Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(12):2184-2190. doi:10.7150/jca.19452 This issue Cite
Research Paper
Loss of Complement Factor H in Plasma Increases Endothelial Cell Migration
Ju Liu1, ![]() , Josephine Hoh2,
, Josephine Hoh2, ![]()
1. Medical Research Center, Shandong Provincial Qianfoshan Hospital, Shandong University, 16766 Jingshi Road, Jinan, Shandong China 250014
2. Department of Epidemiology and Public Health, Yale University, 60 College Street, New Haven, CT 06520, USA.
Received 2017-2-2; Accepted 2017-4-22; Published 2017-7-15
Citation:
Liu J, Hoh J. Loss of Complement Factor H in Plasma Increases Endothelial Cell Migration. J Cancer 2017; 8(12):2184-2190. doi:10.7150/jca.19452. https://www.jcancer.org/v08p2184.htm
Other stylesAbstract
Tumor growth depends on angiogenesis, the growth of new blood vessels. Complement factor H (CFH) is a plasma glycoprotein that functions as a regulator of the complement system. The aim of this study is to delineate the role of CFH in angiogenesis. A conditional null allele of the Cfh gene was generated in C57BL/6J mice by flanking the exon 3 with loxP sites. The Cfhflox/flox mice were crossed with Rosa26-Cre mice to obtain the mice homozygotes of Cfh deletion (Cfh-/-). The Cfh-/- mice were examined by in vivo angiogenesis assays. Mouse endothelial cells were treated with media containing 5% of mouse plasma from the wildtype or Cfh-/- mice and assayed for proliferation, viability and migration. The Cfh-/- mice did not display any obvious abnormalities. They demonstrated a pro-angiogenic phenotype in matrigel plug assay, but not in aorta ring assay. In vitro, loss of Cfh in plasma does not affect proliferation or viability, but significantly increases migration of mouse endothelial cells. Our findings suggest that plasma CFH inhibits angiogenesis by reduction of endothelial cell migration. Thus the mutation of CFH might lead to excessive tumor angiogenesis.
Keywords: complement factor H, angiogenesis, endothelial cell, migration
Introduction
Angiogenesis refers to the formation of new blood vessels from the preexisting vasculature [1]. Since newly-formed capillaries provide nutrients and oxygen to the proliferating tumor cells, angiogenesis is pivotal for maintenance and progression of solid tumors [2, 3]. Tumor cells secret specific growth factors such as vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) to stimulate proliferation and migration of the naturally quiescent endothelial cells, leading to the formation of new vessel structures and blood circulation [4]. The process of angiogenesis is regulated by continuous interactions of its stimulators and inhibitors, including growth factors, enzymes, lipids and chemicals [1, 5]. Anti-angiogenesis therapy has recently emerged as an essential adjunct to the chemotherapy of certain cancers [6-8].
The complement system is an enzyme cascade that helps defend against infection and clear pathogens [9]. Three complement pathways, the classical, the alternative, and the mannose-binding lectin pathway, converge into a final common pathway when C3 convertase cleaves C3 into C3a and C3b [10]. C3 cleavage results in formation of the membrane attack complex (MAC), the cytotoxic component of the complement system [9]. Complement proteins were observed in angiogenic vessels [11], but their roles in angiogenesis has not been fully understood. Activation of the complement system is tightly regulated by complement control proteins, which are present at a high concentration in the blood plasma [12].
Complement factor H (CFH), a serum glycoprotein mainly produced in the liver, is a negative regulator of the alternative pathway of complement activation [13]. Using genome-wide association study (GWAS), we firstly identified that a tyrosine to histidine change at amino acid 402 (Y402H has a T to C substitution at nucleotide 1277 in exon 9) of the CFH gene is significantly associated with age-related macular degeneration (AMD) [14]. Later, a number of other studies proved the strong association of the CFH polymorphism and AMD [15]. AMD, the leading cause of visual disability in the elderly, is characterized by formation of drusen and neovascularization of the choriocapillaris [16]. However, it is not clear whether the mutation of CFH leads to abnormal angiogenesis. In this study, we generated mice with deletion of the Cfh gene and explored the role of CFH on angiogenesis.
Materials and Methods
Animals
The mice with a Cfh floxed allele were generated by Ozgene (Bentley, Australia). Briefly, a FRT-flanked PGK-neomycin selection cassette was inserted downstream of exon 3 of the mouse Cfh gene. The loxP sites were inserted upstream of exon 3 and between exon 4 and the selection cassette. The construct was incorporated into the genomic DNA of embryonic stem (ES) cells from C57BL/6J mice by electroporation following selection. The targeted ES cells were injected into C57BL/6J blastocysts, which were transferred into the uterus of recipient mice to produce chimeric mice. The chimeric mice were crossed to C57BL/6J mice to generate Cfh+/flox-neo mice, which were crossed to Rosa26-FlpE mice (provided by Ozgene) to remove the selection cassette. The Cfh+/flox mice were crossed to Rosa26-Cre mice to create a Cfh null allele. All the mice were maintained on C57BL/6J background. The probes used for Southern blot analysis of ES cell clones and Cfh alleles in mice were summarized in Table 1. Animals were kept in a conventional, pathogen-free facility at the Yale University School of Medicine, and all experiments were carried out in accordance with guidelines prescribed by the Institutional Animal Care and Use Committee at Yale University. Genotyping was performed performed using the REDExtract-N-Amp Tissue PCR Kit (Sigma-Aldrich, St. Louis, MO) according to manufacturer's directions. The following primers were used for PCR genotyping of Cfh alleles: sense, 5'-GCACATAAATAGCTGGATGGACCTG-3'; anti-sense, 5'-CCTTATGTAGGAGTCACGAAAGCAC-3'.
Gene expression analysis
The livers were collected from wildtype and Cfh-/- mice after intra-cardiac perfusion with cold PBS and homogenized in TRIzol® reagent (Invitrogen Life Technologies, Carlsbad, CA). Total RNA was extracted by the RNeasy kit with optional DNaseI treatment (Qiagen, Hilden, Germany). cDNA synthesis was performed using High Capacity RNA-to-cDNA Master Mix (Applied Biosystems, Calsbard, CA) and the newly synthesised cDNA was used as a template for RT‑qPCR. The 20 μl PCR reaction mix consisted of 10 μl 2X SYBR Green (ThermoFisher, Grand Island, NY), 1 μl cDNA template, 1 μl forward and reverse primers, and 8 μl deionised water. qRT-PCR was performed using CFX96 Real-Time PCR Detection System (Bio-Rad). The primer sequences were summarized in Table 2. RNA levels were normalized to the endogenous control gene mouse β-actin. All PCR reactions were repeated in triplicate.
Table 1
The primers used in the production the probes for Southern blotting.
| Probe | Sequence | Size (bp) | Tm (0C) |
|---|---|---|---|
| 5' probe | |||
| Sense | TGACTGATGGACTACAGGAATGAACAG | 510 | 60 |
| Anti-sense | TTACTGAATATAGCTCACCTGGAATTTTCA | ||
| 3' probe | |||
| Sense | CCTTGAGGTCTTAGGTTTGACACAC | 504 | 61 |
| Anti-sense | CAATGGAAGTAGGACACAGCATTTG | ||
| en probe | |||
| Sense | AGCTCACTGCTGATCTCAAATACTTCAAAG | 541 | 59 |
| Anti-sense | GGAGTGCCTAAGCACAAATTGTAGG |
Note: All sequences are in the 5' to 3' orientation
Table 2
RT-PCR primer sequences
| Gene | Sequence | Size (bp) | Tm (0C) |
|---|---|---|---|
| mCFH | |||
| Sense | CTCCTGGTCAGAACAACTATATCCAG | 351 | 61.5 |
| Anti-sense | AGTTCTGTCACAGGTAGACACTTCAC | ||
mβ-actin | |||
| Sense | CGGTTCCGATGCCCTGAGGCTCTT | 100 | 60 |
| Anti-sense | CGTCACACTTCATGATGGAATTGA |
Note: All sequences are in the 5' to 3' orientation
Western blotting
The blood of wildtype and Cfh-/- mice was collected via the retro-orbital sinus, and the serum was prepared in sample buffer (Bio-Rad) with β-mercaptoethanol. The serum samples were electrophoresed on 7% Tris-HCl gradient gels, and the protein bands were transferred onto PVDF membranes (Bio-Rad). The membranes were blocked overnight with 5% non-fat dry milk (EMD Millipore, Billerica, MA) and then incubated overnight with a sheep anti-Factor H antibody (1:500; Abcam, Cambridge, MA) in PBS-T at 4oC. After washing, the membranes were probed with horseradish peroxidase (HRP)-conjugated donkey anti-sheep secondary antibody (1:2500; Santa Cruz Biotechnology, Santa Cruz, CA). The blot signals were detected by enhanced chemiluminescence (ThermoFisher) and exposed on X-ray films.
Aortic ring assay
The two-month old WT and Cfh-/- mice were euthanized mice by perfusion with 10 ml cold PBS. The aortic ring was excised from mice, cut into 1 mm lengths and implanted in growth factor-reduced matrigel (BD Biosciences, San Jose, CA). The aortic explants were incubated with EBM-2 supplemented with 0.5% FBS for 6 days. Capillaries outgrowing from aortas was photographed with a digital camera linked to a Zeiss invert microscope with an objective lens and analyzed using ImageJ software (NIH, Bethesda, MA). Four rings were used for each assay that was repeated at least 3 times.
Matrigel plug assay
After thawing at 4°C overnight, 0.5 ml matrigel (BD Biosciences) was injected subcutaneously into each of the two-month old WT and Cfh-/- mice. At day 10, all the mice were sacrificed, and the matrigel plug was removed and embedded in OCT. The matrigel plugs were sectioned at 7μm and immunostained with a rat anti mouse CD31 antibody (BD Biosciences). The microvascular density count was carried out as previously described [17].
Cell culture
The immortalized mouse brain endothelial cells (bEnd.3) were purchased from ATCC (Manassas, VA) and cultured in basal endothelial cell medium (EBM2) supplemented using the EGM-2-MV bullet kit (Lonza, Basel, Switzerland). The cells were cultured in humidified air at 37oC with 5% CO2.
Proliferation assays
Cell proliferation was evaluated with a MTT assay kit (Cayman Chemical, Ann Arbor, MI). Briefly, cells were seeded at a density of 5x103 cells/well in a 96-well plate. The next day, the culture media was replaced with EBM2 basal media with 5% of mouse plasma from the WT and the Cfh-/- mice. After 24 hrs, cells were washed with PBS and MTT solution (10 μl of 5 mg/mL) was added to each well for 2 hrs. Formazan crystals were solubilized and colorimetric intensity was analyzed using a 96-well plate reader (Molecular Devices, Sunnyvale, CA) at a wavelength of 570 nm. Each experiment was repeated 4 times.
Cell viability assay
The viability of mouse endothelial cells was assessed after 24 hr incubation of EBM2 basal media with 5% of mouse plasma from the WT and the Cfh-/- mice. Cultures were washed and incubated in 0.05% Trypsin for 2 min at 37℃. After disaggregation, cell suspensions were diluted 1:1 in 0.4% Trypan blue (w/v in 0.9% NaCl) (Santa Cruz Technology) and the percentage of dye-free cells was calculated.
Boyden chamber migration assay
Cell migration assay was performed using modified 24-well Boyden chambers (Costar, New York, NY) containing a polycarbonate membrane with 5.0 μm pores. Mouse endothelial cells were starved in basic EBM2 (serum/growth factor-free) for 12 hours at 37°C before being harvested with trypsin and resuspended in EBM2 basal media with 5% of mouse plasma from the WT or the Cfh-/- mice. The single cell suspension was seeded at 1×105 cells/well in the upper chamber, and 0.5 ml EBM2 with 20 ng/ml of VEGF (Lonza) was added to the bottom chamber as chemoattractants. After 12 hrs, the migrated cells on the bottom surface were stained with 0.1% crystal violet and counted under microscope.
Wound-healing assay
A wound-healing assay was used to assess the migration process [18]. Mouse endothelial cells were seeded at a density of 5×105 cells/ml in 6-well flat-bottom plates and allowed to adhere overnight. At 95 % confluence, wounds were made using a sterile 10 μl pipette tip and the wells were washed twice with PBS to remove cellular debris. Then EBM basal medium with 5% of mouse plasma from the WT or the Cfh-/- mice was added, and photographs were taken immediately (time zero) through an inverted microscope (Leica, Wetzlar, Germany). The mouse endothelial cells were allowed to migrate for 12 hr and photographed again. The experiments were carried out 4 times. Measurements were performed on digital images using the ImageJ software (NIH). At least 10 images per treatment were analysed.
Electric Cell-Substrate Impedance Sensing (ECIS) Analysis
The real time wound healing assay was measured using the ECIS technique (ECIS model 1600; Applied Biophysics, Troy, NY) [19]. Briefly, 8-well ECIS arrays (8W10E+) were coated with L-cysteine. Mouse endothelial cells were plated in the wells and allowed to form monolayers. EBM2 basal media with 5% of mouse plasma from the WT or the Cfh-/- mice were added to the wells. Then the AC current was given to the electrodes located at the center of each well, and the cells on the electrodes were killed. The wounded areas of each well were gradually healed by migration of viable cells and the migratory response was measured by recording the trans-endothelial resistance (TER). Data plots are representative of triplicate experiments.
Figure 1
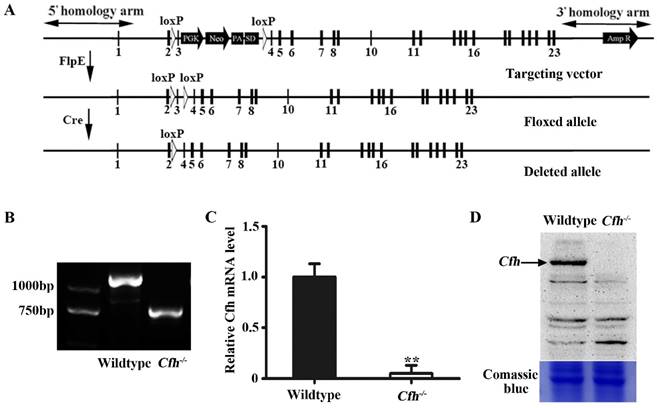
Validation and analyses of mice carrying Cfh null allele. (A) Schematic representation of the floxed and deleted alleles of Cfh conditional null mice. Exons are represented by rectangles. Two loxP sites (white triangles) were inserted between exon 2/3 and 3/4. The floxed sequences were deleted after Cre excision. (B) Genotyping of the ear tissue from WT and Cfh-/- mice. PCR produced a 1180bp band for the WT allele and a 730bp band for the Cfh allele. (C) Quantitative RT-PCR analysis of Cfh gene expression in the liver from WT and Cfh-/- mice. Each bar represents mean SE for 3 individual mice of same genotype. **, p<0.01. (D) Immunoblots of Cfh of the plasma from WT and Cfh-/- mice. Comassie blue staining of the gel was used as loading control.
Statistical analysis
Statistical significances were assessed by a paired-samples t test. A value of p<0.05 was considered significant.
Results
We generated a conditional null allele of Cfh gene in C57BL/6J mice by flanking the exon 3 with loxP sites (Fig. 1A). The Cfhflox/flox mice were crossed with Rosa26-mice to obtain the mice homozygotes of Cfh deletion (Cfh-/-). The genotypes of the Cfh-/- mice were validated by PCR of the DNA extract from the ear tissue (Fig. 1B). The loss of Cfh expression was verified by RT-PCR analyses of the mouse Cfh mRNA from mouse liver and Western blotting of Cfh protein of the mouse plasma (Fig. 1C, D). Consistent to other studies [20], the Cfh-/- mice did not display any obvious phenotype in the vascular system.
Next, the effect of Cfh on angiogenesis in adult mice was examined by aorta ring assay and matrigel plug assay. The vessel growth was similar in the aorta rings from WT and Cfh-/- mice (p=0.22), suggesting that the loss of Cfh gene in vascular cells does not affect anigiogenesis (Fig. 2A, B). However, the number of capillaries penetrated into the matrigel plugs is significantly higher in Cfh-/- mice than those in WT mice (p<0.05) (Fig. 2C, D). CFH is a plasma protein and directly interacts with the endothelium of blood vessels. Thus, plasma Cfh contributes to angiogenesis in the matrigel plug assay but not the aorta ring assay, in which the aorta rings were cultured ex vivo and prevented from interaction with blood plasma.
To define the role of plasma Cfh, the mouse plasma was obtained by cardiac puncture and used for in vitro angiogenesis assays. The mouse endothelial cells were cultured with EBM2 basal media with 5% of mouse plasma from the WT and the Cfh-/- mice. As detected by MTT assay and Trypan blue exclusion assay, loss of Cfh in mouse plasma does not affect proliferation or viability of mouse endothelial cells (p=0.14, p=0.27 respectively; Fig. 3A, B). Cell migration was examined by Boyden chamber-type assays. The number of mouse endothelial cells migrating across the polycarbonate membrane was significantly increased in the groups using the plasma from Cfh-/- mice (p<0.01) (Fig. 4A, B). Cell migration during in vitro wound healing mimics the process of endothelial cell motility in vivo. After treatment with the media containing mouse plasma, the migrated area of mouse endothelial cells was significantly increased in Cfh-/- groups compared to WT groups (p<0.05) (Fig. 4C, D). To capture the subtle alterations of cell migration, we implemented ECIS system, which allows for real-time measurements of resistance caused by migration of endothelial cells. As shown on Fig. 4E, F, the transendothelial resistance was constantly increased in the wound area of mouse endothelial cells cultured with Cfh-/- mouse plasma (p<0.01). Taken together, these in vitro assays indicated that loss of Cfh in plasma increases endothelial cell migration.
Discussion
In this study, we generated a conditional null allele of the Cfh gene in C57BL/6J mice, and obtained Cfh-/- mice which did not display any obvious abnormalities. The Cfh-/- mice demonstrated a pro-angiogenic phenotype in matrigel plug assay. In vitro, the plasma from Cfh-/- mice did not affect endothelial cell growth, but significantly increases endothelial cell migration. By reconstitution of the microenvironment of choroid, Zhang et al. observed that knockdown of CFH in retinal pigmented epithelial cells (ARPE-19 cell line) promotes endothelial cell migration [21]. However, CFH has a relatively large size (139 kD) with a number of protein binding sites [22]. After produced by retinal pigmented epithelium, CFH might be difficult to migrate across peri-vascular tissues, particularly basement membrane, to interact with endothelial cells, the inner layer of the blood vessels. Our model system is unique as it precisely determines the roles of plasma Cfh on angiogenesis both in vitro and in vivo. The plasma Cfh, which is primarily produced from the liver, interacts with other plasma proteins and functions directly on the endothelial cells [23]. In addition, plasma proteins may be targeted by delivering their regulators directly into the blood circulation. In case of their deficiency, injection of recombinant proteins becomes an effective therapy [24]. Based on our findings, recombinant human CFH might be developed as a component for anti-angiogenesis therapy.
Figure 2
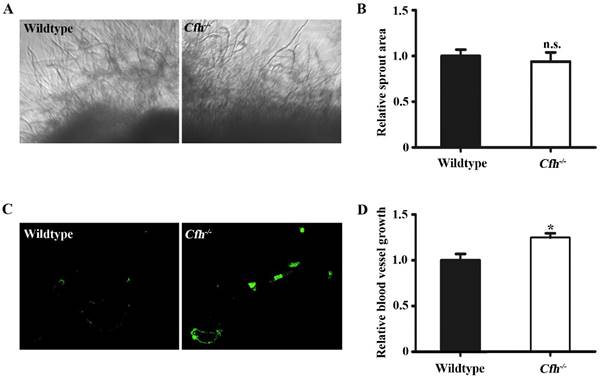
In vivo angiogenesis analyses of the WT and Cfh-/- mice. (A) Representative images of the capillary sprouts from the aorta rings of from WT and Cfh-/- mice. Magnification: 20X. (B) Relative sprout area of aorta rings from WT and Cfh-/- mice. n=6, n.s. non-significant. (C) Representative images of CD31 immunostaining on the sections of matrigel plugs injected into WT and Cfh-/- mice. Magnification: 200X. (D) Relative blood vessel growth of matrigel plugs injected into WT and Cfh-/- mice. n=6, *, p<0.05.
Figure 3
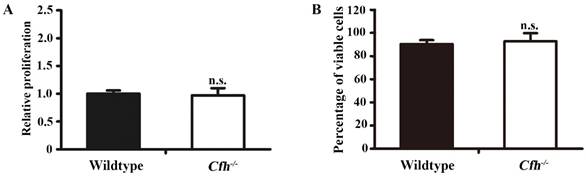
Proliferation and viability assays of mouse endothelial cells treated with plasma from the WT and Cfh-/- mice. MTT assay (A) and trypan blue viability assay (B) of mouse endothelial cells treated with EBM2 basal media with 5% of mouse plasma from the WT and the Cfh-/- mice for 24 hours; n=4; n.s., non-significant
Figure 4
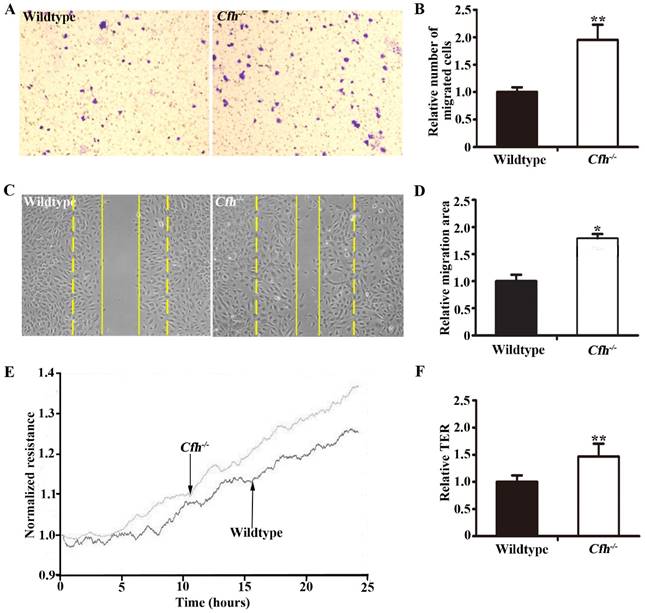
Migration assays of mouse endothelial cells treated with plasma from the WT and Cfh-/- mice. (A) Representative images of cell staining after transwell migration assay. Magnification: 40X. (B) The number of cells migrated through the transwell membrane. n=6, ** p<0.01; (C) Representative image of wound healing assay. Dash line: 0 hr; Solid line: 8 hr; Magnification: 100X. (D) Migrated area after wound healing. n=12, * p<0.05. (E) Representative image of real-time transendothelial resistance (TER) measurement of mouse endothelial cell monolayer; (F) Bar graph of the mean percentage of TER after 24 hr of measurement. n=4; **, p<0.01.
To date, no endothelial cell receptors of CFH have been identified. CFH functions as a negative regulator of complement activation in blood plasma, in part by binding to plasma proteins complement C3, heparin and C-reactive protein [13]. It functions as a cofactor in the inactivation of C3b by factor I, and increases the rate of dissociation of the C3bBb complex and the NBB complex in the alternative complement pathway [13]. Complement components C3a and C5a in drusen promote choroidal neovascularization [25]. C5a stimulates microvascular-like structure formation by endothelial cells [26]. Consistently, our results supported that CFH might block angiogenesis through inhibition of the complement pathway.
Angiogenesis is initiated from proliferation and migration of endothelial cells [27]. In this study, we dissected the effects of plasma Cfh on endothelial cells. Cfh does not affect endothelial cell proliferation, but inhibits migration. Endothelial cell migration is a dynamic and multistep process regulated by chemotactic, haptotactic, and mechanotactic stimuli [28]. Complement components induces migration of several cell types [29]. In addition, CFH might affect cell migration indirectly by regulating angiogenic effectors. In mice, down-regulation of Cfh expression leads to increase of the expressions of VEGF and transforming growth factor-β (TGF-β) [11]. VEGF is a pro-angiogenesis effector that activates endothelial cell proliferation and migration [1]. TGF-β inhibits the proliferation, but not migration of endothelial cells [30]. Thus, the increase of cell migration caused by loss of Cfh in plasma might be a result from the combination effects of VEGF and TGF-β.
In summary, we determined that plasma CFH inhibits endothelial cell migration and reduces angiogenesis. Thus CFH represents a novel target for anti-angiogenic therapy. Future work should elucidate the detailed molecular mechanisms underlying the effects of CFH on endothelial cells.
Acknowledgements
This work was funded by Rosebay Medical Foundation and Yale Medical School Dean's Research Fund to Josephine Hoh. No URL. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We are grateful for the support from Shandong Taishan Scholarship (Ju Liu).
Competing Interests
The authors have declared that no competing interests exist.
References
1. Iruela-Arispe ML, Dvorak HF. Angiogenesis: a dynamic balance of stimulators and inhibitors. Thrombosis and haemostasis. 1997;78:672-7
2. Plate KH, Breier G, Risau W. Molecular mechanisms of developmental and tumor angiogenesis. Brain pathology. 1994;4:207-18
3. Folkman J. Role of angiogenesis in tumor growth and metastasis. Seminars in oncology. 2002;29:15-8
4. Folkman J. Fundamental concepts of the angiogenic process. Current molecular medicine. 2003;3:643-51
5. Liekens S, De Clercq E, Neyts J. Angiogenesis: regulators and clinical applications. Biochemical pharmacology. 2001;61:253-70
6. Gibaldi M. Regulating angiogenesis: a new therapeutic strategy. Journal of clinical pharmacology. 1998;38:898-903
7. Puduvalli VK, Sawaya R. Antiangiogenesis - therapeutic strategies and clinical implications for brain tumors. Journal of neuro-oncology. 2000;50:189-200
8. Sitohy B, Nagy JA, Dvorak HF. Anti-VEGF/VEGFR therapy for cancer: reassessing the target. Cancer research. 2012;72:1909-14
9. Yanai R, Thanos A, Connor KM. Complement involvement in neovascular ocular diseases. Advances in experimental medicine and biology. 2012;946:161-83
10. Khan MA, Assiri AM, Broering DC. Complement and macrophage crosstalk during process of angiogenesis in tumor progression. Journal of biomedical science. 2015;22:58
11. Bora NS, Kaliappan S, Jha P, Xu Q, Sohn JH, Dhaulakhandi DB. et al. Complement activation via alternative pathway is critical in the development of laser-induced choroidal neovascularization: role of factor B and factor H. J Immunol. 2006;177:1872-8
12. Ferreira VP, Pangburn MK, Cortes C. Complement control protein factor H: the good, the bad, and the inadequate. Mol Immunol. 2010;47:2187-97
13. Makou E, Herbert AP, Barlow PN. Functional anatomy of complement factor H. Biochemistry. 2013;52:3949-62
14. Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C. et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385-9
15. McHarg S, Clark SJ, Day AJ, Bishop PN. Age-related macular degeneration and the role of the complement system. Mol Immunol. 2015;67:43-50
16. Ding X, Patel M, Chan CC. Molecular pathology of age-related macular degeneration. Prog Retin Eye Res. 2009;28:1-18
17. Liu J, Deutsch U, Jeong J, Lobe CG. Constitutive notch signaling in adult transgenic mice inhibits bFGF-induced angiogenesis and blocks ovarian follicle development. Genesis. 2014;52:809-16
18. Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329-33
19. Dong F, Zhou X, Li C, Yan S, Deng X, Cao Z. et al. Dihydroartemisinin targets VEGFR2 via the NF-kappaB pathway in endothelial cells to inhibit angiogenesis. Cancer biology & therapy. 2014;15:1479-88
20. Pickering MC, Cook HT, Warren J, Bygrave AE, Moss J, Walport MJ. et al. Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor H. Nat Genet. 2002;31:424-8
21. Zhang Y, Huang Q, Tang M, Zhang J, Fan W. Complement Factor H Expressed by Retinal Pigment Epithelium Cells Can Suppress Neovascularization of Human Umbilical Vein Endothelial Cells: An in vitro Study. PloS one. 2015;10:e0129945
22. Perkins SJ, Fung KW, Khan S. Molecular Interactions between Complement Factor H and Its Heparin and Heparan Sulfate Ligands. Frontiers in immunology. 2014;5:126
23. Pickering MC, de Jorge EG, Martinez-Barricarte R, Recalde S, Garcia-Layana A, Rose KL. et al. Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains. The Journal of experimental medicine. 2007;204:1249-56
24. Tersteeg C, Schiviz A, De Meyer SF, Plaimauer B, Scheiflinger F, Rottensteiner H. et al. Potential for Recombinant ADAMTS13 as an Effective Therapy for Acquired Thrombotic Thrombocytopenic Purpura. Arteriosclerosis, thrombosis, and vascular biology. 2015;35:2336-42
25. Nozaki M, Raisler BJ, Sakurai E, Sarma JV, Barnum SR, Lambris JD. et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. 2006;103:2328-33
26. Kurihara R, Yamaoka K, Sawamukai N, Shimajiri S, Oshita K, Yukawa S. et al. C5a promotes migration, proliferation, and vessel formation in endothelial cells. Inflamm Res. 2010;59:659-66
27. Mullins RF, Khanna A, Schoo DP, Tucker BA, Sohn EH, Drack AV. et al. Is age-related macular degeneration a microvascular disease? Advances in experimental medicine and biology. 2014;801:283-9
28. Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circulation research. 2007;100:782-94
29. Kim YH, He S, Kase S, Kitamura M, Ryan SJ, Hinton DR. Regulated secretion of complement factor H by RPE and its role in RPE migration. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2009;247:651-9
30. Romo P, Madigan MC, Provis JM, Cullen KM. Differential effects of TGF-beta and FGF-2 on in vitro proliferation and migration of primate retinal endothelial and Muller cells. Acta Ophthalmol. 2011;89:e263-8
Author contact
![]() Corresponding author: Ju Liu MD., Ph.D., Medical Research Center, Shandong Provincial Qianfoshan Hospital, Shandong University, 16766 Jingshi Road, Jinan, Shandong, China 250014 Tel: +8613791122228 Email: ju.liuedu.cn. Josephine Hoh Ph.D., Department of Epidemiology and Public Health, Yale University, 60 College Street, New Haven, Connecticut 06520, USA. Tel: 1 (203) 785-2738 Email: Josephine.hohedu
Corresponding author: Ju Liu MD., Ph.D., Medical Research Center, Shandong Provincial Qianfoshan Hospital, Shandong University, 16766 Jingshi Road, Jinan, Shandong, China 250014 Tel: +8613791122228 Email: ju.liuedu.cn. Josephine Hoh Ph.D., Department of Epidemiology and Public Health, Yale University, 60 College Street, New Haven, Connecticut 06520, USA. Tel: 1 (203) 785-2738 Email: Josephine.hohedu
Citation styles
APA
Liu, J., Hoh, J. (2017). Loss of Complement Factor H in Plasma Increases Endothelial Cell Migration. Journal of Cancer, 8(12), 2184-2190. https://doi.org/10.7150/jca.19452.
ACS
Liu, J.; Hoh, J. Loss of Complement Factor H in Plasma Increases Endothelial Cell Migration. J. Cancer 2017, 8 (12), 2184-2190. DOI: 10.7150/jca.19452.
NLM
Liu J, Hoh J. Loss of Complement Factor H in Plasma Increases Endothelial Cell Migration. J Cancer 2017; 8(12):2184-2190. doi:10.7150/jca.19452. https://www.jcancer.org/v08p2184.htm
CSE
Liu J, Hoh J. 2017. Loss of Complement Factor H in Plasma Increases Endothelial Cell Migration. J Cancer. 8(12):2184-2190.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.