Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2010; 1:126-135. doi:10.7150/jca.1.126 This volume Cite
Research Paper
Inhibition of Hypoxia-Induced Cell Motility by p16 in MDA-MB-231 Breast Cancer Cells
Liyuan Li, Yi Lu ![]()
Department of Pathology and Laboratory Medicine, University of Tennessee Center for Cancer Research, University of Tennessee Health Science Center, Memphis, Tennessee, USA
Received 2010-8-3; Accepted 2010-8-31; Published 2010-9-1
Abstract
Our previous studies indicated that p16 suppresses breast cancer angiogenesis and metastasis, and downregulates VEGF gene expression by neutralizing the transactivation of the VEGF transcriptional factor HIF-1α. Hypoxia stimulates tumor malignant progression and induces HIF-1α. Because p16 neutralizes effect of HIF-1α and attenuates tumor metastatic progression, we intended to investigate whether p16 directly affects one or more aspects of the malignant process such as adhesion and migration of breast cancer cells. To approach this aim, MDA-MB-231 and other breast cancer cells stably transfected with Tet-on inducible p16 were used to study the p16 effect on growth, adhesion and migration of the cancer cells. We found that p16 inhibits breast cancer cell proliferation and migration, but has no apparent effect on cell adhesion. Importantly, p16 inhibits hypoxia-induced cell migration in breast cancer in parallel with its inhibition of HIF-1α transactivation activity. This study suggests that p16's ability to suppress tumor metastasis may be partially resulted from p16's inhibition on cell migration, in addition to its known functions on inhibition of cell proliferation, angiogenesis and induction of apoptosis.
Keywords: HIF-1a, hypoxia, migration/motility, p16, breast cancer
Introduction
One general characteristic of fast-growing solid tumors including breast cancer is the development of intratumoral hypoxia, whose existence correlates with a more malignant tumor phenotype and worse diagnosis. Adaptation to the hypoxic environment is critical for tumor cell survival and growth. The hypoxic cells in tumors modify gene expression in order to obtain a blood supply and prevent cellular damage; the main mediator of the hypoxia response is hypoxia-inducible factor-1 (HIF-1) (1). HIF-1 is a transcriptional activator for a group of genes that are responsible for promoting tumor cell angiogenesis and progression (1, 2). HIF-1 is composed of an inducible subunit, HIF-1α and a constitutively expressed subunit, HIF-1ß (3). Under normal oxygen tension, HIF-1α is rapidly degraded by posttranslational ubiquitination-triggered proteolysis. However, under low oxygen tensions (hypoxia), HIF-1α becomes stabilized. After nuclear translocation, HIF-1α heterodimerize with HIF-1ß to form transcriptional activator HIF-1, which binds to HRE within the promoter regions of target genes (such as VEGF) and thus regulates gene transcription (4).
p16, a tumor suppressor gene and cyclin D kinase inhibitor and a negative cell cycle regulator (5, 6), was shown to be able to inhibit breast cancer angiogenesis and metastasis (7). While our previous studies showed that p16 downregulates VEGF gene expression and neutralizes HIF-1α transactivation activity, it is not known whether p16 directly affects one or more aspects of malignant progression such as adhesion and migration of breast cancer cells in order to suppress metastasis. Particularly, cell migration is an important aspect of the tumor metastatic process that transforms the tumor cells from local, noninvasive confined cells to the migrating, metastatic cancer cells. There are many reports showing hypoxia plays an important role in tumor angiogenesis and tumor progression, however, very few studies have been done to date with regard to analyzing how hypoxia specifically affects cell migration. Study on whether HIF-1α is involved in cell migration is rare: while there are reports implying that HIF-1α may stimulate tumor cell migration (8, 9), no substantial evidence is available at present; moreover, no study has been done on whether p16 can inhibit hypoxia-mediated cell migration. In this study, we evaluated whether hypoxia stimulate breast cancer migration and whether HIF-1α is involved in this process; in addition, we were particularly interested in determining whether p16, which was shown to neutralize the function of HIF-1α (6)--the master transcriptional factor in response to hypoxia, directly affects one or more aspects of the malignant process, particularly in the hypoxia-mediated cell migration, of breast cancer cells.
Materials and methods
Cell lines, cell culture conditions, and reagents
Human breast cancer cell lines MDA-MB-231 and mouse mammary carcinoma cell line 4T1 were obtained from American Type Culture Collection (ATCC, Manassas, VA). Murine mammary carcinoma cell line JygMC(A) is a generous gift from Dr. H. Azuma of Osaka Medical College, Osaka, Japan (10). MDA-MB-231 cells and 4T1 cells were grown in RPM1-1640 (Gibco BRL, Gaithersburg, MD) with 10% fetal bovine serum (FBS) (Hyclone Laboratories, Logan, UT), JygMC(A) cells were grown in Dulbecco's Modified Eagle medium (D-MEM) (Gibco BRL) with 10% FBS. All cell lines were grown in medium containing 100 units/ml penicillin, and 100 μg/ml streptomycin. The cell cultures were incubated at 37°C either under nomorxia (5% CO2, 21% O2) or hypoxia (5% CO2, 1% O2, balanced with N2) conditions.
Generation and screen of stably transfected, Dox inducible (Tet-on) p16-expressing breast cancer cell lines
The construction of the Tet-on p16 lentiviral system Lenti-Tet-on p16 (pLenti-Tet-p16-pgkpuro and pcFUW-rtTA3-IRES-puro) was described previously (6). Breast cancer cells (MDA-MB-231, 4T1, and JygMC(A), respectively) were co-transduced by pLenti-Tet-p16-pgkpuro and pcFUW-rtTA3-IRES-puro at multiplicity of infection (moi) of 10 each, together with 6 μg/ml polybrene (Millipore, Bedford, MA). The stably transfected cells were enriched in medium containing 1 μg/ml puromycin (Clontech, Mountain View, CA). The resultant stably transfected cells are named as MDA/Tet-on p16, 4T1/Tet-on p16, and JygMC(A)/Tet-on p16, respectively. Similarly, breast cancer cells stably infected with lentiviral system expressing green fluorescent protein (GFP) reporter gene under inducible Tet-on promoter (Lenti-Tet-on GFP) were generated. For induction of p16 or GFP transgene expression, 1 μg/ml doxycyline (Dox) (Clontech) was used in the medium for incubation.
Immunohistochemistry
For in vitro immunohistochemistry (IHC), the culture cells were grown on SlideFlasks with detachable bottom slides (Nalge Nunc, Naperville, IL) that could be directly used for IHC staining later. The samples (slides) were incubated with the first antibody against human p16 (mouse anti-human p16 antibody, Santa Cruz Biotechnology, Inc., Santa Cruz, CA) for 16 h, then by a corresponding second antibody (goat anti-mouse antibody) and the Universal Elite ABC Kit (Vector Laboratories, Inc., Burlingame, CA) according to the manufacturer's protocol. The reaction was visualized with DAB solution (75 mg 3,3'-Diaminobenzidine and 30 μl 50% H2O2 in 150 ml PBS) for 3-10 min.
Growth proliferation assay
Briefly, the breast cancer cells were plated at 5x104 cells per well in the 6-well culture plate and incubated at 37°C in the absence or presence of 1 μg/ml Dox (for p16 induction). Cellular proliferation was measured by cell counting of the attached cells at day 5 after Dox treatment.
Adhesion assay
The breast cancer cells were incubated in the absence or presence of 1 μg/ml Dox for 72 h. The cells were then harvested and resuspended at 4x105 cells/ml in 1% FBS-medium. The cell suspensions were rested at 37°C in a CO2 incubator for 30 min. A 24-well tissue culture plate, which was precoated with matrigel matrix (BD Biosciences, Bedford, MA) by adding 250 µl of 10 µg/ml Matrigel per well for 2 hours and followed by blocking with 5% BSA in PBS for 1 h at 37°C, were used to seed viable 8×104 cells (in 200 µl cell suspension) in each precoated well of the 24-well plate and incubated for 4 h at 37°C. The nonadherent cells were then removed by washing 4 times with PBS. Then, the adherent cells were fixed with 4% paraformaldhyde for 10 min and stained using 1% toluidine blue for 5 min. After extensive washing with water, cells attached to the plates were solubilized with 1% SDS. The absorbance at 560 nm was used to quantify the cells as cell adhesion ability.
Transactivation assay of VEGF promoter
The MDA/Tet-on p16 cells were incubated in medium containing with or without 1 μg/ml Dox for 2 days prior to cotransfection. A chimeric construct containing a 2.4-kb VEGF promoter and a luciferase reporter gene (pVEGF/Luc) (a generous gift from Dr. BH Jiang of West Virginia University was cotransfected at 10:1 ratio (w:w) with phRLuc-TK (Promega), a plasmid expressing Renilla luciferase as an internal control for normalization of transfection efficiency, by Fugene 6 (Roche, Indianapolis, IN) according to the manufacturer's instruction. Forty-eight h after transfection, the cell extracts were harvested and analyzed for luciferase activity using Dual-Luciferase Assay Kit (Promega) as instructed by the manufacturer's instruction. The normalized luciferase activity was represented as ratio of Firefly luciferase activity (pVEGF/Luc) over Renilla luciferase activity (phRLuc-TK).
To ensure the increased VEGF transactivation under hypoxia is indeed due to the increased activity of HIF-1α, a HIF-1α inhibitor YC-1 (Alexis Biochemicals Corp., San Diego, CA) and two other chimeric luciferase reporter gene constructs, pMAP11WT and pMAP11mut (both generous gifts from Dr. B.H. Jiang of West Virginia University. Ref. 11) were used respectively to replace pVEGF/Luc in a similar transactivation assay. pMAP11WT is a minimal VEGF promoter/Luc chimeric construct that contains only 47 bp of VEGF 5-flanking sequence encompassing a HRE, the hypoxia response element where HIF-1α binds to (2, 8), and pMAP11mut is exactly the same as pMAP11WT except it contains a mutant HRE sequence so HIF-1α binding site is abolished (2, 8). The luciferase activity was normalized with protein concentration.
Cell motility/migration assay
The breast cancer cells were incubated in the absence or presence of 1 μg/ml Dox for 72 h. The cells were then harvested and used for the following migration assay. The cell migration/motility was measured by a modified Boyden's chamber method using BD Falcon Cell Culture Inserts incorporating polyethylene terephthalate (PET) track-etched membrane with a pore size of 8.0 μm (BD Bioscience, Belgium). The inserts were precoated on the undersurface (between upper and lower chambers) with 10 µg/ml fibronectin (Thermo Fisher Scientific) at 370C for 3 h. Above mentioned harvested cells in suspension of serum-free medium (SFM) with 1% BSA were incubated at 37°C for 30 min under nomorxia (5% CO2, 21% O2) or hypoxia (5% CO2, 1% O2, balanced with N2) conditions. Subsequently, cell suspensions were seeded into the upper chamber of an insert at a density of 10,000 cells per well, and 300 µl serum-free medium with 1% BSA was placed in the lower chamber to act as a chemoattractant in the 24-well cell culture plate. The cells were further incubated at normoxia and hypoxia (see above) at 37°C for 3 h. The inserts were then removed and nonmigrating cells remaining on the upper side of the filter were scraped off. The cells that had migrated to the lower surface of the insert were stained using Giemsa staining solution (Sigma, St. Louis, MO). After extensive washing with water, the migrated cells were counted in five different fields under a microscope at x200 magnification. Migratory activity was expressed as the number of cells (that is, the sum of total cell numbers in five randomly selected fields of view) that migrated to the lower side of the filter.
Results
Tet-on inducible transgene system via lentiviral stable transfection in breast cancer cells
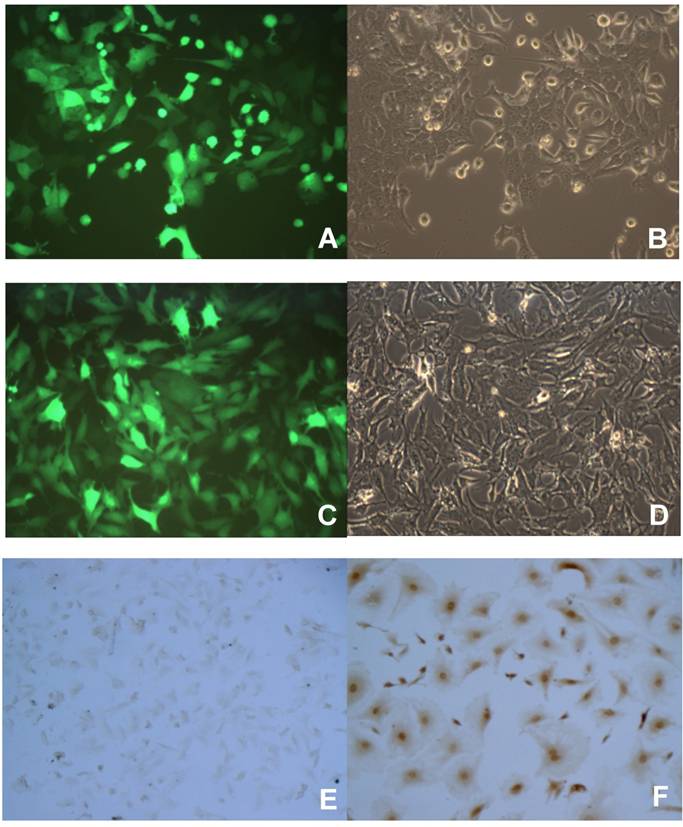
To better study the p16 effects on several aspects of tumor malignant progression, inducible p16 expression is desired in the breast cancer cells in order to compare the p16 effects on the same cells. We designed an inducible gene expression system in lentivirus where the transgene was placed under the control of a Tet-on promoter; the resultant lentiviral vectors would be used to stably transfect the breast cancer cells. To prove that this lentiviral vector works as an inducible transgene expression system in our hands, breast cancer cells were used to transduce with lentivirus expressing an inducible reporter gene GFP, the stably transfected cells were enriched by the selective reagent puromycin. The cells were then incubated with 1 µg/ml Dox to induce the Tet-on promoter controlled transgene GFP expression. As shown in Fig. 1, both breast cancer cells 4T1 and JygMC(A) demonstrated a near 100% transduction rate in terms of transgene GFP expression (Fig. 1A and 1C, respectively). These results indicated that the Tet-on inducible system by lentiviral vectors was able to successfully induce the desired transgene expression in the breast cancer cells by 1 µg/ml Dox. Likewise, another breast cancer cell line MDA-MB-231 stably transfected with lentivirus expressing an inducible human wild-type p16 gene under the control of the Tet-on promoter was generated (MDA/Tet-on p16). The MDA/Tet-on p16 cells showed a 100% p16 protein induction in the presence of 1 µg/ml Dox (Fig. 1F). Importantly, MDA/Tet-on p16 cells in the absence of inducer Dox did not express p16 protein (Fig. 1E), indicating that it is a truly tightly regulated Tet-on inducible system.
Induction of transgene by doxycycline (Dox) in breast cancer cells stably transduced with a retroviral-mediated Tet-on system. 4T1 (A and B) and JygMC(A) (C and D) cells stably infected with Lenti-Tet-on GFP were treated with 1 μg/ml doxycycline (Dox, a tetracycline analogue) for 72 h (A-D). The cells were then observed under a fluorescent microscope for transgene GFP expression. The same image of the cells (4T1, A and B; JygMC(A), C and D, respectively) at both fluorescent (A and C) and light-contrast (B and D) images are shown. E and F. Immunohistochemcal (IHC) staining for p16 protein on MDA-MB-231 cells stably infected with Lenti-Tet-on p16 (MDA/Tet-on p16). MDA/Tet-on p16 cells were treated either without (E) or with (F) 1 μg/ml Dox for 72 h. The cells (E and F) were then IHC stained by primary anti-p16 antibody. The dark brown color indicates p16 protein (F).
Effect of the expression of p16 protein on breast cancer cell growth
Tumor progression and metastasis include primary tumor growth, invasion through basement membrane and extracellular matrix, dissemination to lymphatic and/or blood circulation, migration to distant organs, and colonization at the secondary site (12). All these aspects of tumor progression contribute to the final net result of secondary tumor (metastasis) formation. Accordingly, several corresponding in vitro assays, including proliferation, adhesion, migration, in vitro invasion assays, can be applied to analyze the individual aspect of the tumor progression.
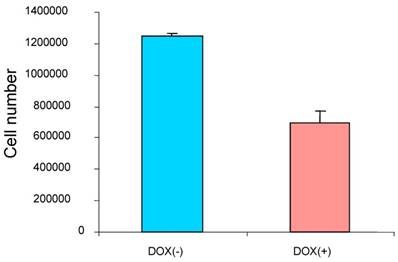
To study the effect of the induced p16 protein expression on breast cancer cell growth, breast cancer cells MDA/Tet-on p16 were treated with or without Dox and followed by measuring cell proliferation after 5 days treatment. A significant reduction of cell growth (about 45.6% inhibition) was observed when p16 expression was induced (Fig. 2), indicating that p16 inhibits breast cancer cell proliferation. Similar results were also observed in other breast cancer cells. The concentration of 1 µg/ml Dox did not have an effect on cell proliferation as evidenced by either treatment with 1 µg/ml Dox in parental breast cancer cells or cells stably expressing Tet-on GFP (not shown).
p16 inhibits breast cancer cell proliferation. The breast cancer cells MDA/Tet-on p16 were incubated with or without 1 μg/ml Dox for 5 days and the cell numbers were counted. The results represent the data from at least two independent experiments, each performed in triplicate.
p16 has no apparent effect on breast cancer cell adhesion ability
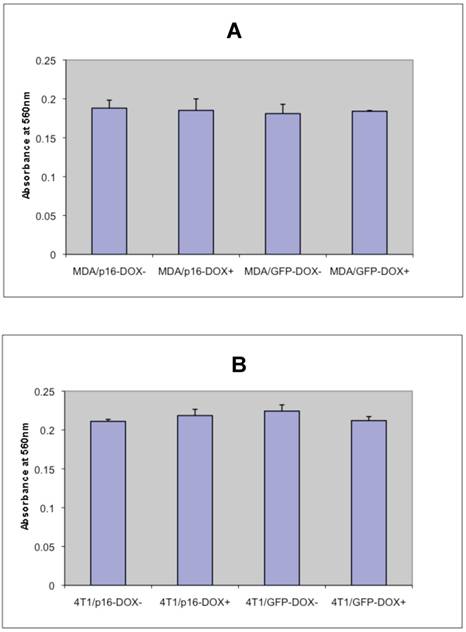
To determine whether p16 modulates cell adhesion of breast cancer cells on extracellular matrix, the cell adhesion assays were preformed on MDA/Tet-on p16 and other breast cancer cells. To detect any potential Dox effect at 1 μg/ml concentration on cell adhesion, MDA/Tet-on GFP was also included. MDA/Tet-on p16 and MDA/Tet-on GFP cells were incubated with or without 1 μg/ml Dox for 3 days, and the treated cells were then plated on 24-well plates precoated with 10 μg/ml matrigel matrix. After 4 h incubation, nonadherent cells were washed off and the adherent cells were stained and read at OD570. As shown in Fig. 3A, similar adhesion abilities were exhibited in both MDA/Tet-on p16 and MDA/Tet-on GFP lines under either presence or absence of Dox induction (Fig. 3A), indicating that (A) 1 μg/ml Dox did not affect cell adhesion behavior and (B) the expression of p16 protein has no significant effect on cell adhesion ability. Similar results were observed in the 4T1/Tet-on p16 and 4T1/Tet-on GFP (Fig. 3B). These combined results indicated that p16 did not affect breast cancer cell adhesion.
p16 does not appear to affect breast cancer cell adhesion. The breast cancer cells MDA/Tet-on p16 (or MDA/p16) and MDA/Tet-on GFP (or MDA/GFP) (A) or 4T1/Tet-on p16 (or 4T1/p16) and 4T1/Tet-on GFP (or 4T1/GFP) (B) were incubated with or without 1 μg/ml Dox for 72 h. The cells were then harvested and used for adhesion assay as described in M & M section. The results represent the data from at least two independent experiments, each performed in triplicate. The differences between with and without Dox treatment groups, as well as between Tet-on p16 and Tet-on GFP groups are not significant (p>0.05).
p16 neutralizes HIF-1α transactivation activity
Our previous study using ectopic expression revealed that HIF-1α increases VEGF gene transcription whereas p16 downregulates it in MDA-MB-231 cells; moreover, p16 neutralizes HIF-1α stimulated transactivation activity (6). Because endogenous HIF-1α can also be induced by hypoxia in MDA-MB-231 cells (7), we intended to investigate whether p16 can also neutralize hypoxia-induced HIF-1α transactivity; if so, we would further analyze whether p16 inhibits HIF-1α/hypoxia induced cell migration, an important aspect of malignant tumor progression.
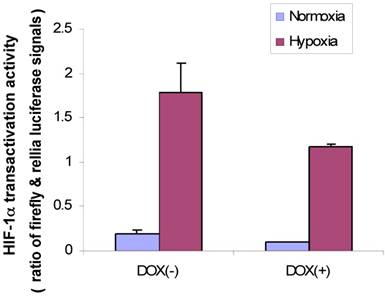
First, we used MDA/Tet-on p16 to cotransfect with a full-length VEGF promoter chimeric luciferase reporter gene construct, pVEGF/Luc (13) and phRLuc-TK, and cells were incubated either with or without Dox to induce p16 expression. The cell extracts were harvested later and analyzed by a Dual-Luciferase Assay Kit for examining whether p16 attenuated HIF-1α transactivity in terms of VEGF promoter activity. As shown in Fig. 4, the p16-expressing cells (Dox+) had reduced luciferase activity compared to that of its p16-nonexpressing counterpart (Dox-) in both normoxia and hypoxia conditions, with more significant effect under the hypoxia. These results indicate that p16 expression can attenuate HIF-1α's transactivation activity.
p16 inhibits HIF-1α transcriptional activity under both hypoxia and normoxia conditions. MDA/Tet-on p16 cells were treated with or without 1 µg/ml Dox and cotransfected with pVEGF/Luc and phRLuc-TK. The Dual-Luciferase Assay Kit was used to analyze the luciferase activity of cell extracts. The normalized luciferase activity was represented as ratio of Firefly luciferase activity (pVEGF/luc) over Renilla luciferase activity (phRLuc-TK). The results represent the data from three independent experiments, each performed in duplicate. Some error bars are too small to be seen in this scale.
Binding of HIF-1α to the HRE is required for the hypoxia mediated HIF-1α transactivation
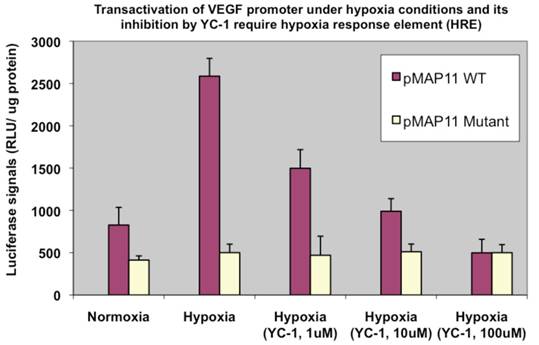
To ensure the elevated transactivation ability in terms of VEGF promoter transactivation under hypoxia we observed above (Fig. 4) is mainly mediated by HIF-1α, a HIF-1α inhibitor agent YC-1 (14) was included in a similar assay. We observed a dose-dependent reduction of HIF-1α transactivation activity by YC-1 on pVEGF/Luc (not shown). To further confirm/determine that result of our transactivation assay is indeed due to the HIF-1α binding that modulates VEGF promoter transactivation (the readout of HIF-1α transactivation), two minimal VEGF promoter chimeric luciferase reporter gene constructs, pMAP11WT (which contains only 47 bp of the VEGF 5-flanking sequence encompassing a HRE where HIF-1α binds to); and pMAP11mut (which is the same as pMAP11WT except it contains a mutant HRE sequence so the HIF-1α binding site is abolished) (11) were also included in a similar study. As shown in Fig. 5, only pMAP11WT, not pMAP11mut, had an increased transactivation under hypoxia compared to that under normoxia; furthermore, while YC-1 exhibited a dose-dependent reduction of HIF-1α mediated transactivation on pMAMP11WT under hypoxia (similar to that of the full-length VEGF promoter, not shown), YC-1 has no apparent effect on pMAP11mut (Fig. 5). These results clearly indicate that the binding of HIF-1α to the HRE is required for both hypoxia-induced transactivation and YC-1 mediated inhibition of VEGF promoter, implying that indeed HIF-1α is mainly responsible for the hypoxia induced transactivation of VEGF promoter and YC-1 mediated inhibition of this transactivation under hypoxia.
Hypoxia induced VEGF promoter transactivation and its inhibition by YC-1 involve HRE, the HIF-1α binding region of the promoter. MDA-MB-231 cells were transfected with pMAP11WT and pMAP11mut, followed by either normoxic or hypoxic conditions, and luciferase activity analysis as described in M & M section. A serial concentration of YC-1, a HIF-1α inhibitor, was added in the medium of cell samples under hypoxia in order to inhibit the hypoxia-induced HIF-1α transactivation activity. The normalized data is presented. The results represent the data from at least two independent experiments, each performed in duplicate.
Hypoxia increases cell migration in breast cancer cells
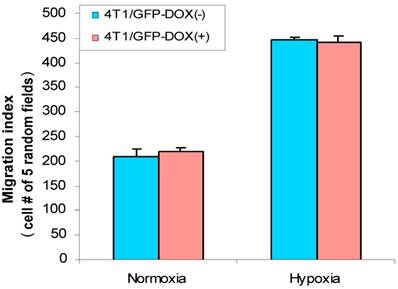
Cell migration is an important aspect of the tumor metastatic process. HIF-1α was implied in stimulation of cancer cell migration (8, 15) and hypoxia induces HIF-1α (7). Thus, we were interested in analyzing whether hypoxia has effect on breast cancer cell migration. First, we used 4T1/Tet-on GFP cells to study whether hypoxia induces cell migration and whether Dox per se had any effect on the cell migration. As shown in Fig. 6, hypoxia induced cell migration significantly, regardless the cells were incubated with or without Dox; on the other hand, 1 µg/ml Dox did not have any effect on cell migration, as evidenced by 4T1/Tet-on GFP with Dox, which did not show altered migration behavior compared to that of the cells without Dox treatment in both normoxia and hypoxia (Fig. 6). These results revealed: (A) hypoxia induced breast cancer cell migration; (B) Dox treatment per se did not have any effect on cell migration (Fig. 6).
Hypoxia increased cell migration/motility in breast cancer cells. The breast cancer cells 4T1/Tet-on GFP cells were incubated with or without 1 μg/ml Dox for 72 h, followed by 24 h in hypoxic or normoxia incubation prior to be used for cell migration assay as described in M & M section. The results represent the data from at least two independent experiments, each performed in triplicate.
p16 inhibits hypoxia-mediated breast cancer migration
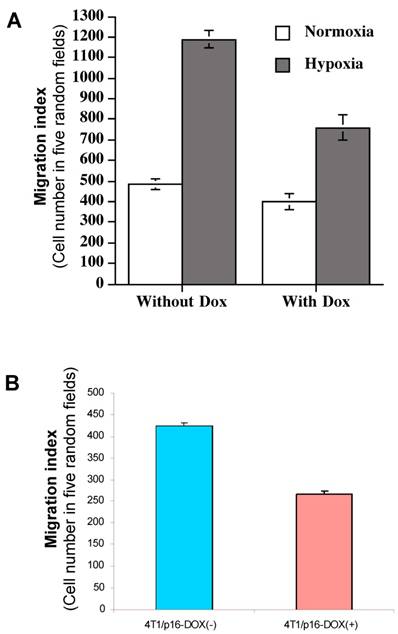
As we showed above, induced HIF-1α by hypoxia is responsible for the elevated VEGF transactivation (Fig. 5) and p16 can neutralize this HIF-1α-mediated action (Fig. 4); we were interested in determining whether p16 modulates cell migration as well, particularly under hypoxia conditions which was shown to increase cell migration (Fig. 6). MDA/Tet-p16 cells were treated with (p16 induction) or without (no p16 expression) Dox for 3 days. The treated cells were seeded in the upper chamber of the transwell that has been precoated with 10 μg/ml fibronectin at the undersurface. After 3 h at 37°C under either normoxia or hypoxia conditions, the cells that had migrated to the undersurface were counted. As shown in Fig. 7A, the cells under hypoxia had more than two fold increase in cell motility than that of normoxia (first two columns from the left, Fig. 7A); contrastingly, inducible p16 expression by Dox significantly inhibited this hypoxia-induced cell migration (column 4, Fig. 7A), with an approximate 36.0% inhibition in comparison to cells under hypoxia without p16 expression (column 2, Fig. 7A). The migration of cells under normoxia were also inhibited by p16 expression (17.1% inhibition), although not as effective as that under hypoxia (compare column 1 and column 3, Fig. 7A), indicating that p16 can inhibit cell migration at both normoxia and hypoxia, with significantly more effect on inhibition of hypoxia-induced cell migration. These data suggest that hypoxia increases (probably through elevated HIF-1α contribution to) MDA-MB-231 cell migration, and p16 is able to inhibit this hypoxia elevated cell migration. A similar result was observed in another breast cancer cell line 4T1/Tet-p16 where the induced p16 expression had a 38.8% inhibition on cell migration under hypoxia (Fig. 7B). These results, taken together with Fig. 4, Fig. 5 and Fig. 6, imply that p16 may inhibit hypoxia/HIF-1α elevated cell migration by neutralizing/attenuating the activity of HIF-1α induced by hypoxia.
p16 inhibits hypoxia-induced cell migration. (A) The breast cancer cells MDA/Tet-on p16 cells were incubated with or without 1 μg/ml Dox for 72 h, followed by cell migration assay as described in M & M section in normoxic and hypoxic conditions. (B) 4T1/Tet-on p16 (or 4T1/p16) cells were incubated with or without 1 μg/ml Dox for 72 h, followed by cell migration assay in hypoxic conditions. The results represent the data from at least two independent experiments, each performed in triplicate.
Discussion
In this report, we found that p16 inhibits breast cancer cell proliferation and migration, but has no apparent effect on cell adhesion. Interestingly, p16 shows much higher inhibition on cell migration in hypoxia than in normoxia. In addition, a parallel inhibition of HIF-1α transactivation by p16 was observed along with the p16-mediated inhibition of hypoxia-induced migration. As hypoxia promotes metastatic process and stimulates HIF-1α activation, these combined results imply a particular aspect of the metastatic process (such as migration) may be contributed, at least in part, by HIF-1α; and that p16 can effectively inhibit that aspect by inactivating HIF-1α. Noticeably, this study is the first report to our knowledge demonstrating that p16 inhibits hypoxia-induced cell migration in breast cancer cells, in parallel with its inhibition of HIF-1a transactivation activity. This study suggests that p16's ability to suppress tumor metastasis may be partially resulted from p16's inhibition on cell migration, in addition to its known functions on inhibition of cell proliferation, angiogenesis and induction of apoptosis.
Cell migration plays a central role in a wide variety of biological phenomena in both normal physiology and pathophysiology. Particularly in a tumor setting, cell migration is the transition process from the local, confined tumor cells to metastatic cancer cells when the cells obtain the ability to dissociate from intracellular adhesions and become motile (16, 17), which is usually driven by complex regulatory signaling cascades that transiently and/or permanently alter the expression of a multitude of genes that act to reorganize the cytoskeletal network (17, 18). Several motility/migration related proteins have been implicated to play important roles in tumor metastatic process (19-24). A previous study showed that vitexin, a HIF-1α inhibitor, significantly inhibited migration of rat pheochromocytoma PC12 cells along with hypoxia-induced activation of c-Jun N-terminal kinase (JNK) (8). A more recent study revealed that hypoxia induced a cell migration in colorectal carcinoma cells and this increased migration was mediated by human enhancer of filamentation 1 (HEF 1), a HIF-1a-regulated gene (9). These reports suggest that hypoxia or HIF-1α may play a role in cancer cell migration. Consistently, our results indicated that hypoxia increases breast cancer cell migration (Fig. 6), and p16 significantly inhibits hypoxia-induced MDA-MB-231 and 4T1 cell migration (Fig. 7A and 7B).
Previous studies demonstrated that binding of HIF-1α to the HRE requires wild-type HRE and mutation in this sequence would abolish such binding (2, 8). By using a pair of minimal VEGF promoter chimeric reporter constructs, one contained with (pMAp11WT) and one without (pMAP11mut) “HIF-1α-binding capable” HRE, we confirmed the requirement of HIF-1α binding to the VEGF promoter (i.e., the presence of HRE) for HIF-1α mediated transactivation (Fig. 5). The parallel inhibition of HIF-1α transactivation (Fig. 4) and hypoxia-induced cell migration by p16 (Fig. 7), together with the fact that p16 neutralizes HIF-1α function and hypoxia induces HIF-1α activation (see above), implies that p16 may inhibit cell migration in breast cancer cells by neutralizing HIF-1α mediated cell migration stimulated by hypoxia. To further verify whether this hypoxia-induced increased cell migration requires HIF-1α (Fig. 2A) and whether this p16-mediated inhibition of cell migration is truly due to the p16's neutralization of HIF-1α's transactivation function, we will examine p16 effects on the migration ability in a HIF-1α knockout breast cancer cells both in normoxia and hypoxia (work in process). Our ongoing study showed that hypoxia stimulated breast cancer cell migration in HIF-1α wild-type breast cancer cells but not in their HIF-1α knockout counterparts, implying hypoxia induced elevated cell migration requires HIF-1α (our unpublished results). In addition, we are also in the process of determining whether p16 has effects on breast cancer cells' invasion abilities, another aspect of tumor progression, by the in vitro invasion assay.
The development of intratumoral hypoxia is a common characteristic for fast-growing solid tumors including breast cancer. The main mediator of cells responding to hypoxia is activation of HIF-1, the master transcriptional factor which stimulates a group of downstream genes including VEGF that are responsible for tumor malignant progression (2, 25). HIF-1α is the inducible and regulatory subunit of the HIF-1; therefore, targeting and inhibiting the activity of HIF-1α should provide a therapeutic strategy to effectively block its regulated downstream signaling pathway and consequently suppress breast cancer malignant progression and metastasis. p16 is a biological agent that can inhibit/neutralize HIF-1α-mediated action on several levels of tumor progression, including growth, migration, angiogenesis which all contribute to the metastatic cascade.
Acknowledgements
This research project was supported by NIH grant (CA107162) (YL). We thank Andrew Lu for reviewing the manuscript.
Abbreviations
Dox: Doxycycline; GFP: green fluorescent protein; HIF-1α: hypoxia-inducible factor-1; HRE: hypoxia response element; IHC: immunohistochemistry; moi: multiplicity of infection; Tet-on promoter: tetracycline-induced regulated promoter; VEGF: vascular endothelial growth factor; WT: wild-type.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721-32
2. Maynard MA, Ohh M. The role of hypoxia-inducible factors in cancer. Cell Mol Life Sci. 2007;64:2170-80
3. Roth U, Curth K, Unterman TG, Kietzmann T. The transcription factors HIF-1 and HNF-4 and the coactivator p300 are involved in insulin-regulated glucokinase gene expression via the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2004;279:2623-31
4. Dachs GU, Patterson AV, Firth JD. et al. Targeting gene expression to hypoxic tumor cells. Nat Med. 1997;3:515-20
5. Shapiro GI, Rollins BJ. p16INK4A as a human tumor suppressor. Biochem Biophys Acta. 1996;1242:165-9
6. Zhang J, Lu A, Li L, Yue J, Lu Y. p16 Modulates VEGF expression via its interaction with HIF-1α in Breast Cancer Cells. Cancer Invest. 2010;28:588-97
7. Zhang J, Lu A, Beech D, Jiang B, Lu Y. Suppression of breast cancer metastasis through the inhibition of VEGF-mediated tumor angiogenesis. Cancer Ther. 2007;5:273-86
8. Choi HJ, Eun JS, Kim BG. et al. Vitexin, an HIF-1alpha inhibitor, has anti-metastatic potential in PC12 cells. Mol Cells. 2006;22:291-9
9. Kim SH, Xia D, Kim SW. et al. Human enhancer of filamentation 1 is a mediator of hypoxia-inducible factor-1alpha-mediated migration in colorectal carcinoma cells. Cancer Res. 2010;70:4054-63
10. Azuma H, Takahara S, Ichimaru N. et al. Marked prevention of tumor growth and metastasis by a novel immunosuppressive agent, FTY720, in mouse breast cancer models. Cancer Res. 2002;62:1410-9
11. Fang J, Xia C, Cao Z. et al. Apigenin inhibits VEGF and HIF-1 expression via PI3K/AKT/p70S6K1 and HDM2/p53 pathways. FASEB J. 2005;19:342-53
12. Heimburg J, Yan J, Morey S. et al. Inhibition of spontaneous breast cancer metastasis by anti-Thomsen-Friedenreich antigen monoclonal antibody JAA-F11. Neoplasia. 2006;8:939-48
13. Forsythe JA, Jiang BH, Iyer NV. et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604-13
14. Yeo EJ, Chun Y-S, Cho Y-S. et al. YC-1: a potential anticancer drug targeting hypoxia-inducible factor 1. J Natl Cancer Inst. 2003;95:516-25
15. Wang Y, Li Z, Zhang H. et al. HIF-1alpha and HIF-2alpha correlate with migration and invasion in gastric cancer. Cancer Biol Ther. 2010 [Epub ahead of print]
16. Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3:362-74
17. Yamazaki D, Kurisu S, Takenawa T. Regulation of cancer cell motility through actin reorganization. Cancer Sci. 2005;96:379-86
18. Kedrin D, van Rheenen J, Hernandez L. et al. Cell motility and cytoskeletal regulation in invasion and metastasis. J Mammary Gland Biol Neoplasia. 2007;12:143-52
19. Komorowski S, Baranowska B, Maleszewski M. CD9 protein appears on growing mouse oocytes at the time when they develop the ability to fuse with spermatozoa. Zygote. 2007;14:119-23
20. Miyake M, Hakomori SI. A specific cell surface glycoconjugate controlling cell motility: evidence by functional monoclonal antibodies that inhibit cell motility and tumor cell metastasis. Biochemistry. 1991;30:3328-34
21. Miyake M, Inufusa H, Adachi M. et al. Suppression of pulmonary metastasis using adenovirally motility related protein-1 (MRP-1/CD9) gene delivery. Oncogene. 2000;19:5221-6
22. Ono M, Handa K, Withers DA, Hakomori S. Motility inhibition and apoptosis are induced by metastasis-suppressing gene product CD82 and its analogue CD9, with concurrent glycosylation. Cancer Res. 1999;59:2335-9
23. Rubinstein E, Ziyyat A, Wolf JP. et al. The molecular players of sperm-egg fusion in mammals. Semin Cell Dev Biol. 2006;17:254-63
24. Zhou H, Huang S. mTOR Signaling in cancer cell motility and tumor metastasis. Crit Rev Eukaryot Gene Expr. 2010;20:1-16
25. Chen WT, Huang CJ, Wu MT. et al. Hypoxia-inducible factor-1alpha is associated with risk of aggressive behavior and tumor angiogenesis in gastrointestinal stromal tumor. Jpn J Clin Oncol. 2005;35:207-13
Author contact
![]() Corresponding author: Yi Lu, Ph.D., Department of Pathology and Laboratory Medicine, University of Tennessee Health Science Center, Cancer Research Building, Room 218, 19 South Manassas Street, Memphis, TN 38163, USA. Telephone: (901) 448-5436; Fax: (901) 448-5496; e-mail: yluedu
Corresponding author: Yi Lu, Ph.D., Department of Pathology and Laboratory Medicine, University of Tennessee Health Science Center, Cancer Research Building, Room 218, 19 South Manassas Street, Memphis, TN 38163, USA. Telephone: (901) 448-5436; Fax: (901) 448-5496; e-mail: yluedu