Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(5):730-736. doi:10.7150/jca.17887 This issue Cite
Research Paper
An investigation of the role of gene copy number variations in sorafenib sensitivity in metastatic hepatocellular carcinoma patients
Ji Yun Lee1,2*, Mineui Hong3,4*, Jeeyun Lee1, Sujin Lee1, Kyoung-Mee Kim3,4, Cheolkeun Park4, Ho Yeong Lim1 ![]()
1. Division of Hematology-Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea;
2. Department of Internal Medicine, Seoul National University Bundang Hospital, Seongnam, Korea;
3. Center for Companion Diagnostics, Innovative Cancer Medicine Institute, Samsung Medical Center, Seoul, Korea;
4. Department of Pathology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
* These authors contributed equally to this work.
Received 2016-10-11; Accepted 2016-12-24; Published 2017-2-25
Citation:
Lee JY, Hong M, Lee J, Lee S, Kim KM, Park C, Lim HY. An investigation of the role of gene copy number variations in sorafenib sensitivity in metastatic hepatocellular carcinoma patients. J Cancer 2017; 8(5):730-736. doi:10.7150/jca.17887. https://www.jcancer.org/v08p0730.htm
Other stylesAbstract
Background: Metastatic hepatocellular carcinoma (HCC) is a highly aggressive tumor with limited treatment options. While sorafenib has recently been shown to provide a survival advantage in patients with advanced HCC, the overall outcomes such as time to progression (TTP) and overall survival (OS) ought to be further improved. To that end, several targeted agents aimed at amplified oncogenes such as HER2 and FGFR2 have recently been developed. In this study, we aimed to identify genetic markers in the form of copy number variations (CNVs) that influence clinical outcomes post-sorafenib treatment in advanced HCC patients.
Methods: We surveyed 38 metastatic HCC patients who were treated with sorafenib for the presence of CNVs using the NanoString nCounter assay.
Results: The median TTP and OS for all patients were 2.7 months (95% confidence interval [CI]: 2.0-3.3 months) and 13.4 months (95% CI: 8.4-18.4 months), respectively. Several genes previously implicated in liver cancer were amplified, including CCND1 (n = 4), CDKN1A (n = 2), KRAS (n = 2), MDM2 (n = 1), and ERBB2 (n = 1). However, we found no correlations between CNVs and survival in our sorafenib-treated patients.
Conclusions: The clinical features and biomarkers that account for sensitivity to sorafenib in HCC are complicated and remain unclear. Further investigation to identify predictive biomarkers and therapeutic strategies, including combining sorafenib with other target agents, are warranted.
Keywords: Hepatocellular carcinoma, metastatic disease, copy number variations, sorafenib.
Introduction
Primary tumors of the liver represent the fifth most frequently diagnosed type of cancer worldwide, and are the second most frequent cause of cancer-related death [1]. More than 75% of cases occur in the Asia-Pacific region, and are largely associated with chronic hepatitis B virus infection [2]. Surgical resection leads to a five-year survival rate of 60-70% for patients with hepatocellular carcinoma (HCC) who present with a solitary tumor and have excellent liver function [3]; however, such resection is only an option for less than 20% of patients [3].
HCC that is diagnosed at an advanced stage or with progression after locoregional therapy has a dismal prognosis owing to the underlying liver disease and lack of effective treatment options [4]. The systemic treatment that can lead to significant improvement in survival in advanced HCC patients is sorafenib, a multi-kinase inhibitor targeting RAF kinase and receptor tyrosine kinases such as platelet-derived growth factor receptor (PDGFR), vascular endothelial growth factor receptor (VEGFR), and c-KIT (a receptor specific for stem cell factor) [5, 6]. Sorafenib simultaneously inhibits two key functions that are reported to play an important role in the pathogenesis of HCC: proliferation (via the Raf-MEK-ERK pathway) and angiogenesis (via VEGFR and PDGFR) [7]. Despite some encouraging data, sorafenib extends survival by only 2-3 months; moreover, the drug has a low partial response rate (3%) [5, 6]. Therefore, it is important to be able to predict sensitivity to sorafenib in advanced HCC patients before commencing treatment.
The mechanisms accounting for HCC sensitivity to sorafenib are complicated and remain unclear. The IC50 values for sorafenib-induced growth inhibition in HCC cell lines in vitro vary widely [8]; one explanation for this is the genetic heterogeneity of HCC [9]. To further elucidate the mechanism of hepatocarcinogenesis, it is useful to reconstruct molecular events at both the gene expression and DNA copy number levels. Copy number variation (CNV) is defined as a gain or loss of copies of DNA segments that are larger than 1 kb in length when compared to a reference genome [10]. CNVs can affect gene expression and have been associated with disease susceptibility [11]; hence, several groups have applied recent technologies to identify CNVs and putative driver genes in HCC [12, 13]. Identifying such markers would be applicable to personalized medicine and/or would enhance existing therapeutic strategies by combining sorafenib with other molecular targeting agents.
To that end, we aimed to identify genetic markers that predict clinical outcomes of sorafenib treatment in advanced HCC patients. We hypothesized that CNVs are found in unique oncogenes and tumor suppressors; hence, we analyzed clinical features of 38 advanced HCC patients who were treated with sorafenib and profiled their tumor specimens for any existing CNVs.
Materials and Methods
Study Design and Samples
HCC formalin-fixed paraffin-embedded samples from 38 patients were collected for CNV detection on the NanoString nCounter platform (NanoString Technologies, Seattle, WA, USA). All cases used in the present study were retrieved during an 8-year period (2005-2012) from Samsung Medical Center. Clinical information including age, sex, etiology, Eastern Cooperative Oncology Group (ECOG) performance status, Barcelona Clinic Liver Cancer (BCLC) staging system, Child-Pugh class, macroscopic vascular invasion, extrahepatic spread, alpha-fetoprotein levels, and previous treatment data were extracted from hospital records. This study was approved by the Institutional Review Board of Samsung Medical Center.
Genomic DNA isolation
Tissue from the needle biopsy was disrupted and homogenized in Buffer RLT plus, Qiagen AllPrep DNA/RNA MiniKit, using the Bullet Blender™ (Next Advance, Inc.). Specifically, tissue was transferred to a microcentrifuge tube containing 600 μL of Buffer RLT Plus and stainless steel beads. The tissue was homogenized in the Bullet Blender at room temperature. The sample was then centrifuged at full speed and the lysate transferred to a Qiagen AllPrep DNA spin column. Genomic DNA purification was conducted as directed by the AllPrep DNA/RNA Mini Handbook (Qiagen). DNA was quantified using the Nanodrop spectrophotometer, and its quality was accessed using the 260/280 nm and 260/230 nm absorbance ratios method.
NanoString nCounter assay
For CNV detection, a panel of 21 gene probes including AURKA, CCND1, CCNE1, CDK4, CDK6, CDKN1A, CDKN2A, EGFR, ERBB2, ERBB3, FGFR1, FGFR2, IGF1R, KLF5, KRAS, MDM2, MET, MITF, MYC, PIK3CA, and TNIK were designed using NanoString nCounter technology and subsequently analyzed on the NanoString nCounter platform [14]. Three probes were designed for each gene. Each assay contained six positive dsDNA control probes, eight negative control probes, and 10 invariant control probes (INVs) designed for autosomal genomic regions predicted not to contain common CNVs.
The NanoString nCounter assay was performed according to NanoString's standard protocol. Briefly, 600 ng of fragmented genomic DNA per assay was hybridized with the capture and reporter probes in 30 μL total volume and incubated at 65°C for a minimum of 16 h. The target and probe complexes were washed and immobilized in the cartridge. Genomic DNA was fragmented into small pieces (200-800 bp) and denatured to produce single strands. The custom CNV CodeSet was then hybridized to the fragmented denatured DNA sample in a single multiplexed reaction (up to 800 genomic loci per CodeSet). Hybridized DNA-CodeSet complexes were purified using the fully automated nCounter prep station, and reporters were counted using the nCounter digital analyzer. The data were normalized to the INVs and to positive and negative controls in each hybridization reaction. Finally, data analysis was performed using the nSolver software.
The copy number was determined by averaging the copy numbers measured by three probes per region. Based on the manufacturer's protocol, the gene was considered to be a single copy if the average copy number was below 1.4, two copies if between 1.5 and 2.4, and three copies if between 2.5 and 3.4.
Copy number gain using real-time quantitative PCR
Based on the CNV results of the NanoString nCounter assay, quantitative PCR (qPCR) analysis was performed on the three most frequent genes using commercially available, predesigned TaqMan Copy Number Assays (Fig. 1). To allow a comparison between both approaches, we selected qPCR probes focused on the immediate region of the NanoString's probes. The reaction mixture contained 2 mL genomic DNA template, 10 mL of Taqman universal PCR master mixture (Applied Biosystems, Foster City, CA, USA), and 0.2 mM of each primer.
To accurately detect copy number gain (CNG), we analyzed three different regions of the CCND1, KRAS, and MDM2 genes (Supplementary Table 1). CNG was determined using the following qPCR sequence: 2 min at 50 ˚C, denaturation at 95 ˚C for 10 min, followed by 40 cycles of 95 ˚C for 15 s and 60 ˚C for 1 min using relative quantification in a 7900 HT fast real-time PCR system in quadruplicate. A RNaseP assay kit (Applied Biosystems) was used as a control. After amplification, the resulting threshold cycle values for the copy number and reference assay were imported into the CopyCaller Software (Applied Biosystems) for post-PCR data analysis as previously described. CNG and the number of copies were determined based on concordance of the results with one or more of the three regions of each probe.
Statistical analysis
Overall survival (OS) was measured from the start of sorafenib administration until the date of death from any cause. The time to progression (TTP) was defined from the start of sorafenib administration until the first documented event of progression (according to RECIST v1.1). Survival curves were constructed using the Kaplan-Meier method for all 38 patients. The associations between clinical features and each of the dichotomized groups were analyzed by the chi-square test or Fisher's exact test. Two-sided P values of less than 0.05 were considered significant. All analyses were performed using SPSS for Windows, version 12.0.
Figure 1
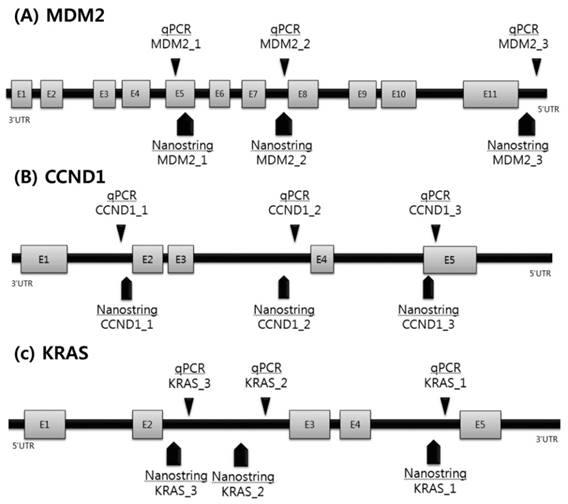
Scheme of validation of copy number variations using qPCR.
Results
Patient characteristics
The characteristics of the study patients at the initiation of sorafenib treatment are summarized in Table 1. The median age was 56 years (range, 35-85 years). The most common etiology for HCC was hepatitis B (74%); 5% of the patients had hepatitis C and the remainder had non-B non-C HCC. Patients had advanced-stage HCC (BCLC stage C: 95%) with preserved liver function (Child-Pugh Class A: 95%) and good ECOG performance status (0: 66%). At baseline, nine patients (24%) had macroscopic vascular invasion and 34 (90%) had extrahepatic spread, with the most common extrahepatic sites being the lymph nodes and lung. Before sorafenib treatment, patients received liver resection (79%), locoregional therapy (transarterial chemoembolization [TACE], 71%; radiofrequency ablation, 40%; radiotherapy, 50%; metastasectomy, 34%; and systemic cytotoxic chemotherapy, 16%).
Table 1
Baseline patient characteristics (n=38).
| Variable | No. (%) |
|---|---|
| Median age, years (range) | 56 (35-85) |
| Male, n (%) | 33 (87) |
| Cause of disease, n (%) | |
| Hepatitis B | 28 (74) |
| Hepatitis C | 2 (5) |
| Unknown | 8 (21) |
| ECOG performance status, n (%) | |
| 0 | 25 (66) |
| 1 | 12 (32) |
| 2 | 1 (2) |
| BCLC stage, n (%) | |
| B | 2 (5) |
| C | 36 (95) |
| Macroscopic vascular invasion, n (%) | 9 (24) |
| Extrahepatic spread, n (%) | 34 (90) |
| Extrahepatic sites, n (%) | |
| Lung | 22 (58) |
| Lymph node | 8 (21) |
| Child-Pugh class, n (%) | |
| A | 36 (95) |
| B | 2 (5) |
| AFP ≥ 200ng/mL, n (%) | 13 (34) |
| Previous therapy, n (%) | |
| Liver resection | 30 (79) |
| Locoregional therapy | |
| Transarterial chemoembolization | 27 (71) |
| Radiofrequency ablation | 15 (40) |
| Radiotherapy | 19 (50) |
| Metastatectomy | 13 (34) |
| Systemic chemotherapy | 6 (16) |
ECOG, Eastern Cooperative Oncology Group; BCLC, Barcelona Clinic Liver Cancer; AFP, alpha-fetoprotein.
CNV landscape in HCC
We surveyed CNVs of 21 genes, and identified amplifications in CCND1 (n = 4), CDKN1A (n = 2), KRAS (n = 2), MDM2 (n = 1), and ERBB2 (n = 1) (Table 2). Because of the small number of patients, further survival analysis was not performed. Of the five genes with amplifications identified by the nCounter assay, we further validated those of CCND1, MDM2, and KRAS using conventional qPCR (Table 3); this confirmed amplification in all four CCND1 samples and the MDM2 sample. However, KRAS amplification results were not consistent between the nCounter assay and qPCR in one of the two patients; the other did not have a sufficient amount of DNA for further analysis by qPCR.
Table 2
The frequency of copy number variations in 38 patients by Nanostring nCounter analysis.
| Gene | Localization | Case no. | Total CNVs frequency no. (%) |
|---|---|---|---|
| CCND1 | chr11:69166461-69173877 | 38 | 4 (11) |
| CDKN1A | chr6:36760296-36761391 | 38 | 2 (5) |
| KRAS | chr12:25261832-25285922 | 38 | 1 (3) |
| ERBB2 | chr17:35112859-35135347 | 38 | 1 (3) |
| MDM2 | chr12:67498111-67518304 | 38 | 1 (3) |
CNVs, copy number variations.
Table 3
Copy number variations validation by qPCR.
| Sample | CNVs by Nanostring | Gene_1 CN Predicted | Gene_2 CN Predicted | Gene_3 CN Predicted | Result |
|---|---|---|---|---|---|
| CCND1 | |||||
| #4 | 181 | 2 | 178 | 7 | concordant |
| #6 | 40 | 15 | 22 | 12 | concordant |
| #18 | 8 | ND | 7 | ND | concordant |
| #25 | 44 | ND | 47 | ND | concordant |
| Negative sample | 3 | 0 | 3 | 0 | |
| MDM2 | |||||
| #11 | 46 | 2 | 0 | 9 | concordant |
| Negative sample | 2 | 2 | 2 | 2 | |
| KRAS | |||||
| #11 | 9 | 2 | 1 | 0 | disconcordant |
| #20 | 90 | ND | ND | ND | failed |
| Negative sample | 2 | 2 | 2 | 2 | |
| Normal control | 2 | 2 | 2 |
Correlation between copy number variation and response to sorafenib
Among 34 patients who were evaluable for response, three (9%) achieved partial response, 10 (26%) had stable disease, and 21 (55%) had progressive disease per RECIST v1.1. Of 38 patients, 4 (11%) had to discontinue sorafenib owing to intolerance or toxicities. The median TTP and OS for the patients were 2.7 months (95% confidence interval [CI]: 2.0-3.3 months) and 13.4 months (95% CI: 8.4-18.4 months), respectively (Fig. 2). No significant correlations were found between clinical features and TTP as well as OS (Supplementary Table 2). At the time of progression, 13 patients received experimental therapy; these included sunitinib (2 patients), everolimus (1 patient), and double-blind randomized placebo-controlled trials of molecular targeted agents such as brivanib, ramucirumab, regorafenib, and E7080 in the context of clinical trials (10 patients).
Of the three responders to sorafenib, two did not have any CNVs detected by the 21-gene NanoString panel. The first of these three patients was a 61-year-old man who was an HBsAg carrier with multiple lung metastases. The patient had an exceptional response to sorafenib, with a persistent reduction in the volumes of multiple lung metastases over 5 years (Fig. 3a). The second responder was an 84-year-old non-B non-C HCC patient with multiple metastases who responded to sorafenib for 30 months (Fig. 3b). This patient did not have any significant amplification in the 21-gene panel. The third patient, a 47-year-old man who was an HBsAg carrier with multiple lung metastases, responded to sorafenib but progressed after 9 months. This patient had KRAS and MDM2 amplifications in his tumor specimen.
Additionally, patient #15 was diagnosed with hepatitis B-related HCC at the age of 31 years and underwent liver sectionectomy. Shortly afterward, the patient had recurrent disease in the liver and was treated with TACE multiple times. The patient then received sorafenib for unresectable recurrent HCC but progressed after two months; this patient's tumor specimen harbored CDKN1A and ERBB2 amplifications.
Discussion
Recently, gene amplifications in various tumors have been highlighted as potential therapeutic targets. Hence, screening for amplified genes using high-throughput technology has become an important diagnostic tool in cancer medicine. In our previous study of gastric cancer specimens, we tested a CNV panel comprising 21 candidate genes for which targeted drugs are available [15]. We demonstrated that the NanoString nCounter assay had a concordance rate of 83.4% for HER2 amplification when compared to conventional immunohistochemistry and fluorescent in situ hybridization methods. In this study, we surveyed HCC specimens with a 21-gene CNV panel to identify potential amplifications in HCC.
Four patients in this study (11%) showed significant CNVs of CCND1. In the TCGA cohort (n = 370), 27 (7.3%) HCC patients were identified to have CCND1 amplification (Fig. 4). This gene, which is located on chromosome 11q13, is amplified in about 4% of HCCs [16]. CCND1 encodes cyclin D1, which interacts with the cyclin-dependent kinases CDK4 and CDK6, resulting in the inactivation of the tumor suppressor RB1 and progression of the cell cycle [17]. Amplification of CCND1 may therefore lead to increased proliferation; in fact, a recent study revealed that lentivirus-mediated shRNA targeting of cyclin D1 inhibits the proliferation of a gastric cancer cell line [18].
Although our study included a small number of patients, tumors from two of three sorafenib responders did not harbor any CNVs. These two patients responded to sorafenib for >30 months and 5 years, respectively. In contrast, one patient with a partial response to sorafenib progressed after nine months; this patient's tumor harbored amplifications in KRAS and MDM2. KRAS amplification in lung cancer has been reported to increase expression levels of p21, suggesting a dysregulated cell cycle [19]. In endometrial cancers, a subset of patients whose tumors had amplifications of the 12p12.1 region harboring KRAS had poorer prognoses [20].
Figure 2
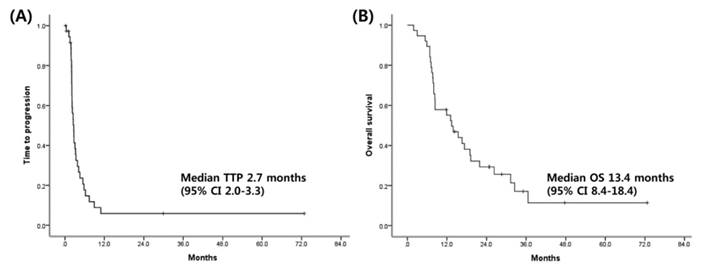
Kaplan-Meier curves for Time to progression (A), overall survival (B).
Figure 3
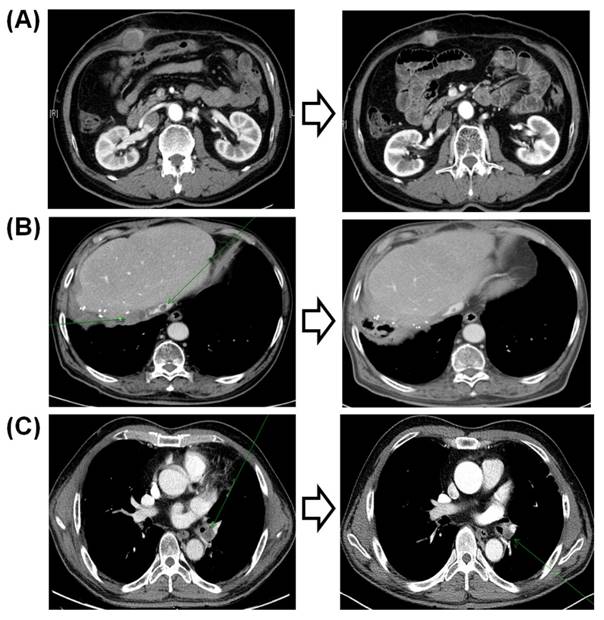
Computed tomography findings of the three responders to sorafenib.
Figure 4
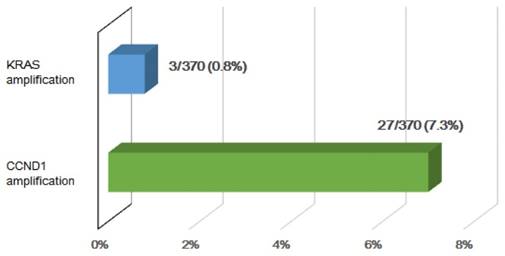
The percentage of KRAS and CCND1 amplification in the TCGA HCC cohort (n = 370).
Of 38 HCC patients, two (5.2%) had MDM2 amplification; in a previous study, MDM2 amplification was detected in approximately 44% of patients with HCC [21]. MDM2 prevents the activity of the tumor suppressor p53; therefore, amplification of MDM2 may be oncogenic [22] and may predict sensitivity to MDM2 inhibitors. However, currently available evidence is inconclusive [23].
Of note, one patient who rapidly progressed following sorafenib had significant CNVs of ERBB2. The overexpression of the HER-2/neu oncogene has been implicated in the development and modulation of many other cancers; however, whether HER-2/neu overexpression plays a similar role in HCC has not been determined [24, 25]. Although phase II trials for lapatinib in HCC revealed a marginal benefit to only a subgroup of patients (in whom predictive markers had not yet been characterized) [26, 27], its role in the treatment of HCC remains to be elucidated [28].
Our study had several limitations. First, the small study population from a single center makes it difficult to draw definitive conclusions concerning the genetic markers that predict clinical outcomes of sorafenib treatment in advanced HCC patients. Second, NanoString probes were designed for only 21 genes. Because of the limited number of CNV gene probes, the discovery of additional CNVs that could impact the outcomes of sorafenib treatment was restricted. Furthermore, high-throughput sequencing and integrative informatics should be needed to uncover new CNVs.
In conclusion, our study did not reveal any candidate predictive markers for sorafenib sensitivity, as there was no correlation between any CNVs and survival; this indicates that any such markers may be of uncertain value. Well-designed prospective clinical studies are required to determine the exact roles of these markers in predicting the response to sorafenib in HCC. Additionally, more preclinical studies are required to clarify the roles of currently known biomarkers in their respective cancer-promoting signal transduction pathways.
Supplementary Material
Supplementary tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA: a cancer journal for clinicians. 2011;61:212-36
2. Kirk GD, Lesi OA, Mendy M, Akano AO, Sam O, Goedert JJ. et al. The Gambia Liver Cancer Study: Infection with hepatitis B and C and the risk of hepatocellular carcinoma in West Africa. Hepatology (Baltimore, Md). 2004;39:211-9
3. Hung H. Treatment modalities for hepatocellular carcinoma. Current cancer drug targets. 2005;5:131-8
4. Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology (Baltimore, Md). 2005;42:1208-36
5. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS. et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. The Lancet Oncology. 2009;10:25-34
6. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF. et al. Sorafenib in advanced hepatocellular carcinoma. The New England journal of medicine. 2008;359:378-90
7. Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H. et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer research. 2004;64:7099-109
8. Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D. et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer research. 2006;66:11851-8
9. O'Connor R, Clynes M, Dowling P, O'Donovan N, O'Driscoll L. Drug resistance in cancer - searching for mechanisms, markers and therapeutic agents. Expert opinion on drug metabolism & toxicology. 2007;3:805-17
10. Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD. et al. Global variation in copy number in the human genome. Nature. 2006;444:444-54
11. Chaignat E, Yahya-Graison EA, Henrichsen CN, Chrast J, Schutz F, Pradervand S. et al. Copy number variation modifies expression time courses. Genome research. 2011;21:106-13
12. Jia D, Wei L, Guo W, Zha R, Bao M, Chen Z. et al. Genome-wide copy number analyses identified novel cancer genes in hepatocellular carcinoma. Hepatology (Baltimore, Md). 2011;54:1227-36
13. Woo HG, Park ES, Lee JS, Lee YH, Ishikawa T, Kim YJ. et al. Identification of potential driver genes in human liver carcinoma by genomewide screening. Cancer research. 2009;69:4059-66
14. Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL. et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nature biotechnology. 2008;26:317-25
15. Ahn S, Hong M, Van Vrancken M, Lyou YJ, Kim ST, Park SH. et al. A nCounter CNV Assay to Detect HER2 Amplification: A Correlation Study with Immunohistochemistry and In Situ Hybridization in Advanced Gastric Cancer. Molecular diagnosis & therapy. 2016;20:375-83
16. Takahashi Y, Kawate S, Watanabe M, Fukushima J, Mori S, Fukusato T. Amplification of c-myc and cyclin D1 genes in primary and metastatic carcinomas of the liver. Pathology international. 2007;57:437-42
17. Witzel II, Koh LF Perkins ND. Regulation of cyclin D1 gene expression. Biochemical Society transactions. 2010;38:217-22
18. Seo JH, Jeong ES, Choi YK. Therapeutic effects of lentivirus-mediated shRNA targeting of cyclin D1 in human gastric cancer. BMC cancer. 2014;14:175
19. Wagner PL, Perner S, Rickman DS, LaFargue CJ, Kitabayashi N, Johnstone SF. et al. In situ evidence of KRAS amplification and association with increased p21 levels in non-small cell lung carcinoma. American journal of clinical pathology. 2009;132:500-5
20. Salvesen HB, Carter SL, Mannelqvist M, Dutt A, Getz G, Stefansson IM. et al. Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase activation. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:4834-9
21. Jablkowski M, Bocian A, Bialkowska J, Bartkowiak J. A comparative study of P53/MDM2 genes alterations and P53/MDM2 proteins immunoreactivity in liver cirrhosis and hepatocellular carcinoma. Journal of experimental & clinical cancer research: CR. 2005;24:117-25
22. Peng Q, Lao X, Chen Z, Lai H, Deng Y, Wang J. et al. TP53 and MDM2 gene polymorphisms, gene-gene interaction, and hepatocellular carcinoma risk: evidence from an updated meta-analysis. PloS one. 2013;8:e82773
23. Liu W, He L, Ramirez J, Ratain MJ. Interactions between MDM2 and TP53 Genetic Alterations, and Their Impact on Response to MDM2 Inhibitors and Other Chemotherapeutic Drugs in Cancer Cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:7602-7
24. Altimari A, Fiorentino M, Gabusi E, Gruppioni E, Corti B, D'Errico A. et al. Investigation of ErbB1 and ErbB2 expression for therapeutic targeting in primary liver tumours. Digestive and liver disease: official journal of the Italian Society of Gastroenterology and the Italian Association for the Study of the Liver. 2003;35:332-8
25. Hsu C, Huang CL, Hsu HC, Lee PH, Wang SJ, Cheng AL. HER-2/neu overexpression is rare in hepatocellular carcinoma and not predictive of anti-HER-2/neu regulation of cell growth and chemosensitivity. Cancer. 2002;94:415-20
26. Bekaii-Saab T, Markowitz J, Prescott N, Sadee W, Heerema N, Wei L. et al. A multi-institutional phase II study of the efficacy and tolerability of lapatinib in patients with advanced hepatocellular carcinomas. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:5895-901
27. Ramanathan RK, Belani CP, Singh DA, Tanaka M, Lenz HJ, Yen Y. et al. A phase II study of lapatinib in patients with advanced biliary tree and hepatocellular cancer. Cancer chemotherapy and pharmacology. 2009;64:777-83
28. Chen YJ, Chi CW, Su WC, Huang HL. Lapatinib induces autophagic cell death and inhibits growth of human hepatocellular carcinoma. Oncotarget. 2014;5:4845-54
Author contact
![]() Corresponding author: Ho Yeong Lim, MD, PhD, Division of Hematology-Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul 135-710, Korea, Tel: +82-2-3410-0918 Fax: +82-2-3410-1757, e-mail: hoylimedu.
Corresponding author: Ho Yeong Lim, MD, PhD, Division of Hematology-Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul 135-710, Korea, Tel: +82-2-3410-0918 Fax: +82-2-3410-1757, e-mail: hoylimedu.
Citation styles
APA
Lee, J.Y., Hong, M., Lee, J., Lee, S., Kim, K.M., Park, C., Lim, H.Y. (2017). An investigation of the role of gene copy number variations in sorafenib sensitivity in metastatic hepatocellular carcinoma patients. Journal of Cancer, 8(5), 730-736. https://doi.org/10.7150/jca.17887.
ACS
Lee, J.Y.; Hong, M.; Lee, J.; Lee, S.; Kim, K.M.; Park, C.; Lim, H.Y. An investigation of the role of gene copy number variations in sorafenib sensitivity in metastatic hepatocellular carcinoma patients. J. Cancer 2017, 8 (5), 730-736. DOI: 10.7150/jca.17887.
NLM
Lee JY, Hong M, Lee J, Lee S, Kim KM, Park C, Lim HY. An investigation of the role of gene copy number variations in sorafenib sensitivity in metastatic hepatocellular carcinoma patients. J Cancer 2017; 8(5):730-736. doi:10.7150/jca.17887. https://www.jcancer.org/v08p0730.htm
CSE
Lee JY, Hong M, Lee J, Lee S, Kim KM, Park C, Lim HY. 2017. An investigation of the role of gene copy number variations in sorafenib sensitivity in metastatic hepatocellular carcinoma patients. J Cancer. 8(5):730-736.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.