Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(7):1255-1262. doi:10.7150/jca.16450 This issue Cite
Research Paper
Evaluation of Attenuated Tumor Antigens and the Implications for Peptide-Based Cancer Vaccine Development
JS Berry1 ![]() , TJ Vreeland2, DF Hale1, DO Jackson3, AF Trappey3, JM Greene3, MO Hardin4, GS Herbert3, GT Clifton5, GE Peoples6
, TJ Vreeland2, DF Hale1, DO Jackson3, AF Trappey3, JM Greene3, MO Hardin4, GS Herbert3, GT Clifton5, GE Peoples6 ![]()
1. Department of Surgery, Division of Colon and Rectal Surgery, Washington University, St. Louis, MO;
2. Department of Surgery, Womack Army Medical Center, Fort Bragg, NC;
3. Department of Surgery, Brooke Army Medical Center, Fort Sam Houston, TX;
4. Department of Surgery, Madigan Army Medical Center, Fort Lewis, WA;
5. Department of Surgical Oncology, University of Texas MD Anderson Cancer Center, Houston, TX;
6. Cancer Vaccine Development Program, San Antonio, TX and Department of Surgery, Uniformed Services University of the Health Sciences, Bethesda, MD.
Received 2016-6-11; Accepted 2017-2-14; Published 2017-5-11
Abstract
INTRODUCTION: Peptide vaccines offer anti-tumor efficacy with very low toxicity. However, repeat stimulation with an immunogenic peptide leads to activation induced cell death (AICD), decreasing efficacy. We engineered variants of an immunogenic peptide (E39) and tested their ability to induce a robust, sustainable immune response.
METHODS: Multiple variants of E39 were created by exchanging 1 or 2 amino acids. We tested the PBMC proliferation, cytokine production and cytolytic activity induced by each variant peptide. RESULTS: Repeated stimulation with E39 likely led to in vitro AICD, while stimulation with E39' led to T-cell proliferation with less evidence of AICD, modest cytokine production and high CTL activity. CONCLUSIONS: E39' appears to be the optimal variant of E39 for inducing effective long-term immunity.
Keywords: Folate binding protein, folate receptor alpha, J65, E39, E39'.
Introduction
Treatment of cancer has significantly expanded beyond the traditional management strategies of chemotherapy, radiation, and surgery, with immunotherapy becoming increasingly important. Within this field, there is considerable interest in developing strategies for prevention of cancer recurrence after standard of care treatment using therapies that induce adaptive immunity against a specific tumor-associated antigen (TAA). We have previously published our successful attempts to induce such a response to a well-known TAA, HER2, using immunogenic epitopes, specifically E75 and GP2. These peptides form the basis of vaccines that strive to stimulate production of cytotoxic T lymphocytes (CTLs) that are specific to the antigen and, thus, capable of targeted lysis of tumor cells.[1-5] In addition to an initial production of CTLs, memory T cells are produced, which maintain immune surveillance and offer the promise of prolonged disease-free survival when these vaccines are given in the adjuvant setting.[6,7] With our most successful peptide vaccine, NeuVax, now in a phase III trial for FDA approval, the identification of additional TAAs to target for similar vaccines is ongoing.
Folate binding protein (FBP), also known as folate receptor alpha, is a near ideal TAA for immunotherapy. FBP is over-expressed in over 90% of ovarian and 20-50% of breast cancers, yet is rarely expressed in normal tissues [8]. Analogous to HER2 in breast cancer, the level of FBP expression has been found to be greater than 20-fold higher in malignant cells compared to normal cells, and its over-expression is linked to more aggressive cancers.[3] Not only is FBP expression increased in cancer cells, it is also inherently immunogenic; it has been recognized by CTLs isolated from ovarian and breast cancer patients and shown to induce tumor-specific cytokine release and cytolysis [4-9]. While the body of literature studying FBP-directed immunotherapies is limited, the combination of frequent over-expression in cancer cells, correlation with aggressive disease, and natural immunogenicity make FBP an attractive TAA for use in peptide vaccines.
We have previously shown that FBP peptides were recognized by HLA-A2+ ovarian and breast tumor associated lymphocytes that were not previously subjected to in vitro antigen stimulation.[4] Upon further investigation, E39 (FBP191-199, EIWTHSYKV) was the most consistently recognized of these HLA-A2 restricted antigenic epitopes within the FBP.[6] Furthermore, E39-stimulated TALs from ovarian cancer patients recognized allogeneic, HLA-A2 positive, FBP-expressing ovarian cancer cells and induced cell lysis of these and other FBP-expressing epithelial tumor cells of the colon and pancreas.[4] Knowing the potential of E39 to function as an immunodominant epitope, we are conducting a phase I/IIa trial, in which HLA-A2 positive patients are given varying doses of E39 with GM-CSF. At the time of interim analysis, the vaccine was very well tolerated and induced a strong, dose-dependent immune response. Early efficacy results showed a statistically significant increase in estimated 2-year DFS for patients receiving an optimal dose of the vaccine over controls and patients receiving lower doses [10].
Given our success with peptide vaccines and promising early results with E39, we anticipate future trials to better assess its clinical efficacy. There are growing concerns, however, that repeated stimulation with one immunogenic peptide, such as E39, may actually impair immune memory, which is the ultimate goal of any peptide vaccine. Although the exact mechanisms are not fully understood, this overstimulation seems to adversely affect CTL selection and proliferation. It has been shown that peptide stimulation produces a polyclonal CTL response, with only some of these CTL populations becoming the effector and memory CTLs necessary for effective long-term immune surveillance. Over-stimulation with an immunogenic peptide may lead to activation induced cell death (AICD) of these highly desired cells [11]. If this process is repeated with continued stimulation, it can lead to weaker responses each time. A strategy of inoculating patients with an attenuated peptide has been proposed as a way to minimize this risk of AICD, but the effect on T cell selection and expansion is unknown. Our early work with the new E39 peptide affords us the opportunity to test these theories.
To this end, we engineered multiple attenuated peptides by substituting one or two amino acids of the wild-type peptide, E39. We then tested the attenuated peptides for in vitro immunologic responses. Our hypothesis was that these attenuated peptides, once bound to the HLA molecule, would lead to altered interactions with the T cell receptor (TCR) and ultimately to proliferation of different populations of CTLs. In this report, we describe the identification of E39' as the ideal attenuated version of E39 through discovery of its in vitro potential as a primer of a robust, sustainable immune response against FBP.
Methods
Peptide Creation
Multiple variants of the E39 peptide were produced by exchanging one or two amino acids; these peptides and their amino acid sequence are displayed in Table 1. DNAStar software predicted that reducing the positive charge of the amino acid in position 5 and removing the phenolic structure at position 7 would improve the flexibility of residues 5-9 and allow for a better fitting of the TCR with the peptide-MHC complex. Specifically, the flexibility of the peptide is improved by reducing the positive charge at the amino acid in position 5 (histidine) through replacement with phenylalanine. Phenylalanine is not charged and its benzene aromatic ring is a close substitution for the imidazole ring of histidine. Then, the phenolic structure of tyrosine was replaced with the aliphatic core chain of threonine. Both tyrosine and threonine contain a hydroxyl side chain group. Thus, the positive charge in position 5 and the rigid structure in position 7 were eliminated in the creation of E39' by changing histidine to phenylalanine at position 5 and tyrosine to threonine at position 7. J77 and J78 each had only one of these key substitutions. The remaining peptides were created with variations on these two changes, some with additional substitutions at position 1 designed to alter the HLA binding. An Applied Biosystems Model 430A peptide synthesizer used solid-phase synthesis to produce the epitopic peptides.
E39 variant peptides and amino acid sequences.
| Peptide | Amino Acid Sequence |
|---|---|
| E39 | EIWTHSYKV |
| J63 | FIWTHSTKV |
| J64 | GIWTHSTKV |
| E39' | EIWTFSTKV |
| J66 | FIWTFSTKV |
| J67 | EIWTHATKV |
| J68 | FIWTFATKV |
| J77 | EIWTHSTKV |
| J78 | EIWTFSYKV |
T2 Stabilization Assay
To evaluate the HLA-A2 binding strength of each peptide, cell line T2 cells were cultured overnight in RPMI 1640 medium in the presence of saturating quantities (100µg/ml) of a single variant peptide (J78 , J77 , E39' , J64 , J63 , J66 , J67 or J68). The cells were then stained with a monoclonal antibody, BB7.2, used to recognize the bound, HLA-A2 molecule. The level of expression of the peptide-bound HLA-A2 complex was measured by the average fluorescent intensity, the mean channel fluorescence (MCF), using flow cytometry.
In vitro Immune Monitoring
Peripheral Blood Mononuclear Cells (PBMCs) were isolated from multiple healthy donors, separated into the plastic-adherent and non-adherent fractions. The different fractions were then prepared as described below. The non-adherent fraction contained 95% CD3 positive cells. The plastic-adherent PBMCs were cultured in complete RPMI 1640 medium containing 10% Fetal Calf Serum (FCS), granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-4 for 5 days. These cell cultures resulted in immature monocyte-derived dendritic cells (iDCs). Plastic-adherent and non-adherent PBMCs were stored frozen in liquid nitrogen until use.
The PBMCs were primed with the E39 or a variant peptide; first with 500,000 iDCs plated in 24 1.0 ml well plates with complete RPMI medium and allowed to attach for 2-3 hours. The complete RPMI medium was removed and replaced with serum-free RPMI medium. Peptides were added to the wells for a final concentration of 10µg/ml. One ml RPMI 1640 medium containing 20% FCS was added to the wells. After 2 hours of incubation at 37C, 3 × 106 of non-adherent fresh PBMCs were added. IL-12 (100 pg/m) was added after two additional hours of incubation. Then, IL-2 was added to a final concentration of 150 IU/ml, 24-36 hours later. Cells were maintained in culture for 7 days in the presence of 100 IU/ml of exogenous IL-2. Thereafter, cells were washed, rested for 24 hours in complete RPMI medium without IL-2. This procedure was repeated for a total of 3 weekly priming sessions for each peptide.
Cell Proliferation
PBMC cultures from six different healthy or naïve, HLA-A2-positive donors were primed with a single dose of E39 or an attenuated E39 variant (J77, J78, E39') weekly as described above. The total number of cells was counted after a week in culture and just prior to the next week's priming.
Interleukin-2 (IL-2) Production
Those peptides able to effectively stimulate PBMC proliferation (J77, E39') were then tested for the ability to stimulate IL-2 production as compared to E39 and a control. PBMCs were primed weekly for three weeks with either a control of no protein (NP), E39, J77, or E39' in the previously described method. For three days after the third priming, IL-2 concentrations were measured in supernatant.
Interferon-γ (IFN-γ) Release Assay
Next, the amount of IFN-γ produced was assessed following PBMC stimulation with different methods. PBMCs from healthy donors were primed once a week for three weeks with E39' or E39 as described above. The PBMCs stimulated with E39' were then targeted against T2 cells loaded with E39, J77, or E39'. PBMCs primed with E39 were only tested against cells loaded with E39. IFN-γ release into the supernatant of each set of PBMC cultures was measured over two days.
Cytotoxicity Assays
Finally, PBMC cultures were tested for their ability to lyse cells loaded with FBP peptides. Primed PBMCs were targeted against 51Chromium labeled, T2 cells loaded with three concentrations (0, 5, 25 µg/ml) of the appropriate peptide. The percent lysis was calculated by the amount of free 51Chromium in supernatant as measured after 5 hours. The PBMC were primed and the T2 cells loaded in the same fashion as the IFN-γ assays; E39'-primed cells were tested against T2 cells loaded with E39, E39', or J77, while E39-primed PBMCs were tested only against E39.
Results
T2 Stabilization Assay
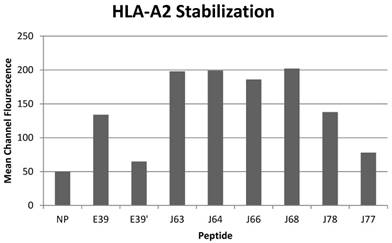
All of the variant peptides were able to bind the HLA-A2 molecule as indicated by a MCF greater than the control (Figure 1). As expected, peptides with a substitution at position 1 (J63, J64, J66 and J68) demonstrated high affinity for the HLA-A2 molecule (198, 199, 186, and 202 MCF, respectively). Those peptides did not undergo further investigation as their higher affinity for the HLA-A2 molecule was not consistent with the goal of an attenuated peptide. Peptides created with only substitutions in positions 5 and 7 (E39', J77 and J78) were all able to bind the HLA-A2 molecule. J78's affinity for HLA-A2 did not differ significantly from that of E39 (127 v 125 MCF). J77 and E39' had much weaker binding, with 78 and 65 MCF, respectively.
HLA-A2 stabilization of E39-derived attenuated epitopes
In vitro Immune Monitoring
Cell Proliferation
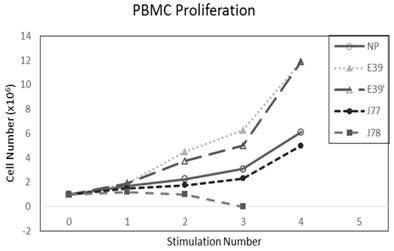
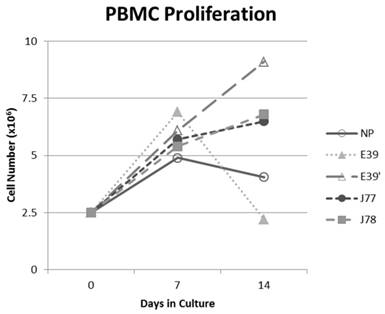
The PBMC proliferation potential of E39', J77, and J78 was measured over six cultures from multiple different, healthy, HLA-A2 positive donors; representative data from two of these donors are shown in Figures 2 and 3, respectively. Figure 2 shows a representative graph of cell counts one week after each of three weekly stimulations. In this case, E39 and E39' resulted in the highest expansion to 11.9 x106 cells and the increase continued to four weeks with both peptides. J77 and J78 did not invoke improved cell proliferation compared to NP. In Figure 3, PBMC proliferation from another donor that underwent similar priming is shown, but the data instead represent cell counts 7 to 14 days after the third priming with each respective peptide. Cells primed with E39' continued to proliferate from 6.1 x106 to 9.1 x106 cells between 7 to 14 days. Conversely, PBMCs primed with the highly immunogenic E39 underwent the opposite response with decrease cell counts between days 7 and 14, likely representing AICD. The J77 and J78 primed PBMCs had a similar, low-level response in this patient with expansion to 6.5 x106 and 6.8 x106 cells, respectively. Because J78 did not exhibit a consistent ability to induce PBMC proliferation, it was precluded from further studies.
PBMC proliferation after stimulation with E39 or E39 derived epitopes; Donor #1.
PBMC proliferation after stimulation with E39 or E39 derived epitopes; Donor #2.
IL-2 Production
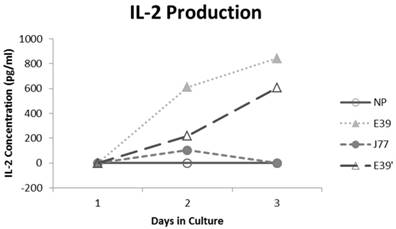
IL-2 production was measured for E39 and the attenuated peptides that consistently resulted in PBMC proliferation (E39' and J77; Figure 4). At three days out from the third stimulation with J77, there was no increase in IL-2 production. In response to both E39 and E39' priming, however, there was a significant increase in IL-2. Importantly, stimulation with E39' produced less IL-2 than E39 (611 and 844 pg/ml, respectively). Because J77 primed PBMCs did not produce IL-2, it was precluded from further testing except as a target peptide of for E39 and E39'-primed PBMCs.
Interleukin 2 (IL-2) levels produced by PBMCs after third priming with E39' E39, and J77.
Interferon-γ Release Assay
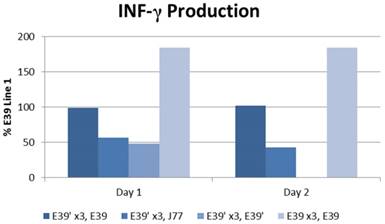
The IFN-γ release after priming with E39 and E39' was measured as described above and is displayed in Figure 5. E39'-primed cells recognized all FBP peptide variants on re-stimulation with peptide-loaded T2 cells as evidence by initial IFN-γ release. Those E39'-primed PBMCs re-stimulated with E39' had the smallest response; 48% and 0% from baseline on days 1 and 2, respectively. The E39'-primed PBMCs stimulated with J77 had a small, yet stable response with a 57% and 43% increase from baseline on days 1 and 2, respectively. The E39'-primed, E39-stimulated (E39', E39) population maintained a stable, moderate response with a 99% and 102% increased from baseline on day 1 and 2. The highest IFN-γ release was seen in E39-primed PBMCs targeted against E39-loaded T2 cells, with an increase of 184% from baseline on days 1 and 2.
IFN-γ levels in PBMCs after three weekly stimulations with E39' or E39, followed by a fourth re-stimulation with either E39', E39, or J77.
Cytotoxicity Assays
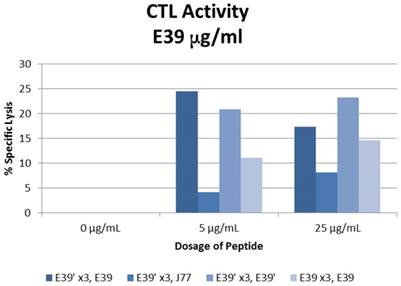
The percent specific lysis of peptide-loaded T2 cells produced by the combinations described above is displayed in Figure 6. As expected, loading cells with 0 µg/ml concentration of peptides produced no response in all sequences. E39'-primed PBMCs recognized and lysed T2 cells loaded with all FBP variants. The lowest response occurred when targeting J77-loaded cells (4.2 and 8.2% at 5 and 25 µg/ml). The other two populations of E39'-primed cells showed impressive cytotoxicity, with 24.5% and 17.4% lysis of E39-loaded cells and 20.9% and 23.2% of E39'-loaded cells at 5 and 25 µg/ml, respectively. The E39-primed PBMCs targeted at E39 did not perform as well as these two groups of E39'-primed cells. E39-primed cells lysed only 11.1% and 14.6% of E39-loaded T2 cells at 5 and 25 µg/ml, respectively.
% specific lysis of CTLs after three weekly E39' or E39 priming doses at different concentrations (0, 5 or 25µg/ml) followed by a single E39', E39, or J77 boosting.
Discussion
In this study of variant peptides created from E39, we found that the candidate peptides, J78, J77 and E39', were able to stably bind the HLA-A2 molecule. When these peptides were tested for the ability to induce PBMC proliferation, E39' consistently showed the best results, particularly over multiple, repeated T cell stimulations. Interestingly, while E39 induced a robust initial proliferation, some populations of cells contracted after continued priming with E39, indicating significant AICD. We then tested cells primed with E39 and E39' for functionality finding that cells primed by E39 produced high amounts of IL-2 and IFN-γ, while those primed by E39' made more moderate amounts of each cytokine. Lastly, E39'-primed cells showed excellent cytotoxicity when targeted to both E39 and E39' loaded T2 cells, and actually performed better than cells primed with the wild type peptide, E39. Cumulatively, these findings suggest that an attenuated version of a strong T cell epitope can help preserve the lytic CTLs in repeatedly stimulated T cell populations.
While there have been great strides made toward the goal of a successful peptide vaccines able to induce a long-term, adaptive anti-tumor response, the final goal is yet to be reached. Multiple investigators have been able to induce anti-tumor T cell responses ex vivo, but clinical responses are often disappointing. Part of the reason for these disappointing results seems to be early elimination of the final effectors of these vaccines, the peptide-specific CTL capable of tumor lysis.[12] There is concern that the repeated stimulation with a strongly immunogenic peptide is actually inducing AICD. A new strategy of inoculating patients with a weaker or attenuated version of a specific peptide has the potential to avoid this issue.
With this strategy in mind, we set out to produce an attenuated version of E39, a known immunogenic peptide, and test the immune response it would induce as compared to E39. Multiple novel peptides were created, with three potential candidates that possessed altered amino acids within the TCR binding site. These three peptides were all shown to bind the HLA-A2 molecule. Next, we tested how these peptides would stimulate PBMC proliferation, particularly through multiple rounds of stimulation. E39' consistently performed better than any other attenuated peptide and nearly as well as E39 at initial stimulation (Figure 2). The more interesting finding, however, was noted after watching continued proliferation two weeks after a third priming (Figure 3). Here, E39-induced PBMC proliferation fell off significantly in the second week, while cells primed by E39'continued to expand in number This graphic is representative of similar findings from additional PBMC populations tested in this study and is a common phenomenon observed with repeated stimulation with one, immunogenic peptide. The late drop-off of PBMCs likely corresponds to the very AICD suspected following continual stimulation with E39. The proliferation shown with E39', however, is very encouraging and indicates that the attenuated peptide did not induce AICD, perhaps by avoiding over-stimulation.
While this proliferation data was encouraging, further testing was needed to determine the functionality of the induced T cell populations. To test this, we first measured cytokine production after PBMC stimulation with E39 and E39'. E39' resulted in blunted cytokine production as compared to E39, as demonstrated in assays of IL-2 and IFN-γ production. These higher cytokine levels in response to E39 may simply occur because E39 is a stronger signal to T cells, but may also be related to selection of certain T cell populations after repeated stimulation.
While E39' led to less AICD, it was unclear how the reduced binding affinity and cytokine response would alter the ability to mediate tumor cell death. The CTL response that is induced by a pathogen or by an immunogenic peptide is understood to be polyclonal, with some cells differentiating to produce cytokines and others, the effector cells, to perform cell lysis. Even within the effector cells, there are short-lived effector cells and memory precursor effector cells.[13] The differentiation, proliferation and apoptosis of these varying T cell populations are controlled by a complex interplay of cytokines and interactions between the TCR and the peptide-HLA complex. While this process is not fully understood, cells with high affinity TCRs seem to be the most effective at lysing target cells and are most likely to become memory T cells.[11,14] Thus, the ability to induce these high TCR affinity T cells specific for the target TAA is key to the success of peptide vaccines. High levels of IFN-γ have been linked to a weaker TCR signal caused by low affinity of the TCR to the HLA-peptide complex and this weak TCR signal has been shown to lead to early contraction of effector CTL populations.[14,15] Similarly, while IL-2 is needed for CTL proliferation, high levels have been linked to production of regulatory T cells that may hinder effective clearance of targeted cancer cells. Multiple, strong stimulations with E39 may, theoretically, have led to AICD of highly cytolytic effector CTLs and instead selected for T cells that focus more on cytokine production, accounting for the high levels of IFN-γ and IL-2.
The cytolytic assays demonstrated that E39'-primed cells were capable of lysing cancer cells loaded with any of the three FBP peptides tested. These cells performed best when targeting cells loaded with E39', but they were also able to lyse a high number of cells loaded with E39. More importantly, these E39'-primed cells had higher peptide-specific cell lysis than E39-primed cells. After over-stimulation with E39, the apoptosis demonstrated by the PBMC proliferation tests seems to have affected mainly effector CTLs, leaving behind cell populations with low TCR affinity that produce high levels of IL-2 and IFN-γ release, but have decreased CTL activity. Less potent stimulation with E39', on the other hand, did not seem to induce the same degree of effector CTL AICD, maintaining populations of CTLs with high TCR affinity. This is ideal, as these cells are thought to not only be effective at target cell lysis, but are also thought to become memory T cells.
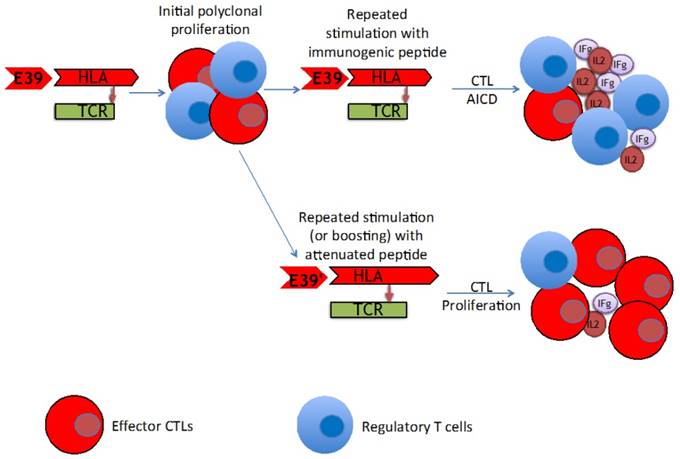
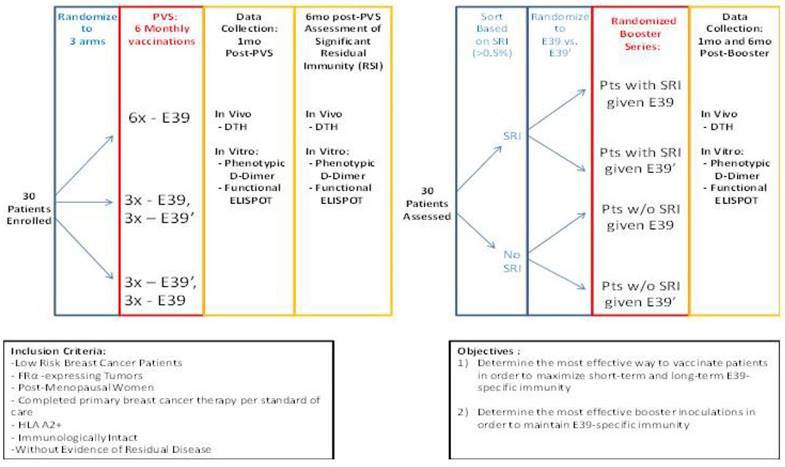
The findings of this trial support the theory that stimulation with an attenuated peptide, E39', could lead to a more effective cancer vaccine (as depicted in Figure 7). Whether this peptide will be most effective alone or in combination with the more immunodominant parent peptide, E39, will be the focus of our next trial. We have started a Phase Ib trial in breast and ovarian cancer patients with FBP-expressing tumors. The trial design incorporates three arms for the primary vaccine series consisting of six inoculations: one arm with all inncoulations being E39, another with the first three inoculations with E39 followed by three with E39', and the last with E39' given first, followed by E39. Once patients have completed their primary vaccine series, their residual immunity will be measured in vivo and in vitro and they will be classified as significant responders or non-responders. Patients will then be randomized again to receive either E39 or E39' in a four arm booster series (Figure 8). The primary objectives are to determine the most effective method to maximize short-term and long-term FBP-specific immunity, to determine the most effective booster inoculations to maintain FBP-specific immunity, and to evaluate the safety of the vaccination series. This currently enrolling study (NCT02019524) will allow us to use different sequences of the wild type peptide (E39) and attenuated peptide (E39') in order to answer these novel and important research questions in the field of immunotherapy.[16] If successful, the application of this novel strategy using a native peptide in combination with an attenuated version could be far-reaching.
Proposed mechanism of improved T cell response after repeated stimulation with attenuated peptide (E39') and parent peptide (E39).
Phase I/2a trial design in breast and ovarian cancer (NCT02019524)16
Conclusion
In summary, these experiments demonstrate that repeated stimulation with highly immunogenic E39 likely leads to AICD of T cells populations in vitro including peptide-specific effector CTLs with high affinity TCRs. E39' appears to be the optimal variant of E39 as it consistently results in T cell proliferation, stimulates T cells to produce modest cytokines and, most importantly, selects for effector CTLs with high TCR affinity that are necessary for tumor cell lysis. The potential of this version of E39 to induce an enduring, cytolytic anti-FBP immune response that results in tumor cell lysis is currently being evaluated in a phase Ib clinical trial in ovarian and breast cancer patients.
Acknowledgements
We greatly appreciate all the work of Dr. Constantin Ioannides and the personnel from his lab which made this project possible.
Disclaimer
The view(s) expressed herein are those of the author(s) and do not reflect the official policy or position of San Antonio Military Medical Center, the U.S. Army Medical Department, the U.S. Army Office of the Surgeon General, the Department of the Army, Department of Defense or the U.S. Government.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Peoples GE, Goedegebuure PS, Andrews JV. et al. HLA-A2 presents shared tumor-associated antigens derived from endogenous proteins in ovarian cancer. J Immunol. 1993;151(10):5481-5491
2. Yoshino I, Goedegebuure PS, Peoples GE. et al. HER2/neu-derived peptides are shared antigens among human non-small cell lung cancer and ovarian cancer. Cancer Res. 1994;54(13):3387-3390
3. Li PY, Del Vecchio S, Fonti R. et al. Local concentration of folate binding protein GP38 in sections of human ovarian carcinoma by in vitro quantitative autoradiography. J Nucl Med. 1996;37(4):665-672
4. Peoples GE, Anderson BW, Lee TV. et al. Vaccine implications of folate binding protein, a novel cytotoxic T lymphocyte-recognized antigen system in epithelial cancers. Clin Cancer Res. 1999;5(12):4214-4223
5. Peoples GE, Anderson BW, Fisk B. et al. Ovarian cancer-associated lymphocyte recognition of folate binding protein peptides. Ann Surg Oncol. 1998;5(8):743-750
6. Kim DK, Lee TV, Castilleja A. et al. Folate binding protein peptide 191-199 presented on dendritic cells can stimulate CTL from ovarian and breast cancer patients. Anticancer Res. 1999;19(4B):2907-2916
7. Ioannides CG, Platsoucas CD, Rashed S. et al. Tumor cytolysis by lymphocytes infiltrating ovarian malignant ascites. Cancer Res. 1991;51(16):4257-4265
8. Garin-Chesa P, Campbell I, Saigo PE. et al. Trophoblast and ovarian cancer antigen LK26. Sensitivity and specificity in immunopathology and molecular identification as a folate-binding protein. Am J Pathol. 1993;142(2):557-567
9. Linehan DC, Goedegebuure PS, Peoples GE. et al. Tumor-specific and HLA-A2-restricted cytolysis by tumor-associated lymphocytes in human metastatic breast cancer. J Immunol. 1995;155(9):4486-4491
10. Greene JM, Schneble EJ, Berry JS. Preliminary Results of the Phase I/IIa Dose Finding Trial of a Folate Binding Protein Vaccine (E39+GM-CSF) in Ovarian and Endometrial Cancer Patients. J Clin Oncol. 2015;33(suppl):abstr e14031
11. Kawano K, Efferson CL, Ioannides CG. Sensitivity of Undifferentiated, High-TCR Density CD8+ Cells to Methylene Groups Appended to Tumor Antigen Determines Their Differentiation of Death. Cancer Research. 2005;65(70):2930-37
12. Chhabra A. Mitochondria-centric activation induced cell death of cytolytic T lymphocytes and its implications for cancer immunotherapy. Vaccine. 2010;28:4566-4572
13. Boulet S, Daudelin JF, Labrecque N. IL-2 Induction of Blimp-1 Is a Key In Vivo Signal for CD8+ Short-Lived Effector T Cell Differentiation. J Immunol. 2014;193:1847-54
14. Stoycheva D, Deiser K, Starck L. et al. IFN-γ Regulates CD8+ Memory T Cell Differentiation and Survival in Response to Weak, but Not Strong, TCR Signals. J Immunol. 2015;194:553-559
15. Matsueda S, Gao H, Ioannides C. N-Terminally LRMK-linked HER-2 peptides, AE-37 and AE-47, Aid Differentiation of E75-TCR+CD8+ Cells to Perforin-positive Cells. Anticancer Research. 2009;29:2427-2436
16. https://www.clinicaltrials.gov/ct2/show/NCT02019524?term=NCT02019524
Author contact
![]() Corresponding authors: George E. Peoples, MD, FACS, Cancer Vaccine Development Program, 600 Navarro Street, Suite 500, San Antonio, TX 77205; e-mail: georgepeoples2com; phone (210)557-4291; Fax (210)349-3666. John S. Berry IV, MD, Department of Surgery, Division of Colon and Rectal Surgery, Washington University, St. Louis, MO. 1 Barnes-Jewish Hospital Plaza, St. Louis, MO 63110 email: berryjwustl.edu; ph: (314)454-7177; cell: (850)766-4291.
Corresponding authors: George E. Peoples, MD, FACS, Cancer Vaccine Development Program, 600 Navarro Street, Suite 500, San Antonio, TX 77205; e-mail: georgepeoples2com; phone (210)557-4291; Fax (210)349-3666. John S. Berry IV, MD, Department of Surgery, Division of Colon and Rectal Surgery, Washington University, St. Louis, MO. 1 Barnes-Jewish Hospital Plaza, St. Louis, MO 63110 email: berryjwustl.edu; ph: (314)454-7177; cell: (850)766-4291.