in the dark. Then, 5 μL of propidium iodide (PI) solution and 145 μL 1× binding buffer were added for detection using flow cytometry. All experiments were repeated three times independently.
Small interfering RNA (siRNA) and transient transfection
PHGDH siRNA was used to silence the PHGDH gene (PHGDH-si). A scrambled sequence siRNA (NC-si) was used as a negative control. The siRNA transfection was optimized using GP-transfectMate (Genepharma, Suzhou, China), according to the manufacturer's instructions.
Western blotting
RIPA lysate (P0013, Beyotime Biotechnology, China) was used to extract the total protein of cells according to the instructions. The protein was separated by 10% SDS/PAGE and transferred onto a PVDF membrane (Millipore, Temecula, CA, USA). After blocking in 10% BSA solution for 1 h, the membranes were incubated with the primary antibody at 4 °C overnight. Subsequently, the membranes were incubated with secondary antibodies at room temperature for 2 h. Protein expression was detected with ECL reagents (G2020, Servicebio, China) and quantified by densitometry using ImageJ. All antibodies were anti-PHGDH (Proteintech, 14719-1-AP), anti-Bcl-2 (Proteintech, 12789-1-AP), anti-Bax (Proteintech, 50599-2-Ig) and anti-β-actin (Proteintech,81115-1-RR).
Statistical analysis
In this study, statistical analyses were performed by GraphPad Prism 9.0. Numerical data is displayed as the mean ± SD. All experiments and analyses were performed in triplicate. The data were assessed for statistical significance by one-way ANOVA and two-tailed Student's t-test according to the test. Statistical significance was defined as p <0.05 (*, p < 0.05; **, p < 0.01; and ***, p < 0.001).
Results
The prognostic signature was constructed from the training cohort.
A total of 422 cases were in the GPL96 platform of GSE37642, and only 348 cases met the requirements and were retained after screening and exclusion. Univariate Cox regression analysis was conducted to ascertain whether gene expression profiles were pertinent to overall survival (OS), and 2160 genes associated with survival prognosis were obtained. Fig.1A plots the coefficients for each gene, and the model showed the prognostic characteristics that were optimal when containing 35 genes (Fig.1B), which can be referred to in more detailed information (Table S1). To further screen for more meaningful genes, we used stepwise multiple Cox regression analysis to finally construct a predictive model composed of 20 genes, and their corresponding coefficients were shown in Table 1, of which 8 genes were risk factors and the remaining 12 were protective factors.
Figure 1
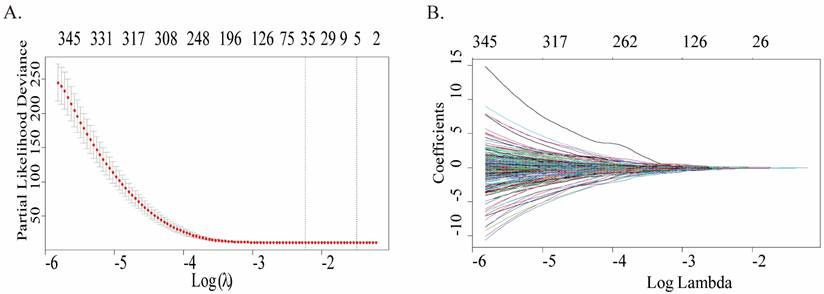
Construction of a prognostic multigene signature by LASSO regression analysis built on the training cohort. (A) Distribution of LASSO coefficient profiles of 35 genes. (B) Coefficient profile plot was created by using the log (lambda) sequence for selecting the best parameter (lambda).
Table 1
Genes in the prognostic multigene signature. Stepwise multiple Cox regression analysis was used to construct the prognostic multigene signature composed of 20 genes, and their corresponding coefficients.
| Gene symbol | Official Full name | Risk coefficient |
|---|---|---|
| TRPC4AP | Transient receptor potential cation channel subfamily C member 4 associated protein | -0.58191 |
| STAR | Steroidogenic acute regulatory protein | -0.10973 |
| ST18 | ST18 C2H2C-type zinc finger transcription factor | -0.17558 |
| SPINT2 | Serine peptidase inhibitor, Kunitz type 2 | 0.13071 |
| SOX1 | SRY-box transcription factor 1 | -0.95504 |
| SLITRK5 | SLIT and NTRK like family member 5 | 0.3104 |
| SLC36A1 | Solute carrier family 36 member 1 | -0.33079 |
| SHANK1 | SH3 and multiple ankyrin repeat domains 1 | -0.6686 |
| PRSS2 | serine protease 2 | 0.20974 |
| PHGDH | Phosphoglycerate dehydrogenase | 0.25727 |
| PCTP | Phosphatidylcholine transfer protein | -0.24132 |
| KDM3B | Lysine demethylase 3B | -0.47246 |
| ITPKA | Inositol-trisphosphate 3-kinase A | -0.63094 |
| FDXR | Ferredoxin reductase | -0.38926 |
| ENPP2 | Ectonucleotide pyrophosphatase/phosphodiesterase 2 | 0.14142 |
| ENAH | ENAH actin regulator | 0.22562 |
| CLU | Clusterin | 0.14254 |
| CENPBD1P1 | CENPB DNA-binding domains containing 2, pseudogene | -0.30834 |
| CALCOCO2 | Calcium binding and coiled-coil domain 2 | -0.28088 |
| ADCY2 | Adenylate cyclase 2 | 0.25403 |
Prognostic value of the multigene signature in the training and validation cohorts
The risk score of each case in both cohorts was calculated using the gene expression levels together with their corresponding regression coefficients, resulting in a median risk score of 2.02. Accordingly, patients were split into two groups: low- (risk score <2.02) and high-risk (risk score ≥2.02). The prognostic value of risk score was assessed through evaluating survival differences in the low- and high-risk groups. Fig.2 displays the distribution of risk scores (Fig.2A, B), survival status (Fig.2C, D), and gene profiles (Fig.2E, F) for this twenty-gene signature in the two cohorts. The high-risk groups possessed more incidents and shorter OS than the low-risk groups. In fact, the heatmap showed that SLITRK5, ENPP2, ADCY2, PRSS2, and PHGDH were overexpressed in high-risk groups, but ST18, STAR, and SLC36A1 were downregulated.
In the training cohort, Kaplan-Meier survival analysis displayed a poor prognosis trend for high-risk patients (p < 0.0001, Fig.2G). It was examined in the validation cohort in order to evaluate the effectiveness of this twenty-gene signature in predicting AML patients' OS (p < 0.0001, Fig.2H). The OS in the high-risk group was noticeably inferior to the low-risk group, along with previous results.
We performed univariate and multivariate Cox analyses in both two cohorts, utilizing accessible variables provided in the dataset, including risk score, age, FAB classification, runx1_mutation, and runx1_runx1t1_fusion, to test the prognostic ability of this twenty-gene signature to clinical features. Both the two analyses of the training cohort found this twenty-gene signature to be a powerful predictor which was highly correlated with overall survival (HR = 1.311, 95% CI = 1.259-1.365, p < 0.001, Figures 3A; HR = 1.307, 95% CI = 1.254-1.362, p < 0.001, Fig.3C). Consistent with the training cohort, the signature showed a significant capability to predict OS in the validation cohort (Fig.3B, D). These findings confirmed that this twenty-gene signature was a powerful and independent variable. Moreover, ROC analysis was conducted to assess the prognostic accuracy of the twenty-gene signature model. Due to the large heterogeneity, rapid progression, and generally worse OS of AML patients, we tested the AUCs of 1-year, 3-year, and 5-year survival in the training cohort, which were 0.86, 0.90 and 0.89, reflecting a strong predictive power of this model (Fig.3E). Correspondingly, the validation cohort also confirmed this finding (AUC=0.77, 0.78, and 0.81, Fig.3F).
In addition, upon comparing the two heatmaps in Fig.2E and F, we found that 5 genes that overexpressed considerably in the high-risk patients of both cohorts included SLITRK5, ENPP2, ADCY2, PRSS2, and PHGDH. Furthermore, we conducted Kaplan‒Meier survival analysis for each of these twenty genes in the training and validation cohorts (Supplementary PDF document), and only SLITRK5, PRSS2, and PHGDH were associated with poorer overall survival in both cohorts (Fig.4). SLITRK5, a transmembrane protein named SLIT and NTRK-like protein-5, was a negative controller of hedgehog signaling in osteoblasts and a therapeutic target to promote bone formation [15]. PRSS2, also known as serine protease 2, was reported to stimulate several solid tumor growth and progression [16, 17]. Compared with these two genes, we are more interested in PHGDH and its role in AML. Therefore, we performed further analysis of PHGDH, one of the partially metabolized enzymes known to be dysregulated in cancer. PHGDH is the first rate-limiting enzyme that catalyzes serine synthesis, and its high expression activates the serine synthesis pathway (SSP) and thus promotes tumour growth [18]. Since tumour cells have exceptional metabolic preferences to meet survival and proliferation needs, it may be a viable therapeutic strategy to treat tumours with PHGDH overexpression by targeting specific enzymes, such as PHGDH [19].
Figure 2
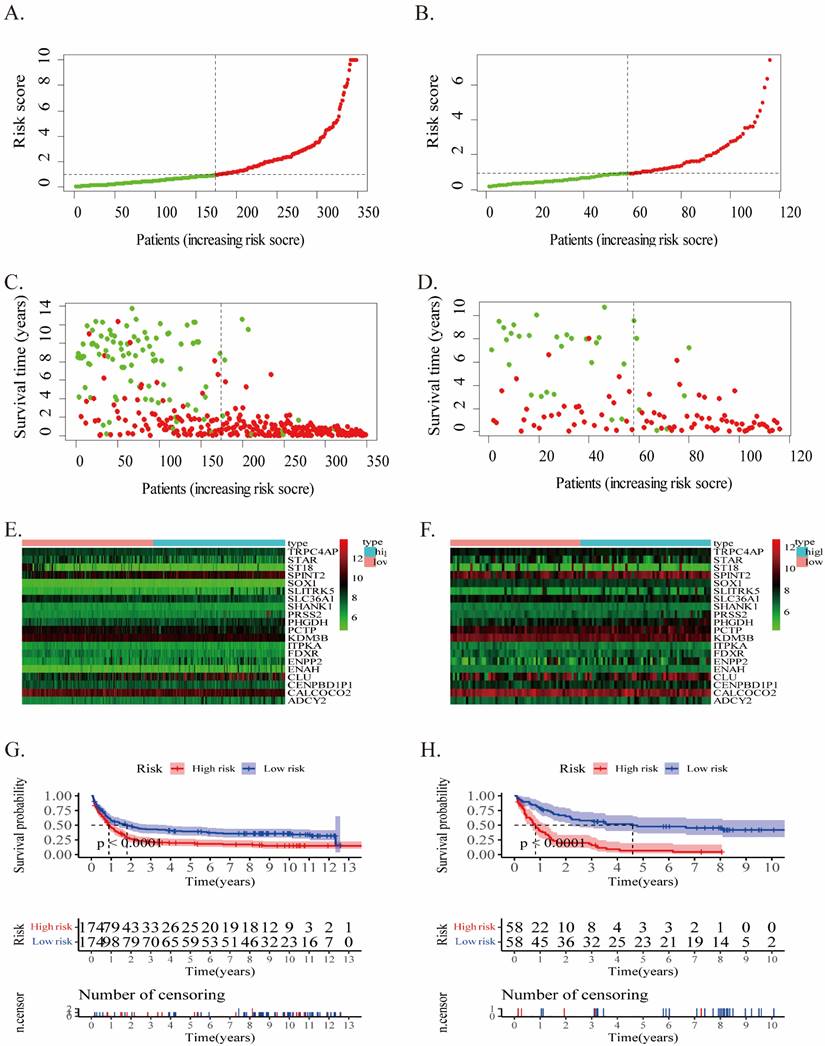
The twenty-gene signature's characteristics in the training (A/C/E/G) and validation (B/D/F/H) cohorts. (A-B) The distribution of the risk score (the red dot represents high risk). (C-D) patient survival time (the red dot represents death). (E-F) Expressions of twenty genes in high- and low-risk groups for the training (G) and validation (H) cohorts.
Figure 3
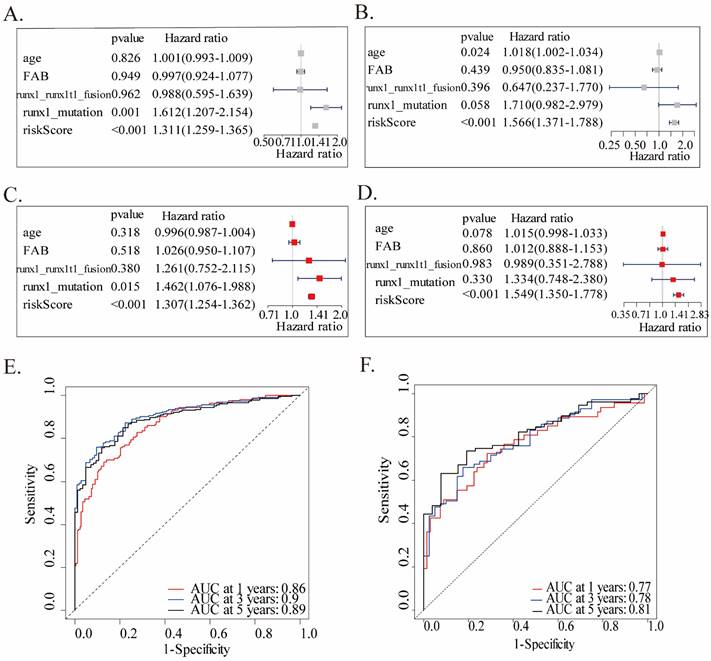
Univariate and multivariate Cox analyses and ROC analysis of the twenty-gene signature. Univariate and multivariate analyses are separately on the basis of this multigene signature and clinical covariates in the training (A, C) and validation (B, D) cohorts. ROC analysis of the overall survival prediction's sensitivity and specificity by 1-, 3-, and 5-year survival time in the training (E) and validation (F) cohorts. AUC stands for the area under the ROC curve.
Figure 4
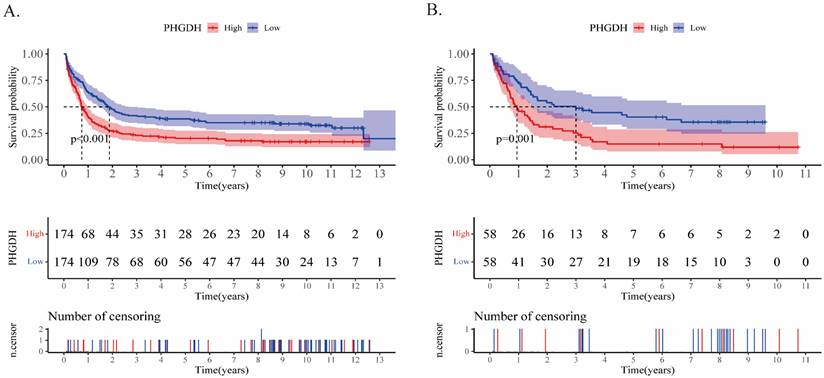
Kaplan‒Meier analyses of PHGDH in the training (A) and validation (B) cohorts. The two-sided log-rank test was used to identify differences between the two curves.
PHGDH expression in AML
In the predictive model, PHGDH was discovered to be overexpressed in high-risk AML patients. Thus, we further compared the expression of PHGDH between healthy donors and AML patients, drug-sensitive and -resistant AML in public datasets (Fig.5). Our analysis of the GSE9476 dataset, comprising 38 healthy donors and 26 AML patients, revealed a significant upregulation of PHGDH expression in AML patients compared to healthy donors (Fig.5A). Similarly, analysis of blood samples from 3 healthy donors and 3 AML patients confirmed these findings by detecting the protein expression level of PHGDH(Fig.5C). The dataset CSE106291 has 235 patients, including drug-resistant and drug-sensitive patients, all of whom received induction therapy based on cytarabine and anthracycline (Fig.5B). In this dataset, we found that the PHGDH level was elevated in the drug-resistant group compared to the drug-sensitive group. These results imply that PHGDH may be a significant factor in AML and a potential therapeutic target.
Effects of PHGDH inhibition in AML cells
To further validate the effects of inhibiting PHGDH in AML, AML cell lines (HL60 and THP1 for FLT3-ITDwt; MV4-11 and MOLM13 for FLT3-ITD+) were treated with different concentrations of the PHGDH inhibitor NCT503. Fig.6A showed that pharmacological inhibition of PHGDH notably inhibited AML cell viability, and the effect was more pronounced in FLT3-ITD+ cells. Therefore, we used MV4-11 and MOLM13 cells for following experiments. The apoptosis test on FLT3-ITD+ cells found that NCT503 induced apoptosis in a concentration-dependent manner (Fig.6B-C). Previous studies confirmed that depletion of PHGDH could induce apoptosis by interacting with the anti-apoptotic protein Bcl-2, reducing the expression and stability of Bcl-2[20-22]. It remained unclear whether similar alterations occurred in FLT3-ITD+ AML cells when PHGDH was inhibited. Under microscopy, it was evident that cells treated with NCT503 exhibited diverse levels of fragmentation, amorphous shapes, and multi-directional morphology (Fig.6D). Consequently, we examined changes in the expression levels of antiapoptotic protein Bcl-2 and proapoptotic protein Bax. The levels of Bcl-2 was also attenuated in a dose-dependent manner, whereas Bax was upregulated in the same manner (Fig.6E). And similar changes have been observed in the cells of different patients with AML (Fig.6F). The results suggested that inhibiting PHGDH pharmacologically could inhibit the proliferation of FLT3-ITD+ AML cells and induce apoptosis and potentially involves activation of the Bcl-2/Bax signaling pathway.
To investigate the involvement of PHGDH in activating the Bcl-2/Bax signaling pathway, we used siRNA to knock down the protein level of PHGDH in AML cells (Fig.6G). Western blot analysis revealed that PHGDH knockdown resulted in up-regulation of the pro-apoptotic protein Bax, while the level of the anti-apoptotic protein Bcl-2 was decreased, which was consistent with previous findings (Fig.6G). In summary, these findings enhanced our understanding of the potential therapeutic implications of targeting PHGDH in FLT3-ITD+ AML and the underlying mechanism may be related to the regulation of Bcl-2/Bax pathway by PHGDH.
Figure 5
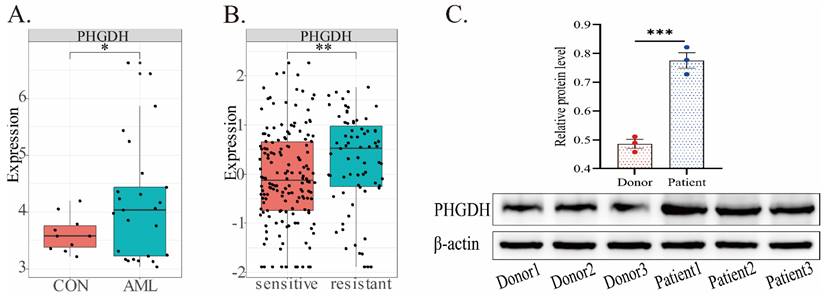
PHGDH expression in AML based on different public datasets. (A) In the GSE9476 dataset, the expression of PHGDH was considerably higher in AML patients than healthy individuals. (B) In the CSE106291 dataset, the drug-resistant group exhibited a higher level of PHGDH than the drug-sensitive group. (C) PHGDH protein expression levels in blood samples from 3 healthy donors and 3 AML patients. (*, p < 0.05; **, p < 0.01; ***, p < 0.001)
Figure 6
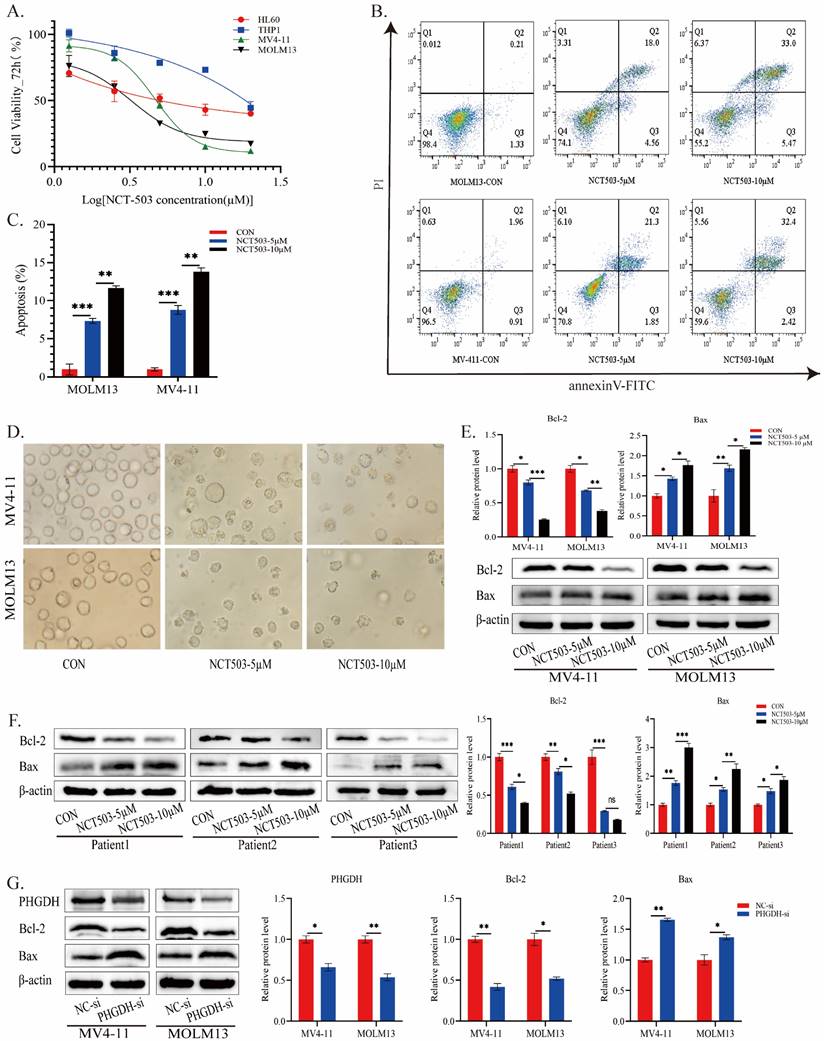
Effects of PHGDH inhibitor on growth inhibition and apoptosis induction in AML cells. (A) Cell viability after treatment with MOLM13 and MV-411 cells. (B-C) MOLM13 and MV-411 cells were treated with various concentrations of NCT-503 for 24 h. Subsequently, apoptosis levels were measured by flow cytometry. (D) Cell morphology changes after treatment with different concentrations of NCT503 for 24 hours(200X). The expression levels of Bcl-2 and Bax in AML cell lines (E) and patient samples (F) were evaluated after treatment with NCT503 for 24 hours. (G) the protein levels of PHGDH, Bcl-2, and Bax were detected following PHGDH knockdown by PHGDH siRNA. Data of three independent experiments were presented as the mean ± standard deviation (SD). (*, p < 0.05; **, p < 0.01)
Discussion
Through comprehensive analysis of GEO databases, we built a twenty-gene signature for the prognostic characteristics of AML. By utilizing this signature in the training and validation cohorts, AML patients in low- and high-risk groups showed statistical significance in Cox regression models, Kaplan-Meier analysis, and ROC curves. All of these results demonstrated the efficiency and applicability of the twenty-gene signature in predicting the prognosis of AML.
Additionally, to identify genes that could be more important for prognostic influence among the twenty genes in the signature, we separately compared their differential expression in both cohorts and the impact on patient prognosis. We found an important gene PHGDH to be expressed higher in patients of the high-risk group than that in low-risk group, and patients with higher levels of PHGDH were related to inferior prognosis. We further verified that PHGDH was overexpressed in AML patients by public databases and patients' blood samples. Consistent with this, another study showed that PHGDH levels were significantly elevated in AML patients and that these patients tended to have a worse prognosis [9]. These results suggest that PHGDH may be abnormally expressed in AML cells and be pivotal to cell metabolism.
PHGDH, a crucial metabolic enzyme of the de novo serine synthesis pathway (SSP), converts glycolysis- or gluconeogenesis-derived 3-PG into serine through a chain of enzymatic reactions together with other rate-limiting enzymes downstream of PHGDH, including phosphoserine aminotransferase 1 (PSAT1) and phosphoserine phosphatase (PSPH)[23, 24]. Subsequently, serine generates glycine through catalysis by serine hydroxy methyltransferases 1/2 (SHMT1/2). Both serine and glycine serve as raw materials for the synthesis of nucleotides, s-adenosylmethionine (SAM), and glutathione (GSH). While serine and glycine participate in nucleotide synthesis, enter one-carbon metabolism, and promote cancer cell proliferation, glutathione acts as a reactive oxygen species (ROS) scavenger to maintain redox balance in cells [25, 26]. This is a highly regulated pathway in response to multiple metabolic stresses, including glucose and glutamine depletion, and the intermediate products NADH and GSH can regulate intracellular redox coordination [27].
In the course of tumour development, metabolic materials, such as amino acids and glucose, usually display aberrant alterations in their metabolic processes to meet the need for uncontrolled proliferation or other demands [28]. One of the most important changes is the activation of SSP, thus increasing the synthesis of serine, which is utilized for protein and nucleotide synthesis, amino acid transport, and folate metabolism, regulating redox homeostasis and cell cycle progression, thus supporting tumour cell survival and proliferation [29-31]. Many studies have confirmed the dysregulation of serine uptake or biosynthesis in tumour cells, as well as abnormal PHGDH expression, in different tumours, including lymphoma, multiple myeloma, glioma, hepatocellular carcinoma, melanoma, colon cancer, breast cancer, and others [32-38]. These studies generally argued that intervention with PHGDH could affect tumour cell growth, but the underlying mechanism was not fully understood.
It is known that metabolic enzymes regulated by key signaling pathways in cancer cells can meet cellular metabolism and growth requirements by performing classical metabolic functions, but a growing number of studies have found that these metabolic enzymes can also support the rapid growth of cancer cells through noncanonical or nonmetabolic functions [39, 40]. In addition to synthesizing serine, PHGDH may also promote tumour proliferation in noncanonical ways. Studies have shown that exogenous serine supplementation does not restore cell growth inhibition caused by PHGDH knockdown, suggesting that PHGDH may have important nonmetabolic functions in tumour development [35]. PHGDH also interacts with the translation initiation factors eIF4E and eIF4A1 to promote translation initiation in the cytoplasm, thereby accelerating tumour development [41]. PHGDH undergoes nuclear translocation after its phosphorylation, binds to c-Jun, and affects the transcription of target genes downstream of c-Jun related to cell growth regulation, thus promoting tumorigenesis [42]. Similarly, PHGDH can promote growth and proliferation of hepatoma cells by activating mitochondrial translation and respiratory metabolism through noncanonical functions [43]. In some other studies, PHGDH may perform noncanonical functions by the mitochondrial apoptotic pathway, facilitating the expression and stability of Bcl-2 and eventually repressing cell apoptosis [20, 22]. Our experiments also demonstrated that inhibition of PHGDH can induce apoptosis in AML cells, which may be related to the Bcl-2/Bax signaling pathway.
In addition, studies demonstrated PHGDH may be involved in cancer and tumour resistance to chemotherapy. Wei et al. found that PHGDH was a key driver of sorafenib resistance in hepatocellular carcinoma (HCC), and the synergistic effect of the PHGDH inhibitor NCT503 and sorafenib can effectively eliminate the growth of HCC in vivo [44]. In multiple myeloma (MM), the level of PHGDH expression was considerably elevated and correlated with inferior survival, and the mechanism involved may be that high levels of PHGDH reduce ROS and DNA damage by increasing intracellular glutathione, promote the survival and proliferation of MM cells, and improve tumour cell resistance to bortezomib [45]. Through a similar mechanism, PHGDH causes triple-negative breast cancer cells to become resistant to doxorubicin treatment [46]. Based on the continuous discovery and improvement of PHGDH inhibitors, their effect in overcoming drug resistance or enhancing chemotherapy efficacy in tumour treatment may gradually become apparent.
These studies show that targeting PHGDH can not only directly intervene in tumour cell survival and proliferation through classical metabolic pathways or nonclassical pathways but also increase the sensitivity of cells to other drugs, indicating that it may be a promising therapeutic target in tumour treatment. To date, the antitumour effects of PHGDH in AML have been poorly studied. In recent years, researchers found that serine biosynthesis was a metabolic vulnerability of FLT3-ITD-driven AML. By inhibiting PHGDH, the proliferation of FLT3-ITD+ AML could be slowed [47]. Our preliminary experiments also confirmed that inhibition of PHGDH significantly inhibited cell proliferation and induced apoptosis in AML cells with or without FLT3-ITD+ mutations. At the same dose, AML cells with FLT3-ITD+ mutations were inhibited to a greater extent. However, further research on its antitumour mechanism needs to be performed.
Conclusions
In summary, our study constructed a multigene signature in AML. The signature was associated with AML OS and strongly identified the prognostic risk factors of AML patients. Remarkably, by analyzing twenty genes in the signature, we discovered a vital metabolism enzyme gene, PHGDH, which is overexpressed in AML patients and is related to inferior prognosis. In this study, we mainly investigated the impact of the noncanonical or nonmetabolic functions of PHGDH on AML. We preliminarily found that pharmacological inhibition of PHGDH can significantly inhibit cell proliferation and induce apoptosis in AML, which the Bcl-2/Bax signaling pathway might be involved in. PHGDH may play a crucial role in development of AML, making PHGDH a potential target for AML therapy.
Supplementary Material
Supplementary methods, figure and tables.
Acknowledgements
Funding
This work was funded by the National Natural Science Foundation of China (82000153), the Science and Technology Foundation of Guangdong Province, China (KTP2020324), and the Natural Science Foundation of Guangdong Province, China (2018A030310091), Guangzhou Clinical High-tech, Major and Characteristic Technology Research Fund, China (2023P-TS16).
Author contributions
JZ and KH organized and wrote the manuscript. SX, AT and JP contributed to the literature search for the manuscript. JZ, KH and YL designed and produced the figures. DN and YL made contributed to the statistical analysis of this manuscript. LM and YL revised the manuscript. All authors reviewed the manuscript and approved the manuscript for publication.
Ethics approval and consent to participate
All the experiment protocol involving human data was in accordance to national/international/institutional guidelines or Declaration of Helsinki in the manuscript.
Availability of data and materials
This study utilized publicly available datasets for the analysis. These data (GSE37642, GSE9476, GSE106291) can be found in GEO (https://www.ncbi.nlm.nih.gov/geo/).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T. et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424-47
2. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND. et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. The New England journal of medicine. 2016;374:2209-21
3. Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM. et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. The New England journal of medicine. 2004;350:1617-28
4. Liersch R, Müller-Tidow C, Berdel WE, Krug U. Prognostic factors for acute myeloid leukaemia in adults-biological significance and clinical use. British journal of haematology. 2014;165:17-38
5. Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ. et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. The New England journal of medicine. 2013;368:2059-74
6. Wang M, Lindberg J, Klevebring D, Nilsson C, Lehmann S, Grönberg H. et al. Development and Validation of a Novel RNA Sequencing-Based Prognostic Score for Acute Myeloid Leukemia. Journal of the National Cancer Institute. 2018;110:1094-101
7. Metzeler KH, Hummel M, Bloomfield CD, Spiekermann K, Braess J, Sauerland MC. et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood. 2008;112:4193-201
8. Chen Z, Song J, Wang W, Bai J, Zhang Y, Shi J. et al. A novel 4-mRNA signature predicts the overall survival in acute myeloid leukemia. American journal of hematology. 2021;96:1385-95
9. Nguyen CH, Glüxam T, Schlerka A, Bauer K, Grandits AM, Hackl H. et al. SOCS2 is part of a highly prognostic 4-gene signature in AML and promotes disease aggressiveness. Scientific reports. 2019;9:9139
10. Wang N, Bai X, Wang X, Wang D, Ma G, Zhang F. et al. A Novel Fatty Acid Metabolism-Associated Risk Model for Prognosis Prediction in Acute Myeloid Leukaemia. Current oncology (Toronto, Ont). 2023;30:2524-42
11. Shi H, Gao L, Zhang W, Jiang M. Identification and validation of a siglec-based and aging-related 9-gene signature for predicting prognosis in acute myeloid leukemia patients. BMC Bioinformatics. 2022;23:284
12. Goeman JJ. L1 penalized estimation in the Cox proportional hazards model. Biometrical journal Biometrische Zeitschrift. 2010;52:70-84
13. Sauerbrei W, Royston P, Binder H. Selection of important variables and determination of functional form for continuous predictors in multivariable model building. Statistics in medicine. 2007;26:5512-28
14. Hu F, Zeng W, Liu X. A Gene Signature of Survival Prediction for Kidney Renal Cell Carcinoma by Multi-Omic Data Analysis. International journal of molecular sciences. 2019 20
15. Sun J, Shin DY, Eiseman M, Yallowitz AR, Li N, Lalani S. et al. SLITRK5 is a negative regulator of hedgehog signaling in osteoblasts. Nature communications. 2021;12:4611
16. Sui L, Wang S, Ganguly D, El Rayes TP, Askeland C, Børretzen A. et al. PRSS2 remodels the tumor microenvironment via repression of Tsp1 to stimulate tumor growth and progression. Nature communications. 2022;13:7959
17. Chen Y, Wang B, Zhao Z, Li M, Wang F. PRSS2 overexpression relates to poor prognosis and promotes proliferation, migration and invasion in gastric cancer. Tissue & cell. 2022;79:101949
18. Mullen AR, DeBerardinis RJ. Genetically-defined metabolic reprogramming in cancer. Trends in endocrinology and metabolism: TEM. 2012;23:552-9
19. Luo J. Cancer's sweet tooth for serine. Breast cancer research: BCR. 2011;13:317
20. Gao X, Wang Y, Lu F, Chen X, Yang D, Cao Y. et al. Extracellular vesicles derived from oesophageal cancer containing P4HB promote muscle wasting via regulating PHGDH/Bcl-2/caspase-3 pathway. Journal of extracellular vesicles. 2021;10:e12060
21. Long B, Shan Y, Sun Y, Wang T, Li X, Huang K. et al. Vitamin C promotes anti-leukemia of DZNep in acute myeloid leukemia. Biochimica et biophysica acta Molecular basis of disease. 2022;1868:166357
22. Zhang Y, Yang L, Dai G, Cao H. Knockdown of PHGDH potentiates 5-FU cytotoxicity in gastric cancer cells via the Bcl-2/Bax/caspase-3 signaling pathway. International journal of clinical and experimental pathology. 2018;11:5869-76
23. Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nature reviews Cancer. 2013;13:572-83
24. Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nature reviews Cancer. 2016;16:650-62
25. Townsend DM, Tew KD, Tapiero H. The importance of glutathione in human disease. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2003;57:145-55
26. Anderson DD, Woeller CF, Chiang EP, Shane B, Stover PJ. Serine hydroxymethyltransferase anchors de novo thymidylate synthesis pathway to nuclear lamina for DNA synthesis. The Journal of biological chemistry. 2012;287:7051-62
27. Sun L, Song L, Wan Q, Wu G, Li X, Wang Y. et al. cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell research. 2015;25:429-44
28. Jiang P, Du W, Wu M. Regulation of the pentose phosphate pathway in cancer. Protein & cell. 2014;5:592-602
29. Labuschagne CF, van den Broek NJ, Mackay GM, Vousden KH, Maddocks OD. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell reports. 2014;7:1248-58
30. Frezza C. Cancer metabolism: Addicted to serine. Nature chemical biology. 2016;12:389-90
31. Geeraerts SL, Heylen E, De Keersmaecker K, Kampen KR. The ins and outs of serine and glycine metabolism in cancer. Nature metabolism. 2021;3:131-41
32. Mattaini KR, Sullivan MR, Vander Heiden MG. The importance of serine metabolism in cancer. The Journal of cell biology. 2016;214:249-57
33. Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ. et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nature genetics. 2011;43:869-74
34. Mullarky E, Mattaini KR, Vander Heiden MG, Cantley LC, Locasale JW. PHGDH amplification and altered glucose metabolism in human melanoma. Pigment cell & melanoma research. 2011;24:1112-5
35. Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K. et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346-50
36. Guo H, Xu J, Zheng Q, He J, Zhou W, Wang K. et al. NRF2 SUMOylation promotes de novo serine synthesis and maintains HCC tumorigenesis. Cancer letters. 2019;466:39-48
37. Nguyen MQ, Teh JLF, Purwin TJ, Chervoneva I, Davies MA, Nathanson KL. et al. Targeting PHGDH Upregulation Reduces Glutathione Levels and Resensitizes Resistant NRAS-Mutant Melanoma to MAPK Kinase Inhibition. The Journal of investigative dermatology. 2020;140:2242-52.e7
38. Mattaini KR, Brignole EJ, Kini M, Davidson SM, Fiske BP, Drennan CL. et al. An epitope tag alters phosphoglycerate dehydrogenase structure and impairs ability to support cell proliferation. Cancer & metabolism. 2015;3:5
39. Xu D, Shao F, Bian X, Meng Y, Liang T, Lu Z. The Evolving Landscape of Noncanonical Functions of Metabolic Enzymes in Cancer and Other Pathologies. Cell metabolism. 2021;33:33-50
40. Kuzuoglu-Ozturk D. PHGDH and cancer: new job for an old enzyme!. The EMBO journal. 2023;42:e113068
41. Ma X, Li B, Liu J, Fu Y, Luo Y. Phosphoglycerate dehydrogenase promotes pancreatic cancer development by interacting with eIF4A1 and eIF4E. Journal of experimental & clinical cancer research: CR. 2019;38:66
42. Ma C, Zheng K, Jiang K, Zhao Q, Sha N, Wang W. et al. The alternative activity of nuclear PHGDH contributes to tumour growth under nutrient stress. Nature metabolism. 2021;3:1357-71
43. Shu Y, Hao Y, Feng J, Liu H, Li ST, Feng J. et al. Non-canonical phosphoglycerate dehydrogenase activity promotes liver cancer growth via mitochondrial translation and respiratory metabolism. The EMBO journal. 2022;41:e111550
44. Wei L, Lee D, Law CT, Zhang MS, Shen J, Chin DW. et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nature communications. 2019;10:4681
45. Wu X, Xia J, Zhang J, Zhu Y, Wu Y, Guo J. et al. Phosphoglycerate dehydrogenase promotes proliferation and bortezomib resistance through increasing reduced glutathione synthesis in multiple myeloma. British journal of haematology. 2020;190:52-66
46. Zhang X, Bai W. Repression of phosphoglycerate dehydrogenase sensitizes triple-negative breast cancer to doxorubicin. Cancer chemotherapy and pharmacology. 2016;78:655-9
47. Bjelosevic S, Gruber E, Newbold A, Shembrey C, Devlin JR, Hogg SJ. et al. Serine Biosynthesis Is a Metabolic Vulnerability in FLT3-ITD-Driven Acute Myeloid Leukemia. Cancer discovery. 2021;11:1582-99
Author contact
![]() Corresponding authors: L. Ma, malipingsysu.edu.cn, Department of Hematology, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, 510120, China. Y. Li, liyiqingsysu.edu.cn, RCID: https://orcid.org/0000-0003-0252-5276, Department of Hematology, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, 510120, China.
Corresponding authors: L. Ma, malipingsysu.edu.cn, Department of Hematology, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, 510120, China. Y. Li, liyiqingsysu.edu.cn, RCID: https://orcid.org/0000-0003-0252-5276, Department of Hematology, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, 510120, China.
Citation styles
APA
Zhong, J., Huang, K., Xie, S., Tan, A., Peng, J., Nie, D., Ma, L., Li, Y. (2024). PHGDH is Key to a Prognostic Multigene Signature and a Potential Therapeutic Target in Acute Myeloid Leukemia. Journal of Cancer, 15(9), 2538-2548. https://doi.org/10.7150/jca.90822.
ACS
Zhong, J.; Huang, K.; Xie, S.; Tan, A.; Peng, J.; Nie, D.; Ma, L.; Li, Y. PHGDH is Key to a Prognostic Multigene Signature and a Potential Therapeutic Target in Acute Myeloid Leukemia. J. Cancer 2024, 15 (9), 2538-2548. DOI: 10.7150/jca.90822.
NLM
Zhong J, Huang K, Xie S, Tan A, Peng J, Nie D, Ma L, Li Y. PHGDH is Key to a Prognostic Multigene Signature and a Potential Therapeutic Target in Acute Myeloid Leukemia. J Cancer 2024; 15(9):2538-2548. doi:10.7150/jca.90822. https://www.jcancer.org/v15p2538.htm
CSE
Zhong J, Huang K, Xie S, Tan A, Peng J, Nie D, Ma L, Li Y. 2024. PHGDH is Key to a Prognostic Multigene Signature and a Potential Therapeutic Target in Acute Myeloid Leukemia. J Cancer. 15(9):2538-2548.
This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See http://ivyspring.com/terms for full terms and conditions.