Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(16):3318-3330. doi:10.7150/jca.20482 This issue Cite
Research Paper
Histone Methyltransferase SETDB1 Promotes the Progression of Colorectal Cancer by Inhibiting the Expression of TP53
Keli Chen1, 2*, Fengjiao Zhang1*, Jie Ding2, Yonghao Liang1, Zetao Zhan1, Yizhi Zhan1, Long-hua Chen1 ![]() , Yi Ding1
, Yi Ding1 ![]()
1. Department of Radiation Oncology, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong Province, China;
2. HuiQiao Medical Center, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong Province, China.
* These authors contributed equally to this work.
Received 2017-4-9; Accepted 2017-9-1; Published 2017-9-16
Citation:
Chen K, Zhang F, Ding J, Liang Y, Zhan Z, Zhan Y, Chen Lh, Ding Y. Histone Methyltransferase SETDB1 Promotes the Progression of Colorectal Cancer by Inhibiting the Expression of TP53. J Cancer 2017; 8(16):3318-3330. doi:10.7150/jca.20482. https://www.jcancer.org/v08p3318.htm
Other stylesAbstract
SETDB1 is a novel histone methyltransferase associated with the functional tri-methylation of histone H3K9. Although aberrant high expression of SETDB1 was experimentally obversed in a variety of solid tumors, its underlying mechanisms in human carcinogenesis are not well known. In this study, we investigated the expression of SETDB1 in a large cohort of colorectal cancer (CRC) samples and cell lines for the first time. Our findings showed that SETDB1 was highly expressed in majority CRC tissues and cell lines; moreover, up-regulation of SETDB1 was negatively correlated with the survival rate of CRC patients. Functionally, over-expression of SETDB1 significantly promoted the proliferation and migration of CRC cells in vitro and in vivo, while knocking down SETDB1 suppressed their growth. Mechanistically, we showed that over-expression of SETDB1 significantly inhibited the apoptosis induced by 5-Fluorouracil in CRC cells, which was closely related to the inhibition of TP53 and BAX expression. Furthermore, we confirmed that SETDB1 could be recruited to the promoter region of TP53, which might contribute its inhibition of apoptosis. For conclusion, our study indicated that SETDB1 is essential for colorectal carcinogenesis, and may be a newly target for treatment and prognostic evaluation in CRC.
Keywords: Histone Methyltransferase, SETDB1, Colorectal Cancer, TP53, Apoptosis.
Introduction
Colorectal cancer (CRC) is the third most common malignancy in men and the second in women worldwide, accounting for 1.36 million new cases and 693, 000 deaths annually [1]. Because the early symptoms are not obvious, many of CRC patients are often diagnosed at advanced stages [2, 3]. Although application of treatments such as surgical resection, chemotherapy [4], radiotherapy [5] and targeted therapy [6] significantly improve overall survival (OS) time, many CRC patients still have poor prognostic outcome. It is generally believed that colorectal carcinogenesis is a multiple-step process and caused by activation of oncogenes and silencing of tumor suppressor genes due to accumulation of various genetic mutations and epigenetic alterations [7, 8]. Some recent evidence highlighted the roles of epigenetic alterations in CRC [9-11], however, the detailed molecular mechanisms remain unclear.
Histone modification is a common epigenetic event in mammalian cells [12], including acetylation and deacetylation, methylation and demethylation, phosphorylation, ubiquitination and so on, which play an important role in maintaining genomic stability [13, 14]. Aberrant histone modifications are associated with many human diseases including various cancers. Methylation is an important form of histone modifications, which usually occurs in lysine residues (K) and arginine residues (R) of histone H3 and H4. Methylation sites of lysine mainly include H3K4, H3K9, H3K27, H3K36, H3K79 and H4K20, and sites of arginine mainly include H3R2, H3R8, H3R17, H3R26 and H4R3 [15]. Generally, methylation of H3K4, H3K36 and H3K79 is associated with gene activation, whereas methylation of H3K9, H3K27 and H4K20 usually leads to gene inactivation [16].
SETDB1 (SET domain bifurcated 1) gene located on human chromosome 1q21.3, a length of about 38.6Kb, encoding SETDB1 protein. SETDB1 protein belong to the SET-domain (Su(var)3-9, E(z), Thrithorax) protein methyltransferase family [17], which is mainly involved in tri-methylation of H3K9 and usually cause gene silencing or transcription inhibition. SETDB1 protein has been considered as a target for treatment in Huntington disease early and is associated with embryo development [18]. In recent years, mounting evidence has linked the aberrant high expression of SETDB1 to the human carcinogenesis, including melanoma, lung cancer, breast cancer, ovarian cancer [19-21]. However, the roles of SETDB1 in CRC are still largely unknown. Therefore, a comprehensive study into the function and mechanism of SETDB1 in CRC is warranted.
In our study, it was shown that the expression level of SETDB1 protein was high in CRC cells and tissues. The high expression of SETDB1 was closely related to poor survival rate of CRC patients. Forced SETDB1 promoted proliferation and migration of CRC cells in vitro and tumor growth in vivo. By contraries, the down-regulation of SETDB1 inhibited their growth. We also showed that the over-expression of SETDB1 reduced the apoptosis induced by chemotherapeutic agent in CRC cells and inhibited the expression of TP53 and BAX. Additionally, our results showed that the inhibition of TP53 might be associated with histone methylation in its promoter region. Therefore, we demonstrated that SETDB1 promoted colorectal carcinogenesis and might be the newly target for treatment and prognostic evaluation of CRC. Inhibitors targeting SETDB1 may benefit the large population of CRC patients with high expression of SETDB1.
Materials and Methods
CRC specimens and cell lines
8 pairs of fresh CRC tissues and corresponding adjacent nontumorous colorectal tissues were offered by Department of Pathology, Nanfang Hospital of Southern Medical University. 30 cases of CRC paraffin sections were collected at Nanfang Hospital (resected during January 2015 to December 2016). 102 cases of CRC paraffin sections with more than 5 years of follow-up data were collected at Department of Pathology, Nanfang Hospital. All tissues and sections were pathologically diagnosed with CRC, and not received radiotherapy and chemotherapy prior to surgery. The acquisition process were agreed and approved by the medical ethics committee of the South Hospital of Southern Medical University. CRC cell lines SW480, SW620, DLD-1, HCT116, LoVo, HT29, RKO, 174ST were offered by the Department of Radiotherapy, Nanfang Hospital.
Mice
All BALB/C nude mice with the animal certificate were purchased from the animal experimental center of Southern Medical University in Guangzhou. All mice reared in SPF grade Animal Center of Nanfang Hospital.
Establishment of SETDB1 over-expression and interference lentiviral expression vector
SETDB1 full length expression plasmid was constructed by the Genepharma (Shanghai) and then packaged in lentiviral vector. Two SETDB1 targeting short hairpin RNA (shRNA) sequences (Table. S1) were cloned into lentiviral vector (GENECHEM, Shanghai).
Cell culture
CRC cell lines SW480, SW620, DLD-1, HCT116, LoVo, HT29, RKO, 174ST were cultured in PRMI1640 medium (Life, USA) supplemented with 10% fetal bovine serum (Gibco, USA), and placed in cell incubator at 37℃ in 5% CO2.
Generation of stably-transfected cell line
The lentiviral SETDB1 over-expression and shRNA vectors were infected into CRC cell lines DLD1 and SW620 respectively. Infected cells were selected with 2~5ug/ml puromycin to obtain stably infected cell lines, and infected efficiency was analyzed by both qRT-PCR and western blotting.
RNA extraction, reverse transcription and real-time PCR analysis
Total RNA from CRC cells were extracted using RNAiso plus reagent (TAKARA, Japan). The reverse transcription was then performed using PrimeScript RT reagent Kit with gDNA Eraser (TAKARA, Japan), according to the protocol. RT-PCR was performed using SYBR Premix Ex Taq (TAKARA, Japan) on PRISM 7500 Fast Real-Time PCR (ABI, USA). The RT-PCR primers of SETDB1 and GAPDH were synthesized by BGI company (Guangzhou, China) and the sequences were as follows: SETDB1F-5'AGGAACTTCGGCATTTCATCG3',R-5'TGTCCCGGTATTGTAGTCCCA3',GAPDHF-5'AGCTGAACGGGAAGCTCACT3',R-5'TGCTTAGCCAAATTCGTTG3'. The expression of GAPDH in each sample were used as control and data were quantified using a relative quantitative method of the 2-△△Ct.
Protein extraction and western boltting
CRC cells were lysed with radio immunoprecipitation assay (RIPA) lysis buffer (keyGen, China), containing protease inhibitor cocktail (Roche Life Science, Switzerland). Protein concentration was determined using the BCA Protein Assay Kit (keyGen bioTech, China). Western blotting was performed with specific primary antibody against SETDB1 (1:1000, Cell Signaling Technology), TP53 (1:1000, Abclonal), Caspase3 (1:1000, Abclonal), BAX (1:1000, Abclonal), Caspase1 (1:1000, Abclonal). The housekeeping gene a-tubulin (1:1000, Proteintech) was used as the loading control.
Immunohistochemistry (IHC)
The CRC tissue was fixed in 10% buffered formalin, embedded in paraffin, and serially sectioned at 4um thickness. The expression of SETDB1 protein was determined by immunohistochemistry (IHC) using rabbit monoclonal antibody specifically against SETDB1 (1:200, Cell Signaling Technology).
CCK8 assay
CCK8 assay was performed using CCK-8 kit (Tongren, China). 800~1000 cells were seeded into 96 well culture plate and incubated for 24 hours, 48 hours, 72 hours, 96 hours, 120 hours respectively in incubator at 37℃ in 5%CO2. 100ul prepared solution (CCK8 reagent l0ul added to 90ul medium) was added into each well and incubated for 2 hours. The absorbance (OD) of each well at 450nm was measured of Model 680 microplate reader. The growth curve was drawn according to OD.
The colony-formation assay
500~600 cells were seeded into 6 well culture plate and incubated for 2 weeks in incubator at 37℃ in 5%CO2 till visible cell clones were observed. The clones were fixed with 4% methanol and stained with Giemsa for 20 min. The number of formed clones were counted using microscope (more than 50 cells were calculated as a clone).
Cell scratch assay
About 5×105 cells were seeded into 6 well culture plate. Cell scratches were performed using 10ul pipet tip when the cell confluence is about 95% (be careful to ensure that each scratch width is the same), and then cell debris were washed off using PBS buffer. Complete medium containing 1% fetal bovine serum was added into wells and cells were incubated in incubator at 37℃ in 5%CO2. Reduced distance of cell scratches were recorded by microscope camera at 0 hours, 24 hours, 48 hours respectively.
Transwell migration assay
Transwell migration assay was performed using transwell chambers (Corning, USA). To induced cell migration, 200ul cell suspension (about 1×105 cells in serum-free PRMI1640 medium) was added in the upper chambers of the wells, while the lower chambers were filled with 500ul PRMI1640 medium containing 10% fetal bovine serum. After cultured at 37℃ in 5% CO2 for 24h, 36h, the chambers were placed into methanol to fix for 30min and then stained with giemsa for 20min. After air dried, the cells in the upper surface of the membrane were removed with a cotton swab. Cells migrated to the lower surface of the membrane were recorded and counted using microscope camera.
Subcutaneous tumor implantation model
200ul cell suspension (about 2×106 cells in serum-free PRMI1640 medium, SETDB1 stable over-expression or interference cells) were subcutaneously injected into BALB/C nude mice at 4 weeks old. After inoculation, the subcutaneous tumor volume was measured with a vernier caliper every other day. Mice were sacrificed at 25 days and the subcutaneous tumors were collected and fixed for further use.
Apoptosis and cell cycle assay
Cell apoptosis assay was performed using Annexin V-APC/PI apoptosis detection kit (KeyGEN, China). 1~5×105 cells were suspended in 500ul Binding buffer, adding 5ul Annexin V-APC, mix well, adding 5ul PI, mix well. Avoiding light, the cell suspension was incubated at room temperature for 5~15min. Flow cytometry assay should be performed in 1h. Cell cycle assay was performed using cell cycle detection kit (KeyGEN, China), according to the protocol.
ChIP assay
ChIP assays were performed using ChIP-seq kit (Chromatrap, USA) according to the protocol. Briefly, 10 million DLD1 cells over-expressing SETDB1 were crosslinked with serum-free PRMI1640 medium containing 1% formaldehyde for 10 min. The chromatin DNA was sheared by enzymatic digestion to lengths of 100-500 bp (Figure S1). Chromatin samples and either anti-SETDB1 antibody (Proteintech, USA) or normal rabbit IgG (Santa Cruz, USA) were mixed well and the IP slurries were incubate on an end to end rotor for 1 hour at 4°C. ChIP-enriched DNA was acquired by reversing cross linking and purified in Chromatrap supply DNA purification columns. Finally IPd or input DNA was quantified by quantitative PCR using specific primers (Table S2, 3) and the PCR products were further examined by agarose gel electrophoresis.
Statistical analysis and Survival analysis
Statistical analyses were performed in SPSS 19.0 software. The results of CCK8 assay were analysed using variance analysis. Two independent samples t-test was used to analyse the results of colony-formation assay, cell scratch assay and transwell migration assay. The 102 CRC patients were enrolled in the survival analysis. The survival curves of patients with high and low expression of SETDB1 were analyzed using Kaplan-Meier method. The difference between two survival curves was compared using the Log-rank test. P < 0.05 was considered statistically significant.
Result
SETDB1 is highly expressed in CRC tissues
The expression of SETDB1 protein was determined using IHC in 30 CRC tissues and western blotting in 8 paired CRC tissue and the adjacent normal tissue proteins. The result of IHC showed that SETDB1 was mainly expressed in nucleus, showed different degrees of brown, and was strongly positive expression in 46.7% (14/30) colorectal cancer tissue (Figure 1A). The expression of SETDB1 was increased gradually with the degree of invasion (Figure 1B). As showed in Figure 1C, in 8 paired tissues, SETDB1 expressed higher in cancerous tissues than in corresponding normal tissues. Collectively, all these results suggested that SETDB1 was highly expressed in CRC tissues.
The expression of SETDB1 in human CRC cell lines
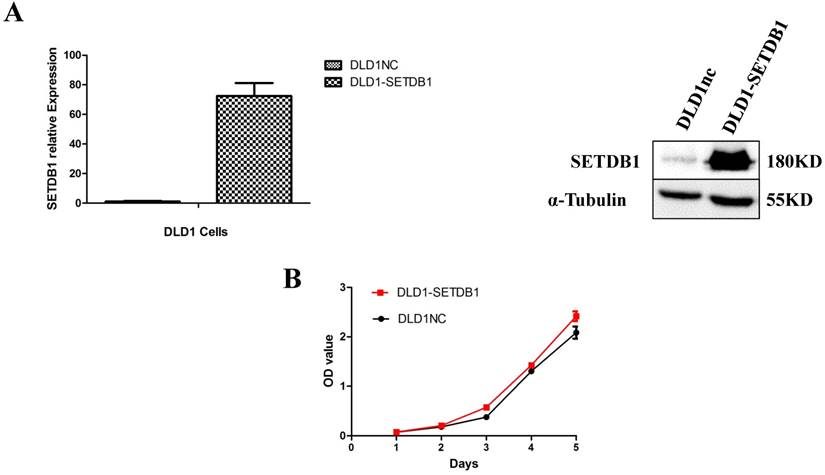
The expressions of SETDB1 were investigated by western blotting and real-time PCR assay in eight human CRC cell lines (SW480, SW620, DLD-1, HCT116, LoVo, HT29, RKO, 174ST). Similar to CRC tissue samples, up-regulation of SETDB1 was detected in multiple CRC cell lines and the expression of SETDB1 protein was highest in the SW480 cell line and lowest in the DLD1 cell line (Figure 1D).
Up-regulation of SETDB1 promoted the proliferation and migration of CRC cells in vitro and tumor growth in vivo
To investigate the functional roles of SETDB1 in CRC, DLD1 cell line was infected with SETDB1 over-expression lentiviral to establish stable SETDB1 over-expression cell line (DLD1-SETDB1) and control lentiviral to establish control cell line (DLD1nc). The efficiency of over-expression was confirmed by western blotting and real-time PCR assay (Figure 2A). The results showed that forced expression of SETDB1 significantly promoted the proliferation (Figure 2B, C) and migration (Figure. 2D, E) of DLD1 cells. In vivo, subcutaneous tumour sizes were significantly increased in mice injected with DLD1-SETDB1 cells compared with DLD1nc cells (Figure S2).
Figure 1
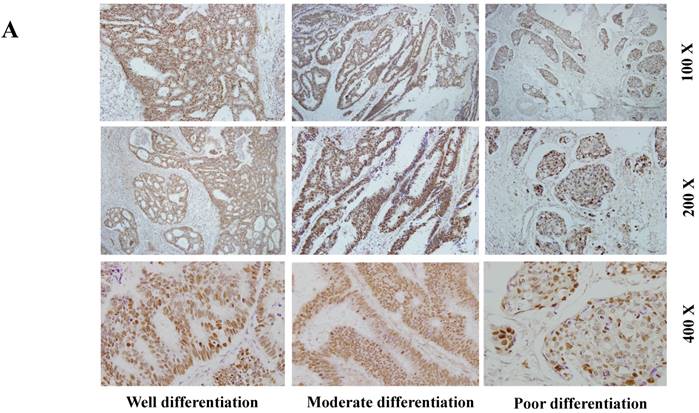
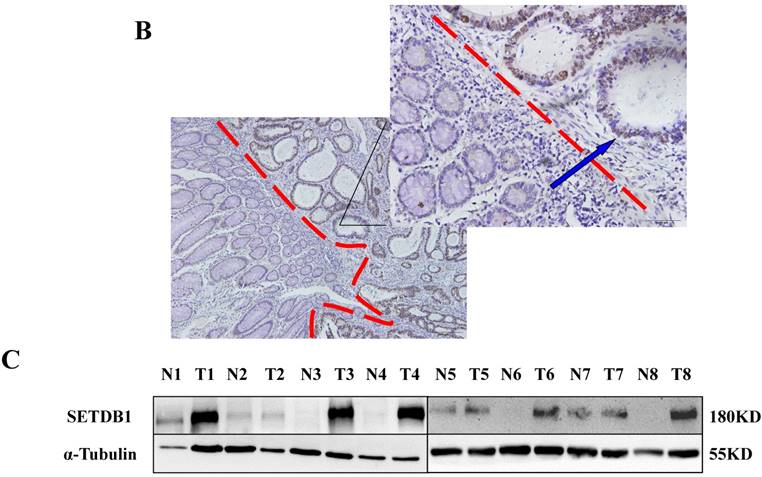
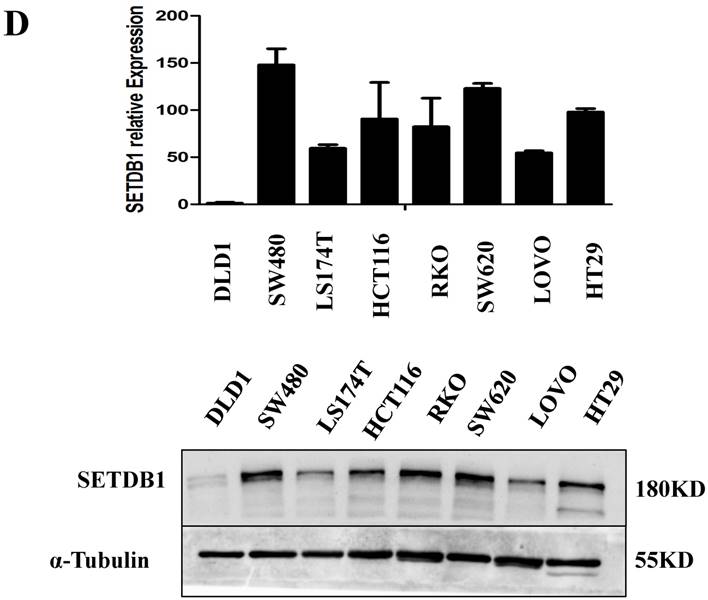
The expression of SETDB1 in CRC tissues and cell lines A. The expression of SETDB1 was analyzed by IHC assay in well, moderate and poor differentiation CRC tissues. B. Representative IHC staining of SETDB1 in CRC tissues (right) and adjacent normal tissues (left). C. Expression levels of SETDB1 protein were analyzed using west blotting in eight pairs human CRC tissues and adjacent normal tissues. D. SETDB1 mRNA and protein expression analysis in a panel of eight human CRC cell lines.
Figure 2


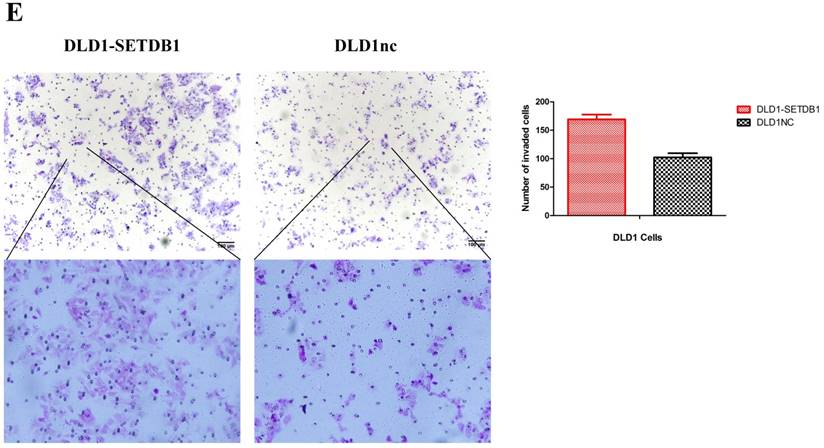
SETDB1 over-expression promotes the proliferation and migration of DLD1 cells A. SETDB1 mRNA and protein expression increased significantly in DLD1 cells after infected with SETDB1 over-expression lentiviral. B-C. CCK8 and colony formation assay in DLD1-SETDB1 and DLD1nc cells. D-E. Cell scratch and transwell migration assay in DLD1-SETDB1 and DLD1nc cells.
Knockdown of SETBD1 inhibited the proliferation and migration of CRC cells in vitro and tumor growth in vivo
We established the SETDB1 stable knockdown model in SW620 cell line. SW620 CRC cell line was infected with SETDB1 interference lentiviral (shSETDB2#) to establish stable SETDB1 knockdown cell line (SW620-shSETDB1) and control lentiviral to establish control cell line (SW620nc). The efficiency of knockdown was confirmed by western blotting and real-time PCR assay (Figure 3A). The results showed that knockdown of SETDB1 significantly suppressed the proliferation (Figure 3B, C) and migration (Figure 3D, E) of SW620 cells. In vivo, subcutaneous tumour sizes were signifcantly reduced in mice injected with SW620-shSETDB1 cells compared with SW620nc cells (Figure S3).
Up-regulation of SETDB1 suppressed apoptosis induced by 5-Fluorouracil in CRC cells
We investigated the apoptosis in CRC cells using flow cytometry assay. The results showed that there were no statistical difference in CRC cells whether SETDB1 over-expression or knockdown (Figure 4A). Interestingly, after treated by 5-Fluorouracil for 48h, significant suppression of apoptosis was observed in DLD1-SETDB1 cells compared with DLD1nc cells. By contraries, the rate of apoptosis was higher in SW620- shSETDB1 cells than SW620nc cells (Figure 4B). An obvious G2 arrest occurred in SW620-shSETDB1 cells compared with SW620nc cells, whereas there was no statistical difference in DLD1 cells after infected with SETDB1 over-expression lentiviral (Figure 4C).
SETDB1 suppressed apoptosis in CRC cells by inhibition the expression of TP53
To investigate the molecular mechanism of apoptosis suppression regulated by SETDB1 in CRC cells, we examined apoptosis related proteins TP53, Caspase1, Caspase3, BAX using western blotting. The results showed that the expression of TP53 was signifcantly decreased in DLD1-SETDB1cells (Figure 5A) after treated with 5-Fluorouracil, whereas, silencing SETDB1 in SW620 cells dramatically increased their protein level of P53 (Figure 5B). Likewise, a little changes of BAX expression was observed (Figure 5A, B). To further investigated whether SETDB1 suppressed TP53 gene by binding directly to its promoter regions, a ChIP-PCR assay was performed to study the putative SETDB1 binding regions. To this end, the TP53 promoter was divided into 6 sections (~200 bp) and specific primers (Table S2) were designed to detect these sections. As predicted, we identified two SETDB1 enrichment sites in the promoter region of TP53 at -200 and -600bp upstream from the start codon (Figure 5C).
Figure 3
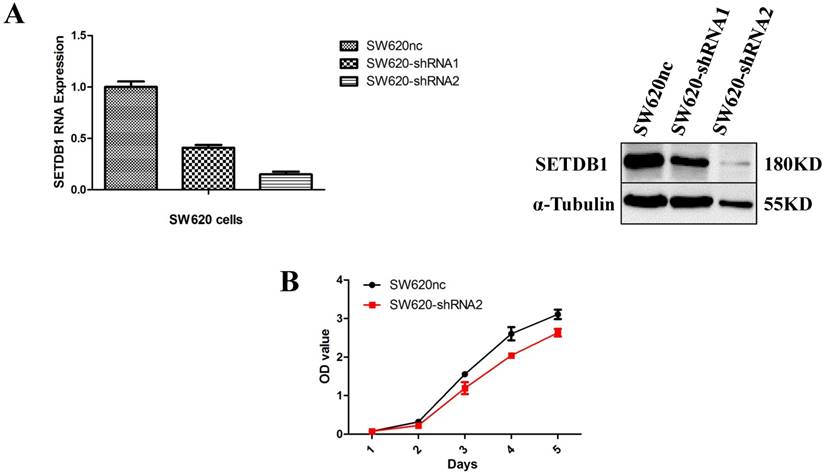
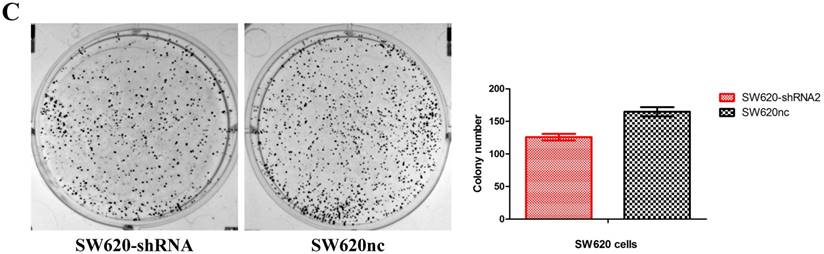
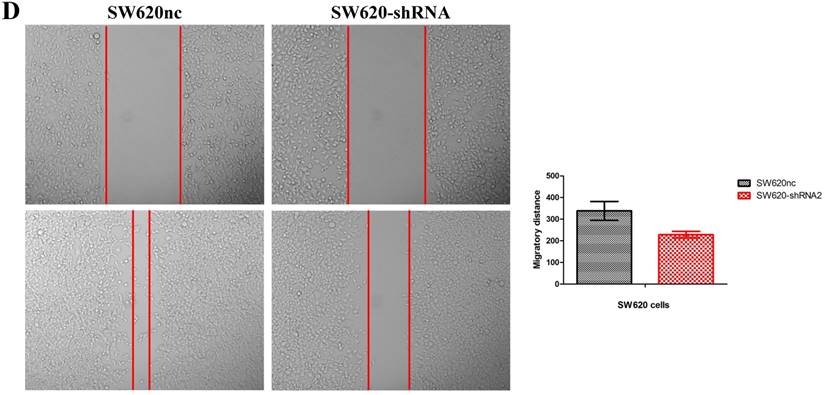
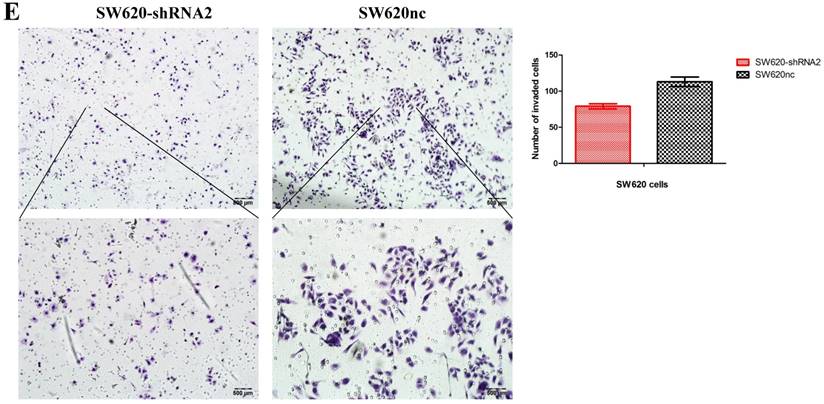
SETDB1 knockdown suppresses the proliferation and migration of SW620 cells A. SETDB1 mRNA and protein expression decreased significantly in SW620 cells after infected with SETDB1 interference lentiviral. B-C. CCK8 and colony formation assay in SW620-shSETDB1 and SW620nc cells. D-E. Cell scratch and transwell migration assay in SW620-shSETDB1 and SW620nc cells.
The relationship between SETDB1 expression and prognosis of CRC patients
We analyzed 102 cases of survival data of CRC patients whose follow-up surveys were made by Department of pathology, Nanfang Hospital of Southern Medical University. These patients were followed up for 1~78 months and the median follow-up period was 52 months, including 36 deaths. The 5 year total survival rate was 47.3% in the sample set. We used Kaplan-Meier analysis to analyze the correlation of survival of CRC patients with SETDB1 expression and Log-rank test to compare the differences in the survival curves between the high and low expression patients group. The results showed that the 5 years survival rate is only 35.3% in SETDB1 high expression group, while it is 76.6% in low expression group. Patients with higher SETDB1 expression associated with a poorer prognosis compared to those with SETDB1 lower expression (Log-rank, P=0.0, Figure 6).
Discussion
Histone methylation plays a significant role in human carcinogenesis. In the majority of colorectal cancer and liver cancer, the high expression of SMYD3 (H3K4 methyltransferase) have been observed, and down-regulation of SMYD3 significantly inhibit the proliferation of cancer cells [22]. EZH2 (H3K27 methyltransferase), regulating BRCA1 gene, is involved in the proliferation and metastasis of breast cancer cells, and over-expression of EZH2 can promote the progression of breast cancer [23]. G9a (H3K9 methyltransferase) promotes the formation of breast cancer stem-like cells through Leptin-STAT3-G9a Signaling [24].
SETDB1, a novel H3K9 methyltransferase, comprises a C-terminal region involved in histone methylation [25] and an N-terminal region involved in DNA silence and transcriptional repression [26]. Previous studies indicated that SETDB1 can form a complex with many partners to modulate transcription, including, KAP-1, HP1, G9a, MBD1, HDAC1/2, Sin3A/B, EZH2, DNMT3A/B and SUV39-h1 [27-29]. Further investigations are ongoing to explore the underlying mechanism of epigenetic modifications by SETDB1. In this study, we identified SETDB1 as an up-regulated methylation regulator in CRC tissues and cell lines. High expression of SETDB1 was validated, for the first time, in an expanded cohort of 102 cases CRC samples by IHC, which significantly associated with a poor outcome of CRC patients. These results suggested that SETDB1 were indeed up-regulated in CRC as well as in other solid tumors [20, 21].
The functions of SETDB1 are involved in many biological processes, such as histone methylation, DNA silence, X-Chromosome inactivation and development of embryos [18, 30, 31]. More importantly, emerging functional roles of SETDB1 in human carcinogenesis were recently highlighted [32, 33]. Although up-regulation of SETDB1 was obversed in CRC in our experiments, its functional roles are still not clear. In further study, we found that forced SETDB1 significantly promoted the proliferation and migration of CRC cells in vitro and tumor growth in vivo, while silencing SETDB1 got opposite results. These findings demonstrated that SETDB1, acting as an oncogene, is essential for proliferation and migration of CRC cells.
Figure 4
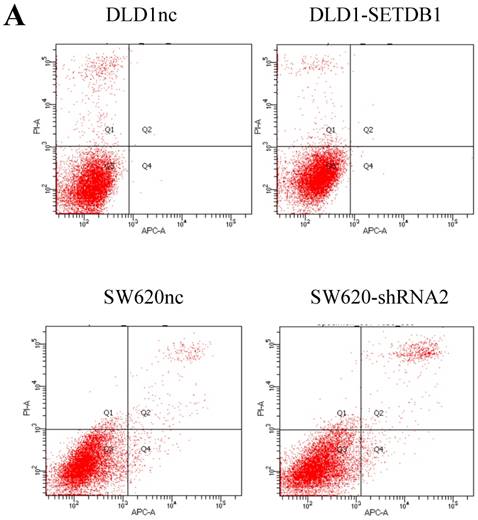
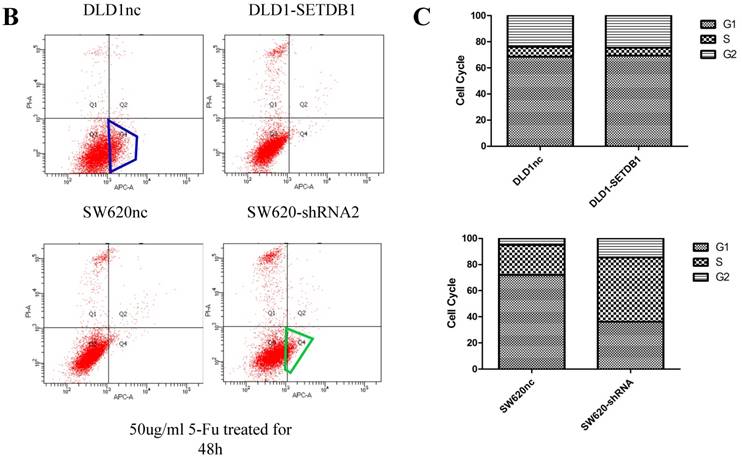
The apoptosis and cell cycle changes in CRC cells after infected with SETDB1 over-expression or knockdown A. Representative flow cytometry assay of apoptosis in CRC cells after infected with SETDB1 over-expression or interference lentiviral. B. Representative flow cytometry assay of apoptosis in CRC cells after treated with 50ug/ml 5-Fluorouracil for 48h. C. cell cycle changes in CRC cells after infected with SETDB1 over-expression or interference lentiviral.
Figure 5
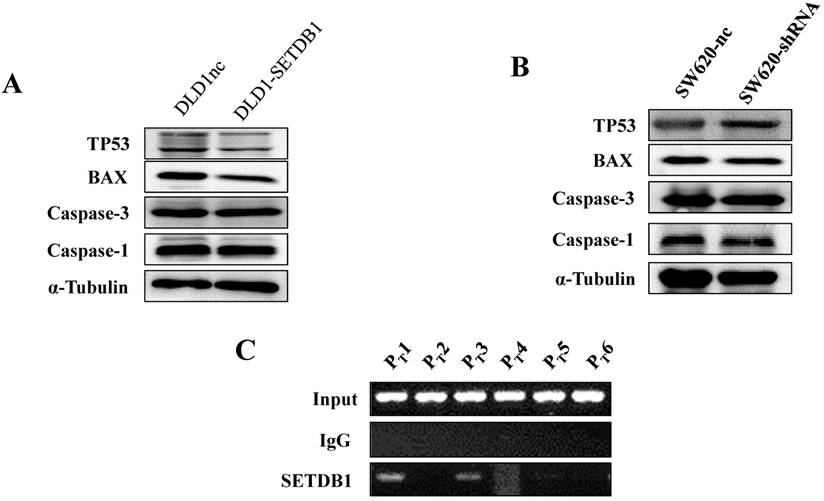
The expression of apoptosis related proteins in CRC cell lines after treated with 50ug/ml 5-Fluorouracil A. The expression of apoptosis related proteins in DLD1-SETDB1 and DLD1nc cells. B. Apoptosis related proteins expression analysis in SW620-shSETDB1 and SW620nc cells. C. Binding of SETDB1 to the promoter region of TP53 was investigated using ChIP assay.
Figure 6
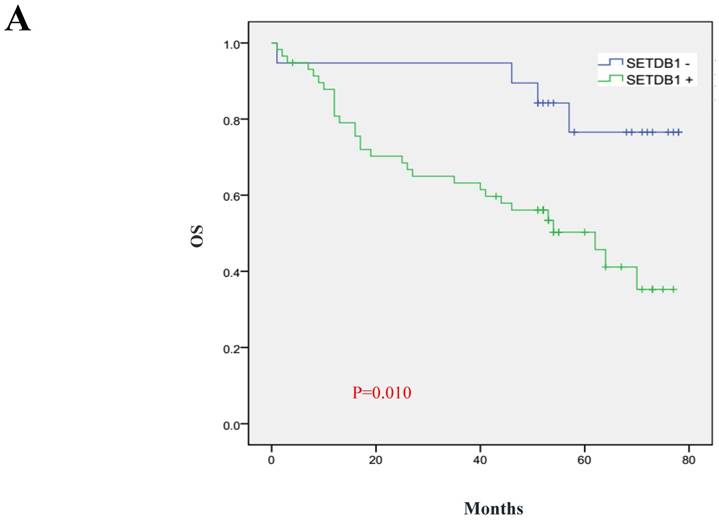
Prognostic significance of SETDB1 in CRC patients A. Kaplan-Meier plots of overall survival (by months) associated with SETDB1 expression in CRC patients: up-regulation of SETDB1 was significantly correlated with poorer survival rate of CRC patients, and p value was calculated by log-rank test.
Many molecular mechanisms contribute to human carcinogenesis promoted by SETDB1. In neuroblastoma, SETDB1 protein affected the proliferation and differentiation of neuroblastoma cells by maintaining the "STEM" of cancer cells. In prostate cancer, the expression of SETDB1 was significantly dependent on hormone, and affected the apoptosis of prostate cancer cells [34]. In non-small cell lung cancer, the expression of SETDB1 was up-regulated, which promoted the proliferation and migration of lung cancer cells through activating WNT/beta-catenin pathway [32]. In liver cancer, SETDB1 was up-regulated at chromosomal, transcriptional and post-transcriptional modification levels, which change biological function of liver cancer cells [33, 35]. In this study, we focused on the functional mechanism of SETDB1 in CRC for the first time. Our findings show that SETDB1 inhibited apoptosis of CRC cells by down-regulation of TP53 associated with its promoter methylation. Similar to our findings in CRC, this mechanism of apoptosis inhibition has also been reported in liver and lung cancers [32, 35]. However, recent evidence by Hyun-A Kim group indicated that SETDB1 also played a CRC suppressor role in mouse [36]. Their findings may be concerned with interaction between NOTCH1 and SETDB1, but the underlying mechanisms need further investigation.
In summary, we demonstrated that SETDB1 promoted colorectal carcinogenesis and might be identified as a potential target for treatment of CRC. Inhibitors targeting SETDB1 may benefit the large population of CRC patients with high expression of SETDB1.
Supplementary Material
Supplementary figures.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (No.81672992); Guangdong Natural Science Funds for Distinguished Young Scholar (Grant No. 2015A030306015); Pearl River Nova Program of Guangzhou, Guangdong Province (Grant No. 2014J2200015); Excellent Young Teachers Program of Higher Education of Guangdong Province (Grant No. YQ2015036); Guangdong Program for Support of Top-notch Young Professionals (Grant No. 2015TQ01R279).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-86
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115-32
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30
4. Ahmed S, Pahwa P, Fields A, Chandra-Kanthan S, Iqbal N, Zaidi A. et al. Predictive Factors of the Use of Systemic Therapy in Stage IV Colorectal Cancer: Who Gets Chemotherapy? Oncology-Basel. 2015;88:289-97
5. Fode MM, Hoyer M. Survival and prognostic factors in 321 patients treated with stereotactic body radiotherapy for oligo-metastases. Radiother Oncol. 2015;114:155-60
6. Troiani T, Napolitano S, Della CC, Martini G, Martinelli E, Morgillo F. et al. Therapeutic value of EGFR inhibition in CRC and NSCLC: 15 years of clinical evidence. ESMO Open. 2016;1:e88
7. Amaro A, Chiara S, Pfeffer U. Molecular evolution of colorectal cancer: from multistep carcinogenesis to the big bang. Cancer Metastasis Rev. 2016;35:63-74
8. Gargalionis AN, Karamouzis MV, Adamopoulos C, Papavassiliou AG. Protein trafficking in colorectal carcinogenesis-targeting and bypassing resistance to currently applied treatments. Carcinogenesis. 2015;36:607-15
9. Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11:726-34
10. Yamauchi M, Morikawa T, Kuchiba A, Imamura Y, Qian ZR, Nishihara R. et al. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut. 2012;61:847-54
11. Okugawa Y, Grady WM, Goel A. Epigenetic Alterations in Colorectal Cancer: Emerging Biomarkers. Gastroenterology. 2015;149:1204-25
12. Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343-57
13. Rothbart SB, Strahl BD. Interpreting the language of histone and DNA modifications. Biochim Biophys Acta. 2014;1839:627-43
14. Strahl BD, Allis CD. The language of covalent histone modifications. Natur. 2000;403:41-5
15. Byvoet P, Shepherd GR, Hardin JM, Noland BJ. The distribution and turnover of labeled methyl groups in histone fractions of cultured mammalian cells. Arch Biochem Biophys. 1972;148:558-67
16. Miao F, Natarajan R. Mapping global histone methylation patterns in the coding regions of human genes. Mol Cell Biol. 2005;25:4650-61
17. Harte PJ, Wu W, Carrasquillo MM, Matera AG. Assignment of a novel bifurcated SET domain gene, SETDB1, to human chromosome band 1q21 by in situ hybridization and radiation hybrids. Cytogenet Cell Genet. 1999;84:83-6
18. Matsui T, Leung D, Miyashita H, Maksakova IA, Miyachi H, Kimura H. et al. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature. 2010;464:927-31
19. Zhang H, Cai K, Wang J, Wang X, Cheng K, Shi F. et al. MiR-7, inhibited indirectly by lincRNA HOTAIR, directly inhibits SETDB1 and reverses the EMT of breast cancer stem cells by downregulating the STAT3 pathway. Stem Cells. 2014;32:2858-68
20. Regina C, Compagnone M, Peschiaroli A, Lena A, Annicchiarico-Petruzzelli M, Piro MC. et al. Setdb1, a novel interactor of DeltaNp63, is involved in breast tumorigenesis. Oncotarget. 2016;7:28836-48
21. Ceol CJ, Houvras Y, Jane-Valbuena J, Bilodeau S, Orlando DA, Battisti V. et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature. 2011;471:513-7
22. Sarris ME, Moulos P, Haroniti A, Giakountis A, Talianidis I. Smyd3 Is a Transcriptional Potentiator of Multiple Cancer-Promoting Genes and Required for Liver and Colon Cancer Development. Cancer Cell. 2016;29:354-66
23. Granit RZ, Gabai Y, Hadar T, Karamansha Y, Liberman L, Waldhorn I. et al. EZH2 promotes a bi-lineage identity in basal-like breast cancer cells. Oncogene. 2013;32:3886-95
24. Chang CC, Wu MJ, Yang JY, Camarillo IG, Chang CJ. Leptin-STAT3-G9a Signaling Promotes Obesity-Mediated Breast Cancer Progression. Cancer Res. 2015;75:2375-86
25. Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M. et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593-9
26. Kang YK. SETDB1 in Early Embryos and Embryonic Stem Cells. Curr Issues Mol Biol. 2015;17:1-10
27. Fritsch L, Robin P, Mathieu JR, Souidi M, Hinaux H, Rougeulle C. et al. A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol Cell. 2010;37:46-56
28. Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FR. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002;16:919-32
29. Li H, Rauch T, Chen ZX, Szabo PE, Riggs AD, Pfeifer GP. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J Biol Chem. 2006;281:19489-500
30. Song YJ, Choi JH, Lee H. Setdb1 is required for myogenic differentiation of C2C12 myoblast cells via maintenance of MyoD expression. Mol Cells. 2015;38:362-72
31. Minkovsky A, Sahakyan A, Rankin-Gee E, Bonora G, Patel S, Plath K. The Mbd1-Atf7ip-Setdb1 pathway contributes to the maintenance of X chromosome inactivation. Epigenetics Chromatin. 2014;7:12
32. Sun QY, Ding LW, Xiao JF, Chien W, Lim SL, Hattori N. et al. SETDB1 accelerates tumourigenesis by regulating the WNT signalling pathway. J Pathol. 2015;235:559-70
33. Wong CM, Wei L, Law CT, Ho DW, Tsang FH, Au SL. et al. Up-regulation of histone methyltransferase SETDB1 by multiple mechanisms in hepatocellular carcinoma promotes cancer metastasis. Hepatology. 2016;63:474-87
34. Sun Y, Wei M, Ren SC, Chen R, Xu WD, Wang FB. et al. Histone methyltransferase SETDB1 is required for prostate cancer cell proliferation, migration and invasion. Asian J Androl. 2014;16:319-24
35. Fei Q, Shang K, Zhang J, Chuai S, Kong D, Zhou T. et al. Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53. Nat Commun. 2015;6:8651
36. Kim HA, Koo BK, Cho JH, Kim YY, Seong J, Chang HJ. et al. Notch1 counteracts WNT/beta-catenin signaling through chromatin modification in colorectal cancer. J Clin Invest. 2012;122:3248-59
Author contact
![]() Corresponding authors: Longhua Chen, Department of Radiation Oncology, Nanfang Hospital, Southern Medical University, Guangzhou510515, People's Republic of China, Tel.:86-20-61642136, E-mail: chenlhsmucom, and Yi Ding, Department of Radiation Oncology, Nanfang Hospital, Southern Medical University, Guangzhou510515, People's Republic of China, Tel.:86-20-61642136, E-mail: dingyi197980com
Corresponding authors: Longhua Chen, Department of Radiation Oncology, Nanfang Hospital, Southern Medical University, Guangzhou510515, People's Republic of China, Tel.:86-20-61642136, E-mail: chenlhsmucom, and Yi Ding, Department of Radiation Oncology, Nanfang Hospital, Southern Medical University, Guangzhou510515, People's Republic of China, Tel.:86-20-61642136, E-mail: dingyi197980com
Citation styles
APA
Chen, K., Zhang, F., Ding, J., Liang, Y., Zhan, Z., Zhan, Y., Chen, L.h., Ding, Y. (2017). Histone Methyltransferase SETDB1 Promotes the Progression of Colorectal Cancer by Inhibiting the Expression of TP53. Journal of Cancer, 8(16), 3318-3330. https://doi.org/10.7150/jca.20482.
ACS
Chen, K.; Zhang, F.; Ding, J.; Liang, Y.; Zhan, Z.; Zhan, Y.; Chen, L.h.; Ding, Y. Histone Methyltransferase SETDB1 Promotes the Progression of Colorectal Cancer by Inhibiting the Expression of TP53. J. Cancer 2017, 8 (16), 3318-3330. DOI: 10.7150/jca.20482.
NLM
Chen K, Zhang F, Ding J, Liang Y, Zhan Z, Zhan Y, Chen Lh, Ding Y. Histone Methyltransferase SETDB1 Promotes the Progression of Colorectal Cancer by Inhibiting the Expression of TP53. J Cancer 2017; 8(16):3318-3330. doi:10.7150/jca.20482. https://www.jcancer.org/v08p3318.htm
CSE
Chen K, Zhang F, Ding J, Liang Y, Zhan Z, Zhan Y, Chen Lh, Ding Y. 2017. Histone Methyltransferase SETDB1 Promotes the Progression of Colorectal Cancer by Inhibiting the Expression of TP53. J Cancer. 8(16):3318-3330.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.