Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(8):1453-1459. doi:10.7150/jca.18762 This issue Cite
Hypothesis
ZEB1 Mediates Drug Resistance and EMT in p300-Deficient CRC
Darina Lazarova, Michael Bordonaro ![]()
Department of Basic Sciences, Geisinger Commonwealth School of Medicine, 525 Pine Street, Scranton, PA 18509, USA
Received 2016-12-14; Accepted 2017-2-27; Published 2017-5-12
Citation:
Lazarova D, Bordonaro M. ZEB1 Mediates Drug Resistance and EMT in p300-Deficient CRC. J Cancer 2017; 8(8):1453-1459. doi:10.7150/jca.18762. https://www.jcancer.org/v08p1453.htm
Other stylesAbstract
We discuss the hypothesis that ZEB1-Wnt-p300 signaling integrates epithelial to mesenchymal transition (EMT) and resistance to histone deacetylase inhibitors (HDACis) in colorectal cancer (CRC) cells. The HDACi butyrate, derived from dietary fiber, has been linked to CRC prevention, and other HDACis have been proposed as therapeutic agents against CRC. We have previously discussed that resistance to butyrate likely contributes to colonic carcinogenesis, and we have demonstrated that butyrate resistance leads to cross-resistance to cancer therapeutic HDACis. Deregulated Wnt signaling is the major initiating event in most CRC cases. One mechanism whereby butyrate and other HDACis exert their anti-CRC effects is via Wnt signaling hyperactivation, which promotes CRC cell apoptosis. The histone acetylases (HATs) CBP and p300 are mediators of Wnt transcriptional activity, and play divergent roles in the downstream consequences of Wnt signaling. CBP-mediated Wnt signaling is associated with cell proliferation and stem cell maintenance; whereas, p300-mediated Wnt activity is associated with differentiation. We have found that CBP and p300 differentially affect the ability of butyrate to influence Wnt signaling, apoptosis, and proliferation. ZEB1 is a Wnt signaling-targeted gene, whose product is a transcription factor expressed at the invasive front of carcinomas where it promotes malignant progression and EMT. ZEB1 is typically a transcriptional repressor; however, when associated with p300, ZEB1 enhances transcription. These changes in ZEB1 activity likely affect the cancer cell phenotype. ZEB1 has been shown to promote resistance to chemotherapeutic agents, and expression of ZEB1 is upregulated in butyrate-resistant CRC cells that lack p300 expression. Since the expression of ZEB1 correlates with poor outcomes in cancer, ZEB represents a relevant therapeutic target. Here we propose that targeting the signaling network established by ZEB1, Wnt signaling, and p300 signaling can reverse HDACi resistance and inhibit EMT.
Keywords: ZEB1, Wnt, epithelial to mesenchymal transition, histone deactylase inhibitors, colorectal cancer, Wnt signaling, p300
Introduction
Wnt signaling and histone deacetylase inhibitors
The histone deacetylase inhibitor (HDACi) butyrate, derived from dietary fiber, exerts a strong preventive effect against colorectal cancer (CRC) [1-13], and other HDACis are possible cancer therapeutic agents [14-16]. Therefore, neoplastic cell resistance to the effects of butyrate likely contributes to CRC, as well as to cross-resistance to HDACis proposed for CRC therapy [2]. Mutations resulting in deregulated Wnt signaling are a major initiating event in most sporadic CRC cases [17-26]. One mechanism whereby butyrate and other HDACis exert their anti-CRC effects is via hyperactivation of the deregulated Wnt signaling in neoplastic colonic cells, promoting apoptosis and repressing cell proliferation [1, 2,16].
The histone acetylases (HATs) CBP and p300 associate with the Wnt signaling factor beta-catenin and mediate the transcriptional activity of the Wnt pathway. Although CBP and p300 share considerable sequence homology, they play divergent roles in the downstream consequences of Wnt activity. CBP-mediated Wnt signaling is associated with cell proliferation and stem cell maintenance; whereas, p300-mediated Wnt activity is associated with differentiation [27-37]. CBP and p300 affect the ability of butyrate to influence Wnt signaling, apoptosis, and proliferation [38-41].
To understand the phenomenon of butyrate resistance, we developed a HCT-116 CRC cell line (HCT-R) resistant to the effects of physiologically relevant concentrations of butyrate [2]. These cells are also cross-resistant to structurally distinct and clinically relevant HDACis [2, 42], and exhibit repressed expression of p300 [39]. HCT-R cells exhibit (a) increased expression of the epithelial to mesenchymal transition (EMT) factor ZEB1 and (b) modified expression of other characteristics of EMT, such as decreased expression of E-cadherin and increased expression of vimentin [3]. Coincidentally, p300 knockout cells, also derived from the HCT-116 cell line, also exhibit physiological changes consistent with EMT, such as decreased cell-cell/matrix adhesion and increased migration [43].
ZEB1, CBP/p300, and EMT
EMT, associated with cancer cell metastasis, results in altered gene expression and changes stationary and polarized epithelial cells into mesenchymal cells with enhanced motility and invasiveness. The reverse process, mesenchymal to epithelial transition (MET), occurs after the metastatic cells have settled into a new location. ZEB1 is expressed at the invasive front of carcinomas, where it affects gene expression to induce EMT [44,45] (and refs therein). ZEB1 upregulates expression of vimentin and downregulates expression of E-cadherin, which is a key event for EMT and metastasis [44]. Since ZEB1 promotes tumorigenesis and metastasis [44-52], its expression is correlated to poor outcomes in cancer, including resistance to chemotherapy [48 and refs. therein]. ZEB proteins most frequently repress transcription through both passive and active mechanisms [44]. Passively, ZEB1 displaces transcriptional activators from promoters; actively, ZEB proteins recruit transcriptional corepressors. In addition, ZEB can associate with transcriptional coactivators to upregulate expression [44]. ZEB proteins can act through a variety of transcriptional cofactors; for example, ZEB1 binds to the HATs p300 and PCAF [45,46], and this complex activates transcriptional activity. Alternatively, ZEB1 acts as a repressor when it interacts with BRG1, an ATPase that forms part of the SWI/SNF chromatin remodeling complex, or with CtBP, which forms a complex that may include HDACis [44 and refs. therein]. The expression levels of p300 and CtBP influence ZEB1 activity; in particular, high levels of CtBP result in an inverse relationship between ZEB1 and E-cadherin expression [51], consistent with the role of ZEB1/CtBP in repressing E-cadherin expression. ZEB1 is a Wnt signaling-targeted gene, and its product can modulate gene expression in a Wnt activity-dependent manner [46-50].
The miR-200 family of microRNAs (miRNAs) influence EMT and MET; these miRNAs are downregulated during EMT and upregulated during MET, likely reflecting their functional contribution to the cell phenotypes along the epithelial to mesenchymal continuum [46,47]. ZEB proteins and miR-200 members negatively modulate each other's expression, consistent with the pro-EMT role of ZEB factors and the anti-EMT role of miR-200 family members [46,47]. The downregulation of ZEB expression is mediated by miR-200 binding to ZEB RNA 3' UTR and blocking translation [46,47]. Complexes composed of ZEB1, p300, and PCAF can bind to the miR200c/141 promoter, resulting in acetylation of ZEB1 and reversal of ZEB1 repression of miR200c/141 expression [46]. This interaction decreases ZEB1 expression and therefore inhibits EMT. The HDACi trichostatin A (TSA), which synergizes with HAT activity, shifts cells toward a more epithelial phenotype, demonstrating the effect of net acetylation [46,47]. These reports are consistent with our observation that cells resistant to the HDACi butyrate downregulate p300 and exhibit an EMT-like profile that includes increased expression of ZEB1. Treatment of human airway epithelial cells with the CBP-Wnt inhibitor ICG-001, which enhances p300-Wnt activity [27-37], suppresses EMT induced by TGFbeta1 [53]. This is also consistent with findings that p300 knockout cells have an EMT-like phenotype [43], since CBP and p300 compete for binding to beta-catenin. In summary, decreased p300 favors CBP-Wnt activity and promotes EMT-type gene expression and phenotype. However, low expression of p300 is not sufficient for acquisition of an EMT-like phenotype. For example, HCT-15 cells that are naturally p300-deficient have an epithelial phenotype [55]; however, these cells exhibit low ZEB1 expression [56]. Thus, expression of ZEB1 and associated factors [46-58] might be essential for EMT following p300 downregulation.
Intriguing connections have also been drawn between ZEB1, EMT, dedifferentiation of cancer cells to cancer stem cells (CSCs), resistance to therapy, and HDACis [59-65]. EMT likely promotes the emergence of a drug-resistant, relatively dedifferentiated mesenchymal cancer cell phenotype [59], which contributes to HDACi-resistance during colonic tumorigenesis. Therefore, the phenomenon of butyrate resistance [66-75] and resistance to other HDACis, may be in part mediated by ZEB1, possibly through altered gene expression and cell signaling [76-81]. A summary of the impact of ZEB1 on CRC [45, 47-50, 58, 82-87] is shown in Table 1.
Table 1
Summary of Functional impact of ZEB1 in CRC
| ZEB1 FUNCTION | EXPERIMENTAL MODEL | REFERENCE |
|---|---|---|
| Represses stemness-inhibiting microRNAs | CRC and pancreatic cancer cells | 82 |
| Inhibits senescence | SW480 and SW620 CRC cells | 83 |
| Promotes metastasis and loss of cell polarity | SW480, HCT-116 | 84 |
| Not required for EMT | LS174T CRC cells | 85 |
| Regulates miR-200c in EMT | human CRC sample analyses | 86 |
| ZEB1-hTERT complex inhibits E-cadherin expression | HCT-116 CRC cells | 58 |
| ZEB1 and TCF4 reciprocally modulate each other's transcriptional activity | SW480, SW620, and HCT-116 CRC cells | 50 |
| Regulates the plasminogen proteolytic system by inducing uPA and inhibiting PAI-1 | SW480, HCT116, and Colo320 CRC cells | 49 |
| Promotes vasculogenic mimicry through EMT induction | HCT-116 CRC cells | 87 |
| Promotes metastasis and loss of cell polarity | HCT-116, SW480 CRC cells | 84 |
| Represses E-cadherin expression, induces EMT | SW480 CRC cells | 47 |
| Promotes EMT | HCT-116 CRC cells | 45 |
| Promotes tumor invasiveness | HCT-116 and SW480 CRC cells | 48 |
Butyrate resistance, EMT, and colonic tumorigenesis
Studies of butyrate resistance [66-75] provide more information on signaling crosstalk affecting CRC development and behavior. HCT15 cells, which are p300 negative, are more resistant to the effects of butyrate than other CRC cells such as SW480 [70], and this report is consistent with what we have observed when evaluating these cell lines, as well as other, p300-deficient, cells [39 and unpublished data]. HCT-15 cells, which have been further selected for resistance through chronic exposure to butyrate, exhibit an EMT phenotype (albeit one relatively more epithelial-like [55]), and show greater ability for tumor growth, angiogenesis, and metastasis in an in vivo nude mouse model [70]. These observations suggest that presence of butyrate in the colonic microenvironment may select for phenotypically more aggressive, therapy-resistant tumor types during neoplastic progression. However, the overall incidence of colon cancer is reduced with a high fiber dietary intake, which may indicate a major preventive effect of butyrate at the initiating stage of neoplastic development. Thus, on the one hand, butyrate likely reduces the incidence of CRC; however, on the other hand, if CRC does develop despite the presence of higher levels of butyrate in the colonic microenvironment, the resulting tumors may be more aggressive and therapy-resistant.
Possible associations between HDACi resistance and EMT is also indicated by the finding that treatment of certain CRC cell lines with HDACis increases EMT-like phenotypes, as measured by changes in gene expression and cellular physiology [75]. Thus, there may be a connection between butyrate resistance developed as a result of chronic butyrate exposure, EMT, and the relative levels of CBP-Wnt activity vs. p300-Wnt activity. Of direct relevance to the connection between ZEB1 and HDACi resistance, a breast cancer cell line resistant to the HDACi phenylbutyrate exhibited increased expression of ZEB1, and ZEB1 inhibited the expression of genes (e.g., RAB25, ESRP1) that enhance phenylbutyrate sensitivity in these cells [76].
Hypothesis
Our central hypothesis is that ZEB1 cooperates with the downregulation of p300 and p300-Wnt activity in promoting both resistance to therapeutics and EMT in CRC. This hypothesis can be divided into two parts. First, we hypothesize that ZEB1 upregulation promotes both EMT and HDACi resistance in CRC cells. This is based on the observation that butyrate-resistant cells exhibit changes in gene expression consistent with EMT, such as increased expression of ZEB1 and vimentin and downregulated expression of E-cadherin [3]. ZEB1 expression and EMT are also associated with cancer cell dedifferentiation, which promotes drug (e.g., HDACi) resistance. Second, we posit that ZEB1 is a downstream effector of CBP-Wnt signaling that controls HDACi resistance and EMT, and is repressed by p300-Wnt signaling partially through miR-200 activity. ZEB1 is upregulated in butyrate-resistant cells that exhibit repressed expression of p300. Further, the association of ZEB1 with p300 changes ZEB1 function from that of a repressor to that of an activator of transcription [45,46]. Therefore, the relative levels of CBP- vs. p300-mediated Wnt signaling [28-31,38-41, 77] may affect both ZEB1 expression and function, influencing HDACi resistance and EMT [78] through altered gene expression in neoplastic colonic cells. In addition, we propose that there is reciprocal repression between ZEB1 and the miR-200 family of miRNAs, particularly miR-200c, and this modulation of expression influences EMT [79]. Furthermore, miR-200 family members, particularly miR-200a, downregulate Wnt signaling [80,81]; therefore, miR-200a and miR-200c may contribute to the ability of ZEB1 to integrate Wnt signaling, resistance to HDACis, and EMT. This hypothetical interaction network is summarized in Fig. 1.
Figure 1
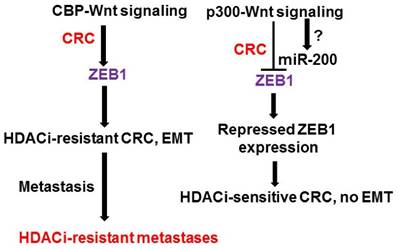
Proposed ZEB1 interactions in CRC. Deregulated Wnt signaling modulates gene expression, thus promoting CRC. CBP-Wnt signaling (left) is linked to proliferation, neoplasia, and cancer stem cell maintenance; whereas, p300-Wnt signaling (right) is associated with differentiation. We hypothesize that ZEB1 is upregulated in p300-deficient CRC cells through CBP-Wnt signaling. This results in enhanced EMT and altered gene expression leading to resistance to HDACis. The end result of these interactions is an HDACi-resistant metastatic CRC phenotype. Members of the miR-200 family repress ZEB expression, and are in turn repressed by ZEB factors, and some miR-200 members can repress Wnt signaling. The exact relationship between miR-200 family members and p300-mediated Wnt activity is unknown.
Testing the hypotheses
The first experimental objective would be to establish how altered expression of ZEB1 integrates HDACi-resistance and EMT in CRC cells. In addition, we would need to ascertain whether altered ZEB1 expression mediates effects of CBP-Wnt activity vs. p300-Wnt activity on HDACi-resistance and EMT, and the role of miRNA in these interactions.
The role of ZEB1 in promoting resistance to HDACis and mediating the effects of altered CBP-Wnt vs. p300-Wnt activity, can be determined by ZEB1 knockout in p300-deficient butyrate-resistant cells by CRISPR. The reverse experiment, in which ZEB1 is overexpressed in butyrate-sensitive, p300-expressing HCT-116 cells, can be achieved through stable transfections with a ZEB1 expression vector. Cells with ZEB1 knockout or overexpression would be assayed in absence or presence of butyrate and the clinically relevant HDACis vorinostat and LBH589 for: (a) Wnt activity, measured by reporter assays and levels of active (dephosphorylated) beta-catenin; (b) proliferation; (c) apoptosis; (d) clonal cell growth; (e) expression of vimentin and E-cadherin via Western blot analysis; (f) matrigel growth [43]; and (g) invasiveness.. HDACi-resistance would be evaluated by metrics a,b,c,d and EMT by metrics e,f,g,h.
To examine the role of p300 and relative levels of CBP-Wnt activity vs. p300-Wnt activity in affecting ZEB1 expression levels and activity, a number of methodologies can be used. One experimental model could be the p300 knockout CRC cells [43] that have EMT-like characteristics and that reverse their EMT phenotype by “rescue” transfection with a p300 expression vector. In addition, specific inhibitors that repress CBP-Wnt [28-31] and p300-Wnt [77] activity could be employed. CRISPR could also be utilized to create CBP knockout cells analogous to the p300 knockout lines.
In addition, in cells with modified CBP-Wnt activity vs. p300-Wnt activity, coimmunoprecipitation followed by Western blot can be utilized to measure the degree of association of ZEB1 with its corepressive (CtBP, BRG1) or coactivating (p300, PCAF) cofactors, to correlate this association with cell phenotype. ZEB1-cofactor association may mediate the effects of ZEB1 expression on cell physiology. For example, in biliary tract cancer cells, SMAD4 mediates the ability of the HDACi vorinostat to suppress EMT and reduce chemoresistance to gemcitabine; in this experimental model, vorinostat inhibited the nuclear translocation of SMAD4 and its interaction with ZEB1 and other EMT factors [78].
In this experimental approach, the levels of expression of relevant miR-200 family members would be measured in the relevant experimental cell models described above, and correlated to ZEB1 levels and cell physiology. A ZEB1 3' UTR expression vector, which is sensitive to translational control by miRNAs, can be utilized as a high throughput luciferase reporter to identify conditions (e.g., CBP-Wnt vs. p300-Wnt activity, HDACis) that affect the ability of miR200 family miRNAs to modulate ZEB1 expression. Thus, changes in ZEB1 expression that are mediated through interactions of miR-200 members with ZEB1 mRNA would alter expression from the ZEB1 3' UTR vector. If changes in 3' UTR reporter activity suggestive of involvement of miR-200 family members are observed, miRNA expression vectors (e.g., miR-200a and miR-200c) can be used to downregulate ZEB1 expression. This approach is expected to reverse the butyrate/HDACi-resistance and EMT characteristics observed in HCT-R and p300 knockout CRC cells.
Figure 2
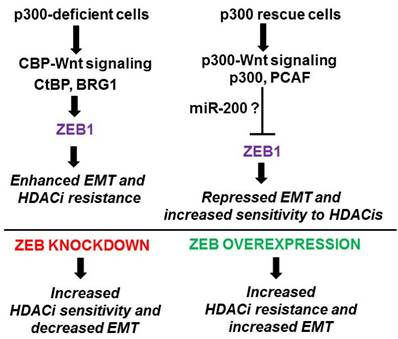
Expected results from proposed experiments. Cells deficient in p300 expression (e.g., HCT-R and p300 knockout HCT-116 cells) (top left) are expected to have relatively higher CBP-mediated Wnt activity, leading to higher levels of ZEB1 and association of ZEB1 with CtBP and BRG1. The resulting activity of ZEB1 as a transcription repressor would enhance EMT and resistance to HDACis. p300 rescue cells (top right) would exhibit the opposite pattern with increased p300-mediated Wnt signaling, decreased expression of ZEB1, and association of ZEB1 with p300 and PCAF. Lower levels of ZEB1, along with increased activity of ZEB1 as a transcription activator, would promote sensitivity to HDACis and repress EMT. We expect that the effects of p300-mediated Wnt signaling in repressing ZEB1 levels will be at least partially mediated by miR-200 family miRNAs. At bottom we show expected outcomes for knockdown or overexpression of ZEB1 in p300-deficient and p300 rescue cells, respectively.
Expected results
We expect (Fig. 2) ZEB1 knockout to decrease HDACi resistance and the EMT-like phenotype of butyrate-resistant cell lines; whereas, ZEB1 overexpression may induce a degree of butyrate/HDACi-resistance in butyrate-sensitive cells, along with a more pronounced EMT-like phenotype. The ZEB1 knockout cells should also be more sensitive to the pro-apoptotic and growth suppressing effects of HDACis, and should exhibit increased expression of E-cadherin and decreased expression of vimentin. The opposite will be expected in butyrate-sensitive cells with stable overexpression of ZEB1.
We expect that p300 rescue cells would exhibit increased sensitivity to HDACis, and decreased ZEB1 expression coupled to a lesser degree of EMT-like phenotype (measured by changes in vimentin/E-cadherin expression, matrigel-growth and invasion). If this is observed, we would determine whether exogenous overexpression of ZEB1 at least partially reverses these effects of restored p300 expression. This outcome would strongly suggest that ZEB1 is a major mediator of CBP-Wnt activity vs. p300-Wnt activity, and their downstream physiological outcomes.
In summary, CBP-Wnt signaling would be associated with increased resistance to HDACis, EMT, and expression of ZEB1; p300-Wnt signaling is expected to promote the opposite: HDACi sensitivity, less pronounced EMT behavior, and decreased expression of ZEB1. Enhanced CBP-Wnt signaling is expected to increase ZEB1 association with repressors such as CtBP and BRG1 and promote EMT; whereas, enhanced p300-Wnt signaling is expected to increase ZEB1 association with p300 and PCAF. We expect that the effects of p300-Wnt signaling on ZEB1 expression, and, consequently, on EMT and HDACi-resistance, would be at least partially mediated by miR-200 family miRNAs, particularly miR-200 and miR-200c.
Conclusion
This line of inquiry may lead to a comprehensive understanding of how ZEB1 modulates CBP- vs. p300-mediated Wnt signaling to influence EMT and resistance to HDACis. Expression of ZEB1 is correlated to poor outcomes in human cancer; thus ZEB represents a therapeutic target. The findings of the proposed studies can be utilized to design approaches that reverse HDACi resistance and inhibit EMT.
Abbreviations
CRC: colorectal cancer; EMT: epithelial to mesenchymal transition; HDACi: histone deactylase inhibitor; miRNA: microRNA; TSA: trichostatin A; CSCs: cancer stem cells.
Acknowledgements
MB and DL are supported by funding from Geisinger Commonwealth School of Medicine.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lazarova DL, Bordonaro M, Carbone R. et al. Linear relationship between WNT activity levels and apoptosis in colorectal carcinoma cells exposed to butyrate. Internat J Cancer. 2004;110:523-31
2. Bordonaro M, Lazarova DL, Sartorelli AC. The activation of beta-catenin by WNT signaling mediates the effects of histone deacetylase inhibitors. Exp Cell Res. 2007;313:1652-66
3. Chiaro C, Lazarova DL, Bordonaro M. Tcf3 and cell cycle factors contribute to butyrate resistance in colorectal cancer cells. Biochem Biophys Res Commun. 2012;428:121-6
4. Richter M, Jurek D, Wrba F. et al. Cells obtained from colorectal microadenomas mirror early premalignant growth patterns in vitro. Euro J Cancer. 2002;38:1937-45
5. Kautenberger T, Beyer-Sehlmeyer G, Festag G. et al. The gut fermentation product butyrate, a chemopreventive agent, suppresses glutathione S-transferase theta (hGSTT1) and cell growth more in human colon adenoma cells (LT97) than tumor (HT29) cells. J Cancer Res Clin Oncol. 2005;131:692-700
6. Bingham SA, Day NE, Luben R. et al. Dietary fibre in food and protection against colorectal cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC): an observational study. Lancet. 2003;361:1496-501
7. Peters U, Sinha R, Chaterjee N. et al. Dietary fibre and colorectal adenoma in a colorectal cancer early detection programme. Lancet. 2003;361:1491-5
8. Bingham SA, Norat T, Moskal A. et al. Is the association with fiber from foods in colorectal cancer confounded by folate intake? Cancer Epid Biom Prev. 2005;14:1552-6
9. Cummings JH, Pomare EW, Branch WJ. et al. Short chain fatty acids in human large intestine, portal, hepatic, and venous blood. Gut. 1987;28:1221-7
10. Belcheva A, Irrazabal T, Robertson SJ. et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell. 2014;158:288-99
11. Heerdt BG, Houston MA, Augenlicht LH. Potentiation by specific short-chain fatty acids of differentiation and apoptosis in human colonic carcinoma cell lines. Cancer Res. 1994;54:3288-94
12. Hague A, Elder DJ, Hicks DJ. et al. Apoptosis in colorectal tumor cells: induction by the short chain fatty acids butyrate, propionate and acetate and by the bile salt deoxycholate. Internat J Cancer. 1995;60:400-6
13. Medina V, Young GP, Edmonds B. et al. Induction of caspase-3 protease activity and apoptosis by butyrate and another inhibitor of histone deacetylase: dependence on protein synthesis and synergy with a mitochondrial/cytochrome c-dependent pathway. Cancer Res. 1997;57:3697-707
14. Marks PA, Dokmanovic M. Histone deacetylase inhibitors: discovery and development as anticancer agents. Expert Opin Investig Drugs. 2005;12:1497-511
15. Lin HY, Chen CS, Lin SP. et al. Targeting histone deacetylase in cancer therapy. Med Res Rev. 2006;26:397-413
16. Bordonaro M, Mariadason JM, Aslam F. et al. Butyrate-induced apoptotic cascade in colonic carcinoma cells: modulation of the beta-catenin-Tcf pathway and concordance with effects of sulindac and trichostatin A but not curcumin. Cell Growth Differ. 1999;10:713-20
17. Li VSW, Ng SS, Boersema PJ. et al. WNT signaling through inhibition of β-catenin degradation in an intact Axin1 complex. Cell. 2012;149:1245-56
18. Cook D, Fry M, Hughes K. et al. Wingless inactivates glycogen synthase kinase-3 via an intracellular signaling pathway which involves a protein kinase C. EMBO J. 1996;15:4526-36
19. Rubinfeld B, Souza B, Albert I. et al. Association of the APC gene product with β-catenin. Science. 1993;262:1731-4
20. Su LK, Vogelstein B, Kinzler KW. Association of the APC tumor suppresser protein with catenins. Science. 1993;262:1734-7
21. Munemitsu S, Albert I, Souza B. et al. Regulation of intracellular β-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci USA. 1995;92:3046-50
22. Behrens J, Von Kries JP, Kuhl M. et al. Functional interaction of β-catenin with the transcriptional factor LEF-1. Nature. 1996;381:638-42
23. Molenaar M, Van De Wetering M, Osterwegel M. XTcf-3 transcription factor mediates β-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86:391-9
24. Korinek V, Barker N, Morin PJ. et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997;275:1774-7
25. Morin J, Sparks AB, Korinek V. et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1777-90
26. Bienz M, Clevers H. Linking colorectal cancer to WNT signaling. Cell. 2000;103:311-20
27. Emami KH, Nguyen C, Ma H. et al. A small molecule inhibitor of β-catenin/CREB-binding protein transcription. Proc Natl Acad Sci USA. 2004;101:12681-7
28. McMillan M, Kahn M. Investigating WNT signaling: a chemogenomic safari. Drug Disc Today. 2005;10:1467-74
29. Teo JL, Ma H, Nguyen C. et al. Specific inhibition of CBP/β-catenin interaction rescues defects in neuronal differentiation caused by presenilin-1 mutation. Proc Natl Acad Sci USA. 2005;102:12171-6
30. Ma H, Nguyen C, Lee KS. et al. Differential roles for the coactivators CBP and p300 on TCF/β-catenin-mediated survivin gene expression. Oncogene. 2005;24:3619-31
31. Miyabayashi T, Teo JL, Yamamoto M. et al. WNT/β-catenin/CBP signaling maintains long-term murine stem cell pluripotency. Proc Natl Acad Sci USA. 2007;104:5668-73
32. Teo JL, Kahn M. The WNT signaling pathway in cellular proliferation and differentiation: a tale of two coactivators. Adv Drug Delivery Rev. 2010;62:1149-55
33. Iyer NG, Ozdag H, Caldas C. p300/CBP and cancer. Oncogene. 2004;23:4225-31
34. Hecht A, Vleminckx K, Stemmler MP. et al. The p300/CBP acetyltransferases function as transcriptional coactivators of beta-catenin in vertebrates. EMBO J. 2000;19:1829-50
35. Bryan EJ, Jokubaitis VJ, Chamberlain NL. et al. Mutation analysis of EP300 in colon, breast and ovarian carcinomas. Int J Cancer. 2002;102:137-141
36. Levy L, Wei Y, Labalette C. et al. Acetylation of beta-catenin by p300 regulates beta-catenin-Tcf4 interaction. Mol Cell Biol. 2004;24:3404-14
37. Takahashi-Yanaga F, Kahn M. Targeting WNT signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. 2010;16:3153-62
38. Lazarova DL, Chiaro C, Wong T. et al. CBP activity mediates effects of the histone deacetylase inhibitor butyrate on WNT activity and apoptosis in colon cancer cells. J. Cancer. 2013;4:480-90
39. Lazarova DL, Wong T, Chiaro C. et al. p300 influences butyrate-mediated WNT hyperactivation in colorectal cancer cells. J. Cancer. 2013;4:491-501
40. Lazarova DL, Lee A, Wong T. et al. Modulation of Wnt Activity and cell physiology by butyrate in LT97 microadenoma cells. J. Cancer. 2014;5:203-13
41. Bordonaro M, Lazarova DL. CBP, p300, butyrate, and Wnt signaling in colorectal cancer. World J Gastroenterology. 2015;21:8138-48
42. Bordonaro M, Drago E, Atamna W. et al. Comprehensive suppression of all apoptosis-induced proliferation pathways as a proposed approach to colorectal cancer prevention and therapy. PLoS One. 2014;9:e115068
43. Krubasik D, Iyer NG, English WR. et al. Absence of p300 induces cellular phenotype changes characteristic of epithelial to mesenchyme transition. Br J Cancer. 2006;94:1326-32
44. Sanches-Tillo E, Siles L, de Barrios O. et al. Expanding roles of ZEB factors in tumorigenesis and tumor progression. Am J Cancer. 2012;2:897-912
45. Postigo AA. Opposing functions of ZEB proteins in the regulation of the TGFβ/BMP signaling pathway. EMBO J. 2003;22:2243-52
46. Mizuguchi Y, Specht S, Lunz III JG. et al. Cooperation of p300 and PCAF in the control of microRNA 200c/141 transcription and epithelial characteristics. PLoS One. 2012;7:e32449
47. Sanchez-Tillo E, Lazaro A, Torrent R. et al. ZEB1 represses E-cadherin and induces EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene. 2010;29:3490-500
48. Sanchez-Tillo E, de Barrios O, Siles L. et al. β-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc Natl Acad Sci USA. 2011;108:19204-9
49. Sanchez-Tillo E, de Barrios O, Siles L. et al. ZEB1 promotes invasiveness of colorectal carcinoma cells through the opposing regulation of uPA and PAI-1. Clin Cancer Res. 2013;19:1071-81
50. Sanchez-Tillo E, de Barrios O, Valls E. et al. ZEB1 and TCF4 reciprocally modulate their transcriptional activities to regulate Wnt target gene expression. Oncogene. 2015;34:5760-70
51. Pena C, Garcia JM, Garcia V. et al. The expression levels of the transcriptional regulators p300 and CtBP modulate the correlations between SNAIL, ZEB1, E-cadherin and vitamin D receptor in human colon carcinomas. Int J Cancer. 2006;119:2098-104
52. Aigner K, Dampier B, Descovich L. et al. The transcription factor ZEB1 (δEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene. 2007;26:6978-88
53. Moheimani F, Roth HM, Cross J. et al. Disruption of β-catenin/CBP signaling inhibits human airway epithelial-mesenchymal transition and repair. Int J Biochem & Cell Biol. 2015;68:59-69
54. Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science. 2015;349:1482-8
55. Buck E, Eyzaguirre A, Barr S. et al. Loss of homotypic cell adhesion by epithelial-mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition. Mol Cancer Ther. 2007;6:532-41
56. Ono H, Imoto I, Kozaki K. et al. SIX1 promotes epithelial-mesenchymal transition in colorectal cancer through ZEB1 activation. Oncogene. 2012;31:4923-34
57. Hu Z, Wang J. Histone deacetylase inhibitor induces the expression of select epithelial genes in mouse utricle sensory epithelia-derived progenitor cells. Cell Reprogram. 2014;16:266-75
58. Qin Y, Tang B, Hu CJ. et al. A hTERT/ZEB1 complex directly regulates E-cadherin to promote epithelial to mesenchymal transition (EMT) in colorectal cancer. Oncotarget. 2015;7:351-61
59. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741-51
60. Zaravinos A. The regulatory role of microRNAs in EMT and cancer. J Oncol. 2015:articleID865806
61. Jolly MK, Jia D, Boareto M. et al. Coupling the modules of EMT and stemness: A tunable 'stemness window' model. Oncotarget. 2015;6:25161-74
62. Roca H, Hernadez J, Weidner S. et al. Transcription Factors OVOL1 and OVOL2 Induce the Mesenchymal to Epithelial Transition in Human Cancer. PLoS One. 2013 DOI: 10.1371/journal.pone.007633
63. Ring A, Kim Y-M, Khan M. Wnt/catenin signaling in adult stem cell physiology and disease. Stem Cell Rev. 2014;10:512-25
64. Sellerio AL, Ciusani E, Ben-Moshe NB. et al. Overshoot during phenotypic switching of cancer cell populations. Sci Rep. 2015;5:15464
65. Medema JP, Vermeulen L. Microenvironmental regulation of stem cells in intestinal homeostasis and cancer. Nature. 2011;474:318-26
66. Lopez de Silanes I, Olmo N, Turnay J. et al. Acquisition of resistance to butyrate enhances survival after stress and induces malignancy of human colon carcinoma cells. Cancer Res. 2004;64:4593-4600
67. Gill RK, Dudeja PK. A novel fact to consider for the effects of butyrate on its target cells. Focus on “The short-chain fatty acid butyrate is a substrate of breast cancer resistance protein.” Am J Physiol Cell Physiol. 2011;301:C93-9
68. Goncalves P, Gregorio I, Martel F. The short-chain fatty acid butyrate is a substrate of breast cancer resistance protein. Am J Physiol Cell Physiol. 2011;301:C983-94
69. Shi J, Jiang X, Yu Z. et al. ZNRF3 contributes to the growth of lung carcinoma via inhibiting Wnt/b-catenin pathway an is regulated by miR-93. Tumour Biol. 2016;37:3051-7
70. Serpa J, Caiado F, Carvalho T. et al. Butyrate-rich microenvironment is a relevant selection factor for metabolically adapted tumor cells. J Biol Chem. 2010;285:39211-23
71. Kim AY, Kwak JH, Je NK. et al. Epithelial-mesenchymal transition is associated with acquired resistance to 5-flurouracil in HT-29 colon cancer cells. Toxicol Res. 2015;31:151-6
72. Lazarova DL, Bordonaro M. Extreme fluctuations in Wnt/beta-catenin signaling as an approach for colon cancer prevention and therapy. Advanced Studies in Biology. 2012;4:351-62
73. Bordonaro M, Tewari S, Cicco CE. et al. A switch from canonical to noncanonical Wnt signaling mediates drug resistance in colon cancer cells. PLoS One. 2011;6:e27308. doi: 10.1371/journal.pone.0027308
74. Lazarova DL, Bordonaro M, Sartorelli AC. Transcriptional regulation of the vitamin D3 receptor by ZEB. Cell Growth Diff. 2001;12:319-6
75. Ji M, Lee EJ, Kim KB. et al. HDAC inhibitors induce epithelial-mesenchymal transition in colon carcinoma cells. Oncol Rep. 2015;33:2299-308
76. Kikuchi M, Yamashita K, Waraya M. et al. Epigenetic regulation of ZEB1-RAB25/ESRP1 axis plays a critical role in phenylbutyrate treatment-resistant breast cancer. Oncotarget. 2016;7:1741-53
77. Higuchi Y, Nguyen C, Yasuda SY. et al. Specific direct small Molecule p300/β-catenin antagonists maintain stem cell potency. Curr Mol Pharmacol. 2016;9:272-9
78. Sakamoto T, Kobayashi S, Yamada D. et al. A histone deacetylase inhibitor suppresses epithelial-mesenchymal transition and attenuates chemoresistance in biliary tract cancer. PLoS One. 2016;11:e0145985
79. Burk U, Schubert J, Wellner U. et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9:581-588
80. Su J, Zhang A, Shi Z. et al. MicroRNA-200a suppresses the Wnt/β-catenin signaling pathway by interacting with β-catenin. Int J Oncol. 2012;40:1162-70
81. Ghahhari NM, Babahah S. Interplay between microRNAs and WNT/β-catenin signaling pathway regulates epithelial-mesenchymal transition in cancer. Euro J Cancer. 2015;51:1638-49
82. Wellner U, Schubert J, Burk UC. et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487-95
83. de Barrios O, Győrffy B, Fernández-Aceñero MJ. et al. ZEB1-induced tumourigenesis requires senescence inhibition via activation of DKK1/mutant p53/Mdm2/CtBP and repression of macroH2A1. Gut. 2017;66:666-82
84. Spaderna S, Schmalhofer O, Wahlbuhl M. et al. The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res. 2008;68:537-44
85. Jägle S, Dertmann A, Schrempp M. et al. ZEB1 is neither sufficient nor required for epithelial-mesenchymal transition in LS174T colorectal cancer cells. Biochem Biophys Res Commun. 2017;48:1226-1232
86. Muto Y, Suzuki K, Kato T. et al. Heterogeneous expression of zinc-finger E-box-binding homeobox 1 plays a pivotal role in metastasis via regulation of miR-200c in epithelial-mesenchymal transition. Int J Oncol. 2016;49:1057-67
87. Du J, Sun B, Zhao X. et al. Hypoxia promotes vasculogenic mimicry formation by inducing epithelial-mesenchymal transition in ovarian carcinoma. Gynecol Oncol. 2014;133:575-83
Author contact
![]() Corresponding author: Geisinger Commonwealth School of Medicine, 525 Pine Street, Scranton, PA 18509. Tel: 570-504-9646; Fax: 570-504-9636; Email: mbordonaroedu
Corresponding author: Geisinger Commonwealth School of Medicine, 525 Pine Street, Scranton, PA 18509. Tel: 570-504-9646; Fax: 570-504-9636; Email: mbordonaroedu
Citation styles
APA
Lazarova, D., Bordonaro, M. (2017). ZEB1 Mediates Drug Resistance and EMT in p300-Deficient CRC. Journal of Cancer, 8(8), 1453-1459. https://doi.org/10.7150/jca.18762.
ACS
Lazarova, D.; Bordonaro, M. ZEB1 Mediates Drug Resistance and EMT in p300-Deficient CRC. J. Cancer 2017, 8 (8), 1453-1459. DOI: 10.7150/jca.18762.
NLM
Lazarova D, Bordonaro M. ZEB1 Mediates Drug Resistance and EMT in p300-Deficient CRC. J Cancer 2017; 8(8):1453-1459. doi:10.7150/jca.18762. https://www.jcancer.org/v08p1453.htm
CSE
Lazarova D, Bordonaro M. 2017. ZEB1 Mediates Drug Resistance and EMT in p300-Deficient CRC. J Cancer. 8(8):1453-1459.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.