Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2013; 4(6):491-501. doi:10.7150/jca.6582 This issue Cite
Research Paper
p300 Influences Butyrate-Mediated WNT Hyperactivation In Colorectal Cancer Cells
Darina L Lazarova, Terrence Wong, Christopher Chiaro, Eric Drago, Michael Bordonaro ![]()
Department of Basic Sciences, The Commonwealth Medical College, 525 Pine Street, Scranton, PA 18509, USA.
Received 2013-4-29; Accepted 2013-6-13; Published 2013-7-18
Abstract
Deregulated WNT/catenin pathway, usually resulting from mutations in the adenomatous polyposis coli and beta-catenin genes, drives colorectal tumorigenesis. Dietary fiber has been shown to have a protective role against colorectal cancer (CRC). We have previously demonstrated that the histone deacetylase inhibitor (HDACi) butyrate, a fermentation product of dietary fiber, induces WNT/catenin hyperactivation, which promotes CRC cell apoptosis. Therefore, the ability of butyrate to induce WNT hyperactivation and thus promote CRC cell apoptosis may in part explain the preventive function of fiber against CRC. The association between beta-catenin and the transcriptional coactivator p300 may influence WNT/catenin signaling and, therefore, colonic cell physiology. p300 functions as a histone acetylase (HAT); therefore, the modulation of WNT/catenin activity by p300 may influence the ability of the HDACi butyrate to hyperinduce WNT signaling and apoptosis in CRC cells. Our findings indicate that p300 affects the hyperinduction of WNT activity by butyrate. Knockdown of p300 levels represses butyrate-mediated WNT/catenin activity; but still allows for butyrate-mediated apoptosis. Overexpression of p300 stimulates basal and butyrate-induced WNT signaling in some, but not all, CRC cell lines. We also evaluate the role of p300 in therapeutic approaches that target CBP. The small molecule ICG-001, in clinical trial, is a specific inhibitor of CBP-mediated WNT signaling, and previous studies have suggested that p300 is required for the activity of ICG-001. However, we report that ICG-001 maintains full activity against CBP-mediated WNT signaling in p300-deficient cell lines, including the butyrate-resistance line HCT-R. In addition, our findings evaluating combinatorial treatment of ICG-001 and butyrate in HCT-R cells may have important therapeutic implications for the treatment of butyrate-resistant CRCs.
Keywords: p300, colorectal cancer, WNT, butyrate, fiber, ICG-001.
Introduction
Deregulated WNT/beta-catenin (WNT/catenin) signaling, resulting from mutations in the adenomatous polyposis coli (APC) and beta-catenin genes (1-3), promotes colonic cell proliferation and tumorigenesis. Dietary factors, particularly fiber, have protective effects against colorectal cancer (CRC) (4-11). We have previously demonstrated that the histone deacetylase inhibitor (HDACi) butyrate, a fermentation product of dietary fiber (12,13), hyperactivates WNT signaling and promotes CRC cell apoptosis (4,5). This is consistent with the reported activity of HDACis in inducing cell cycle arrest, differentiation, and/or apoptosis of CRC cells (14-19). Therefore, the ability of butyrate to induce WNT hyperactivation and apoptosis may in part explain the preventive function of fiber against CRC (4,5,20). These findings are consistent with the observation that relatively high and relatively low levels of WNT/catenin activity lead to CRC cell apoptosis, while moderate levels of deregulated WNT/catenin activity promotes proliferation (4,5,20,21,22). Thus, by inducing excessive levels of WNT/catenin activity, butyrate promotes CRC cell death, reducing CRC risk.
The association between beta-catenin and the transcriptional coactivators CREB-binding protein (CBP) and p300, both of which are HAT factors, influences WNT signaling in CRC cells (23-28). Since both HDACis and HAT factors result in net protein acetylation, it is reasonable to hypothesize that CBP and p300 potentiate the effects of butyrate on WNT signaling and on apoptosis in CRC cells. On the other hand, CBP/beta-catenin and p300/beta-catenin have been reported to have distinct functions in mediating aspects of cell physiology (e.g., proliferation vs. differentiation) (23-28). Thus, WNT signaling and CRC cell apoptosis might be modulated differentially by CBP and p300.
The small molecule inhibitor ICG-001, which binds to CBP but not to p300, can be used to specifically evaluate the role of CBP/beta-catenin association on WNT signaling and cell physiology (23). In a recent study (29), we utilized ICG-001 to demonstrate that CBP-mediated WNT signaling is essential for the effects of butyrate on WNT hyperactivation, and we also observed important cell type-specific effects of ICG-001/butyrate cotreatment on the induction of apoptosis.
In the present study, we extend those studies to evaluate the role of p300 in the activity of butyrate and ICG-001 on WNT hyperactivation and CRC cell physiology. We demonstrate that p300 affects the hyperinduction of WNT activity by butyrate. Knockdown of p300 levels represses butyrate-mediated WNT/catenin activity, but butyrate-mediated apoptosis is maintained. In addition, overexpression of p300 stimulates basal and butyrate-induced WNT signaling in some, but not all, CRC cell lines, emphasizing the importance of p300 for WNT hyperactivation. We also demonstrate that ICG-001 maintains full activity against CBP-mediated WNT signaling in p300-deficient cell lines, including butyrate-resistant HCT-R cells. These findings may have important implications for chemopreventive and therapeutic approaches against CRC.
Materials and Methods
Reagents
Sodium butyrate was obtained from Sigma, ICG-001 and IQ-1 was provided by Dr. Michael Kahn (USC, Los Angeles, CA); ICG-001 was also obtained from Selleckchem.
Cell lines, Plasmids, siRNA, Transfection, reagent treatment
Cell lines were obtained from the ATCC and cultured as described previously (4,5). Expression vectors for p300 and CBP were obtained from Addgene. The TOPFlash and FOPFlash vectors were described previously (1,2,4,5). p300 siRNA sequences were as previously described (23). Control siRNA (sc-37007 or sc-44230) were from Santa Cruz. HCT-116 and HCT-R cells were transfected with the TOPFlash (wild-type) and FOPFlash (mutant) luciferase reporter vectors along with the normalization vector pRLTK (Promega) using Lipofectamine 2000 as previously described (4,5). Cells were treated with 5 mM sodium butyrate or mock-treated for 17.5 hr along with small molecule inhibitors ICG-001 and/or IQ-1 (23-27), or DMSO vehicle at the indicated for each experiment concentrations. Cells were lysed and assayed for reporter expression by the Dual Luciferase system (Promega) as previously described (4,5).
Apoptosis
Apoptotic analyses were performed using the caspase 3/7 Caspase-Glo luciferase system (Promega) (23). 20,000 HCT-R or HCT-15 cells/well in a 96 well plate were plated and the next day treated with 75 μM ICG-001, 5 mM sodium butyrate, both agents, or mock treated 24 hr, a time point which we have found yields optimal butyrate-induced apoptosis subsequent to WNT hyperactivation (4,5). Caspase 3/7 activity was measured with the Caspase Glo kit (Promega) according to manufacturer's instructions. Statistical analyses of caspase data was performed on background corrected raw data.
Western blot
Western blotting was performed as previously described (4,5), using anti-p300 antibody (sc-584, Santa Cruz Biotechnology) and ACTIN (A-5441, Sigma). For siRNA transfection for western blot 350 pmoles p300 siRNA per 1 million cells were nucleofected into HCT-116 CRC cells, compared to scrambled control siRNA (control). Total protein lysates were analyzed by western blot analysis, using p300 or CBP antibodies, with ACTIN antibody used as a control.
Proliferation assays and cell cycle analysis
Proliferation assays were performed with the QuickCell Proliferation kit (Biovision), according to manufacturer's instructions. Cell cycle analysis was performed with propidium iodide treatment; for flow cytometry, the propidium iodide was excited at 488 nm, and its fluorescence collected between 620 and 650 nm.
Statistics
Student's T-test was utilized to determine statistical significance (P < 0.05).
Results
Role of p300 in the hyperactivation of WNT/catenin signaling by butyrate
ICG-001, a specific inhibitor of CBP/beta-catenin association (23-28), influences butyrate-mediated WNT/catenin activity and colonic cell physiology (29). In a previous study (29), we demonstrated that ICG-001 treatment of HCT-116 and SW620 CRC cells diminished basal WNT/catenin activity and markedly repressed the hyperactivation of WNT signaling by butyrate. Further, while ICG-001 and butyrate inhibited proliferation in these cell lines, we demonstrated important cell type-specific effects of ICG-001/butyrate combinatorial treatment on CRC cell apoptosis. Thus, combining ICG-001 and butyrate interferes with the apoptotic response in SW620, but not HCT-116, cells, with implications for the use of ICG-001 as a chemotherapeutic agent in the human patient (29).
Given that CBP and p300 may play different roles in cell signaling and downstream physiological responses (23-28), it is important to evaluate the role of p300 in butyrate-mediated WNT hyperactivation and consequent effects on cell proliferation and apoptosis. In the absence of an available pharmacological inhibitor of p300/beta-catenin association, we utilized multiple approaches to ascertain the role of p300 in the hyperactivation of WNT/catenin signaling by butyrate.
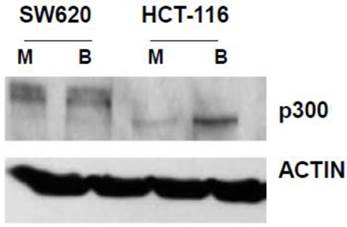
In our main approach, we used siRNA to knock down p300 levels to evaluate how reduced expression of p300 influences the hyperactivation of WNT/catenin signaling by butyrate. We first confirmed that SW620 cells express p300 (Fig. 1). We also confirmed that HCT-116 cells express a truncated form of p300, which retains normal HAT activity (30-32), and that this expression is upregulated by butyrate (Fig. 1).
Expression of p300 in CRC cells. SW620 and HCT-116 cells, mock treated (M) or treated for 17.5 hr with 5 mM butyrate (B) were lysed, and total protein analyzed by western blot, utilizing an anti-p300 antibody, with ACTIN antibody used as a control. Representative data are shown.
We then confirmed that a p300 siRNA previously shown to efficiently downregulate expression of p300, and to have a minimal effect on CBP expression (23), functions similarly under our experimental conditions (Fig.2A).
Repression of p300 influences WNT signaling hyperactivation by butyrate. (A) Confirmation of activity of p300 siRNA previously utilized to downregulate p300, but not CBP, expression (30). (B) 15 pmoles/well control (mock, M) or p300 siRNA (p300si) were cotransfected into HCT-116 cells (96-well plate) along with the TOPFlash or FOPFlash vectors and pRLTK control. Cell media was changed after 5 hours, and after another 24 hr, cells were mock treated (M) or treated with 5 mM butyrate (B) for another 17 hr. Data are from three separate experiments. Bars, SDs. * = statistical significance. (C) SW620 cells were treated as in (B). Data are from three separate experiments. Bars, SDs. * = statistical significance. (D) HCT-116 cells were transfected with Ctl or p300 siRNA, mock treated or treated with 5 mM butyrate for 24 hr, and proliferation assayed as described in Materials and Methods. Data are from four separate experiments. Bars, SDs. (E) SW620 cells were treated with p300 siRNA and butyrate as in (D). Data are from four separate experiments. Bars, SDs.
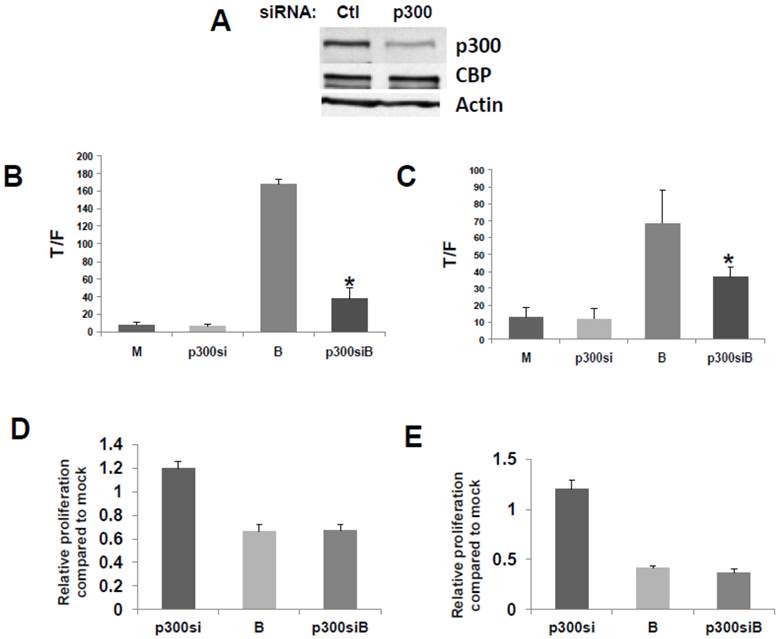
To evaluate the effects of p300 silencing on the ability of butyrate to hyperactivate WNT signaling, HCT-116 cells were cotransfected with the TOP/FOP reporter vectors and with control or p300 siRNA. Knockdown of p300 repressed the enhancement of WNT/catenin activity by butyrate (Fig. 2), analogous to the disruption of CBP-WNT activity by ICG-001 (23-29). Thus, whereas p300 knockout did not influence WNT/catenin activity in mock-treated cells, in the presence of butyrate, WNT signaling levels were sharply repressed by p300 siRNA (P<0.001) (Fig. 2B). However, even though p300 knockdown reduced the final levels of butyrate-mediated WNT/catenin activity, butyrate still retained the ability to induce WNT hyperactivation. Thus, HCT-116 cells transfected with p300 siRNA and treated with butyrate exhibited 6-fold higher levels of WNT/catenin activity compared to similarly transfected cells not treated with butyrate (P<0.02). The second p300-expressing CRC cell line used in this study, SW620 cells, exhibited similar repression of butyrate-induced WNT/catenin activity after p300 knockdown (P<0.03). In addition, butyrate retained the ability to upregulate WNT/catenin activity in SW620 cells transfected with p300 siRNA (P<0.002). These data are consistent with p300 contributing to the hyperactivation of WNT/catenin activity by butyrate. In addition, p300 knockdown mimics ICG-001 treatment in that while final levels of butyrate-mediated WNT/catenin activity are reduced, butyrate remains capable of inducing a several-fold increase in WNT signaling.
Knockdown of p300 levels with siRNA did not affect the ability of butyrate to upregulate caspase activity in either the HCT-116 or SW620 cells; for both cell lines, levels of butyrate-mediated caspase activity were similar in the presence or absence of p300 siRNA (data not shown). However, unlike what was observed with ICG-001-mediated disruption of CBP-WNT activity (23-29), p300 knockdown did not reduce HCT-116 or SW620 cell proliferation in the presence or absence of butyrate (Fig. 2D,E). In both cell lines, in the absence of butyrate, p300 knockdown resulted in a modest (20%) increase of cell growth, which did not reach the level of statistical significance.
Activity of butyrate and ICG-001 in p300 deficient CRC cell lines
The role of p300 in the effects of butyrate on WNT signaling was also evaluated utilizing p300 deficient CRC cells. Previous studies (23-28) suggest that expression of p300 may be required for the activity of ICG-001; thus, it has been suggested that the repressive role of ICG-001 on WNT signaling and CRC cell growth is due to inhibition of CBP/beta-catenin complexes and promotion of p300/beta-catenin complexes. If p300 is required for ICG-001 activity, then the effects of ICG-001 would be attenuated or abrogated in p300-deficient CRC cells.
We have developed a butyrate-resistant version of HCT-116 cells, by exposing that cell line to increasing levels of butyrate. Thus, HCT-R cells are resistant to the effects of butyrate (and other HDACis) on WNT/catenin activity, apoptosis, and growth suppression (2). While HCT-R cells express CBP, we establish here that they do not express p300 (Fig. 3A). Thus, we evaluated the effects of ICG-001 treatment on HCT-R cells, to ascertain if p300 plays a required role in the activity of this clinically relevant (33) chemotherapeutic agent.
ICG-001 modulates WNT/catenin activity and apoptosis in p300 deficient HCT-R CRC cells. (A) Western blot data confirming expression of CBP and lack of p300 expression in HCT-R cells, mock treated (M) or treated with 5 mM butyrate (B). In the p300 immunoblot, non-specific background bands are present, but p300 expression is not observed. (B) ICG-001 inhibits butyrate-induced WNT/catenin activity in HCT-R cells. Experiments were performed as described in ref. 29. Data are from three separate experiments. Bars, SDs. * = statistical significance. (C) Butyrate interferes with upregulation of apoptosis by ICG-001 in HCT-R cells. Experiment performed as in the Materials and Methods section. Data are from four separate experiments. Bars, SDs. * = statistical significance.
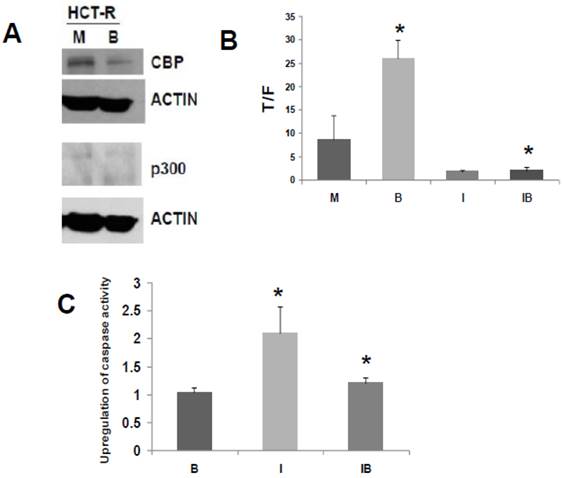
Similar to what was observed for HCT-116 cells, treatment of HCT-R cells with 75 μM ICG-001 reduced WNT/catenin activity, as measured by the T/F ratio (Fig. 3B). However, given the variability in WNT/catenin activity in mock-treated HCT-R cells (routinely maintained in butyrate), the repression of WNT activity by ICG-001 was slightly above the level of statistical significance (P=0.08). As expected, ICG-001 markedly reduced WNT/catenin activity in HCT-R cells cultured in butyrate (P<0.001). Importantly, treatment with ICG-001 eliminated the low level of induction of WNT/catenin activity in butyrate-exposed HCT-R cells: WNT/catenin activity was modestly enhanced (P<0.01) after 17.5 hr exposure to 5 mM butyrate; cotreatment with ICG-001 resulted in the loss of this induction (P<0.001). Therefore, HCT-R cells differ from HCT-116 and SW620 cells in that ICG-001 completely eliminates the ability of butyrate to induce WNT/catenin activity.
Next, we evaluated how ICG-001 influences the ability of butyrate to induce apoptosis in HCT-R cells, as measured by caspase activity (Fig. 3C). HCT-R cells are resistant to the apoptotic effects of butyrate compared to parental HCT-116 cells (5). Consistent with previous findings (2), treatment of HCT-R cells with butyrate did not upregulate apoptosis. In contrast to HCT-116 cells (29), treatment of HCT-R cells with ICG-001 significantly upregulated caspase activity (P<0.01). Cotreatment with butyrate and ICG-001 repressed caspase activity compared to ICG-001 alone (P<0.03). Compared to mock treatment, exposure of HCT-R cells to both butyrate and ICG-001 resulted in a modest 20% increase in caspase activity (P<0.01). Thus, in butyrate-resistant HCT-R cells, the greatest level of caspase activity was achieved by treatment with ICG-001 alone, and butyrate interfered with the pro-apoptotic action of ICG-001.
HCT-R cells do not express detectable levels of p300 in presence or absence of butyrate (Fig. 3A), and these cells exhibit marked differences in global gene expression compared to the parental HCT-116 cells (34). Therefore, HCT-R cells, which were developed in our laboratory, may not fully reflect the characteristics of CRC cells that are p300-deficient due to spontaneous mutation in vivo. Therefore, we also assessed effects of ICG-001 in HCT-15 CRC cells. The HCT-15 line is WNT activity-positive and APC mutant; these cells lack expression of p300, but express CBP (30). The lack of p300 expression is due to an in vivo mutation during the process of tumorigenesis, and it is believed that this mutation results in nonsense mediated decay of p300 mRNA (30-32). Therefore, HCT-15 cells represent p300-deficient tumors that naturally arise in vivo; such tumors may exhibit differential sensitivities to butyrate and to agents such as ICG-001. The lack of p300 expression and the presence of CBP expression in HCT-15 cells (30) was confirmed under our experimental conditions, and we established that this pattern of CBP and p300 expression is not altered in presence of 5 mM butyrate (Fig. 4A).
ICG-001 modulates WNT/catenin activity and apoptosis in p300 deficient HCT-15 CRC cells. (A) Western blot data confirming expression of CBP and lack of p300 expression in HCT-15 cells, mock treated (M) or treated with 5 mM butyrate (B). (B) ICG-001 inhibits butyrate-induced WNT activity in HCT-15 cells. Experiments were performed as in Fig 3B. Data are from three separate experiments. (C) Butyrate and ICG-001 influence apoptosis in HCT-15 cells. Experiment performed as in Fig. 3C. Data are from three separate experiments. Bars, SDs. * = statistical significance.
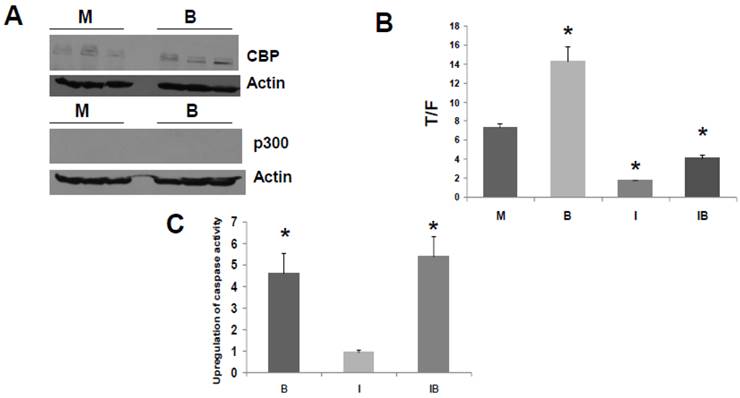
Titration experiments indicated that the WNT/catenin activity in HCT-15 cells was downregulated effectively by 50 to 100 μM ICG-001, with maximal downregulation at 75 μM (data not shown). We therefore used 75 μM ICG-001 for all subsequent experiments with these cells. HCT-15 cells exhibited a marked repression of WNT/catenin activity in presence of ICG-001 (Fig. 4B, P<0.001). Interestingly, HCT-15 CRC cells did not markedly induce WNT/catenin activity in the presence of butyrate (Fig. 4B, P<0.003); this is similar to butyrate-resistant HCT-R cells. The observation that both p300-deficient cell lines exhibit low-fold induction of WNT activity by butyrate supports a role for p300 in WNT hyperactivation.
We then examined the effects of ICG-001 on butyrate-induced apoptosis of HCT-15 cells (Fig. 4C). Butyrate induced caspase activity compared to mock treated controls (P < 0.001), while there was no statistically significant difference between caspase activity in HCT-15 cells treated with 75 μM ICG-001 compared to control. ICG-001 did not interfere with the ability of butyrate to upregulate caspase activity; the combination of butyrate and ICG-001 resulted in the largest induction of caspase activity compared to control (P<0.01). However, compared to butyrate alone, cotreatment with butyrate/ICG-001 did not further increase caspase activity to a statistically significant extent.
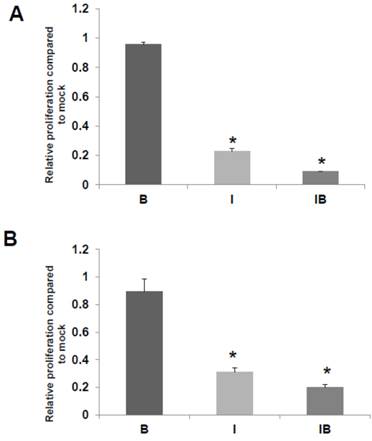
We also assessed the effects of butyrate and/or ICG-001 treatment on HCT-R and HCT-15 cell growth. As expected HCT-R cell proliferation was unaffected by treatment with butyrate, but was sharply decreased by treatment with ICG-001 (P<0.002), as well as by cotreatment with both agents (P<0.001) (Fig. 5A). In HCT-15 cells, butyrate did not result in a statistically significant decrease in the number of viable cells, while cell growth was diminished by ICG-001 (P<0.05) and cotreatment with both butyrate and ICG-01 (P<0.03) (Fig. 5B).
HCT-R and HCT-15 cell proliferation. Effects of butyrate and ICG-001 on HCT-R (A) and HCT-15 (B) cell proliferation were assayed as described in Materials and Methods. Data are from three separate experiments. Bars, SDs. * = statistical significance.
Overexpression of p300 in CRC cells
The p300 siRNA data (Fig. 2) clearly show that p300 is involved in the hyperactivation of WNT activity by butyrate, a finding supported by the low level of WNT/catenin hyperactivation observed in p300-deficient CRC cells (Figs. 3,4). To further confirm that p300, like CBP, influences butyrate-mediated WNT/catenin activity, we performed overexpression experiments. If p300 positively influences WNT hyperactivation, then exogenous overexpression of p300 should upregulate butyrate-induced WNT signaling. Since p300 contributes to butyrate-mediated WNT hyperactivation in CRC cells (Figs. 3,4), overexpressed p300 and ICG-001 may have opposing effects on WNT/catenin activity in CRC cells.
To confirm the positive role of p300 on WNT hyperactivation, and to assess interactions between p300 and the action of ICG-001, we exogenously overexpressed p300 in CRC cell lines. We then evaluated effects of p300 overexpression on basal WNT/catenin activity, butyrate-induced WNT hyperactivation, and ICG-001-mediated changes in WNT activity. CRC cells were cotransfected with the TOPFlash or FOPFlash reporters, and an expression vector for p300 and treated with butyrate and/or ICG-001. Overexpression of p300 in HCT-116 cells upregulated WNT/catenin activity in the presence and absence of butyrate (Fig. 6A). Treatment with ICG-001 reduced WNT/catenin activity resulting from expression of p300. Thus, expression of p300 in HCT-116 cells enhanced WNT activity (P<0.05), and this statistically significant increase was eliminated by treatment with ICG-00l. Overexpression of p300 also augmented WNT/catenin activity in butyrate-treated HCT-116 cells (P<0.005). This fold increase in WNT signaling was maintained in the presence of ICG-001 (compare IB and p300IB, P<0.001). However, the absolute levels of WNT/catenin activity in p300-transfected cells exposed to butyrate was decreased by ICG-001 (compare p300B with p300IB, P<0.001). In contrast, overexpression of p300 did not have any significant effect on WNT/catenin activity in mock and butyrate-treated SW620 CRC cells (data not shown).
Overexpression of p300 levels influences WNT signaling in CRC cells. Overexpression of p300 enhances WNT/catenin activity in HCT-116 (A), butyrate-resistant HCT-R (B), and p300-mutant HCT-15 (C) CRC cells. Cells were transfected with TOP/FOPFlash reporter vectors (0.1 μg/well), and an expression vector for p300 or a control vector (0.4 μg/well), and with pRLTK for normalization of transfection efficiency. After 5 hours cells were left untreated (M and p300) or treated with 75 μM ICG-001 (I), 5 mM butyrate (B) or both agents (IB) overnight. Data for Fig. 6 are from three to four separate experiments; Bars, SDs. * = statistical significance.
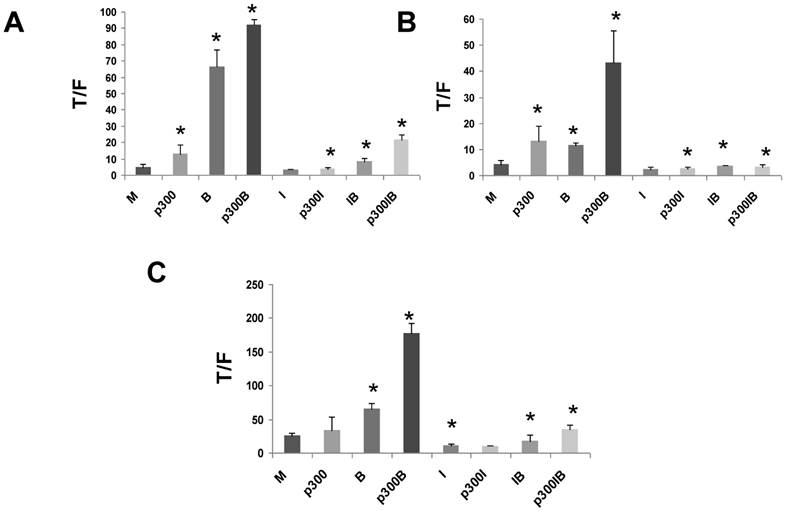
Since HCT-R and HCT-15 cells do not express p300 protein (Figs. 3,4), it was important to evaluate the effects of exogenous p300 expression in these cell lines. We observed that HCT-R cells transfected with p300 exhibited enhanced WNT/catenin activity, and a higher level of induction of this activity by butyrate (Fig. 6B). Similar to HCT-116 cells, treatment of HCT-R cells with ICG-001 eliminated the upregulation of WNT/catenin activity resulting from endogenously expressed p300 (compare p300I with p300, P<0.02). The high levels of WNT activity in butyrate-treated p300-transfected HCT-R cells were markedly repressed by cotreatment with ICG-001 (compare p300B with p300IB, P<0.002). In addition, in contrast to HCT-116 cells, ICG-001 treatment of HCT-R cells also eliminated the fold-increase in WNT activity mediated by p300 in the presence of butyrate (compare p300IB to p300I). Exogenous expression of p300 did not significantly alter levels of WNT/catenin activity in mock-treated p300-deficient HCT-15 cells (Fig. 6C). However, exogenous p300 markedly augmented the ability of butyrate to upregulate WNT/catenin signaling (P<0.001). Similar to HCT-116 and HCT-R cells, the high final levels of WNT/catenin activity observed with p300 expression and butyrate treatment were reduced by ICG-001 (P<0.001). Finally, ICG-001 did not eliminate the fold-increase of WNT/catenin activity observed from p300-transfected, butyrate-treated HCT-15 cells (compare p300IB to p300I).
In certain CRC cell lines, treatment with ICG-001 promotes p300/beta-catenin complex formation, while inhibiting CBP/beta-catenin binding (23). The small molecule IQ-1 has the ability to block increased p300/beta-catenin complex formation that is promoted by ICG-001 treatment (27). To evaluate whether the repression of WNT/catenin activity by ICG-001 is due to enhanced p300/beta-catenin association, we co-treated CRC cells with both ICG-001 and IQ-1, and observed no significant difference compared to treatment with ICG-001 alone (data not shown). This finding further argues against a repressive role for p300/beta-catenin complexes in the hyperactivation of WNT signaling by butyrate, and is consistent with our data that p300 positively contributes to butyrate-induced WNT/catenin signaling (Figs. 2-4).
In contrast to the p300 findings we did not detect an effect of overexpressed CBP on basal or butyrate-mediated WNT signaling in CRC cells (data not shown).
Discussion
Our findings demonstrate that p300-mediated WNT/catenin activity contributes to the upregulation of WNT signaling by butyrate, as demonstrated by suppression and overexpression experiments (Figs. 2,6), and the observation that CRC cell lines deficient in p300 exhibit low fold induction of WNT/catenin activity by butyrate (Figs. 3,4).
While modulation of CBP-WNT activity has been more extensively studied, p300-WNT activity represents another potential target for therapeutic intervention. Previous studies indicated that CBP-mediated and p300-mediated WNT signaling have distinct functions in colonic cell physiology, with CBP enhancing cell proliferation and p300 promoting differentiation (23-28). Consistent with this, while the CBP-WNT inhibitor ICG-001 sharply decreased CRC cell proliferation (23,29), silencing of p300 did not have a statistically significant effect on cell growth (Fig. 2). However, our findings suggest that, similar to CBP (29), p300 contributes to the hyperactivation of WNT/catenin activity by butyrate (Figs. 2-4,6).
Further, even though ICG-001 specifically represses CBP and beta-catenin association (23, 29), we observed that ICG-001 treatment interfered with the ability of p300 to enhance butyrate-mediated WNT hyperactivation (Fig. 6). However, the HAT activity of CBP and p300 is also mediated through interactions with factors other than beta-catenin. For example, in addition to acetylation of histones, CBP and p300 acetylate non-histone proteins, including those involved in WNT signaling, thus influencing transcriptional activity (35-38). Knockdown or overexpression of p300 may have more pleiotropic effects compared to the more targeted action of ICG-001 against CBP. Thus, increased levels of p300 may influence WNT/catenin signaling directly, as well as indirectly by enhancing CBP-WNT activity. If p300 indirectly enhances CBP-mediated WNT signaling, then effects of overexpressed p300 on WNT/catenin activity would be sensitive to ICG-001, as observed (Fig. 6). The mechanism by which interference between ICG-001 and overexpressed p300 occurs is currently under investigation in our laboratory.
Although some studies suggest a role for p300 in the activity of ICG-001, our findings with the p300-deficient HCT-15 and HCT-R CRC cell lines demonstrate that p300 is not required for the effects of ICG-001 on WNT hyperactivation and cell proliferation. The p300-deficient HCT-15 cell line exhibits similar characteristics as the p300-expressing HCT-116 cell line with respect to response to ICG-001. Thus, similar to HCT-116 cells (29), ICG-001 treatment of HCT-15 cells reduces butyrate-induced WNT/catenin activity; in addition, butyrate retains the ability to upregulate WNT/catenin activity even in the presence of ICG-001 (Fig. 4B). Further, and also similar to HCT-116 cells, the upregulation of HCT-15 cell apoptosis induced by butyrate is not repressed by ICG-001 (Fig. 4C).
In contrast, butyrate could not upregulate WNT/catenin activity in HCT-R cells treated with ICG-001; WNT/catenin activity observed after butyrate/ICG-001 cotreatment was statistically equivalent to that observed with ICG-001 alone (Fig. 3B). HCT-R cells also exhibit significant interference between ICG-001 and butyrate in the induction of caspase activity: treatment with ICG-001 alone markedly upregulates caspase activity and this increased activity is suppressed upon cotreatment with butyrate (Fig. 3C). Previous work from our laboratory indicated that HCT-R cells exhibit abnormal regulation of genes related to WNT/catenin signaling, cell cycle progression, and apoptosis (34). These changes in gene expression may in part explain the differences between HCT-R cells and other CRC cell lines in the fold-induction of WNT signaling and of apoptosis with butyrate/ICG-001 cotreatment.
Since HDACi-resistant HCT-R CRC cells exhibited the greatest apoptotic response to ICG-001 (Fig. 3C) compared to other CRC cell lines (Fig. 4, ref. 29), we posit that ICG-001-like agents might be highly effective in inducing apoptosis in colon cancers that are resistant to the effects of butyrate and other HDACis (1,2). Right-sided colonic tumors may exhibit such resistance, since in patients with high dietary fiber intake, such neoplasms develop despite the high levels of butyrate in the proximal colon (39). In CRC cells in which butyrate does not increase WNT/beta-catenin signaling levels and therefore, does not induce apoptosis, ICG-001-like agents can potentiate apoptosis through suppression of WNT activity (Fig. 3). The therapeutic choice of hyperactivating or suppressing WNT/beta-catenin activity in colonic neoplasms could be based upon assays of this signaling in patients' cells obtained through biopsy. Further, particular mutation profiles associated with the HDACi-resistant and HDACi-sensitive phenotypes may allow for more informed therapeutic choices in the future. Determination of the expression levels of CBP and p300 in patient samples may also contribute to the decision of whether ICG-001/HDACi combination therapy would be cooperative or antagonistic with respect to apoptosis. However, we need to further characterize the molecular properties of CRC cells that facilitate a particular apoptotic response to combination therapy, and such studies are currently underway in our laboratory.
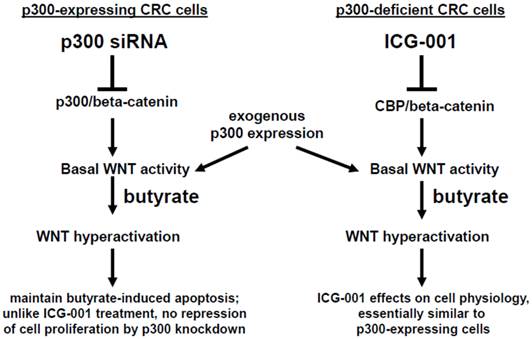
In summary, p300 plays an important role in mediating the hyperactivation of WNT/catenin activity observed after treatment of CRC cells with the HDACi butyrate. However, although p300 may be required in mediating some effects of ICG-001 in CRC cells (23-28), p300 expression is not required for effects of ICG-001 on cell growth or on butyrate-mediated WNT hyperactivation. We summarize the possible role of p300 in the activity of butyrate and ICG-001 in Fig. 7. In addition, the strong promotion of apoptosis in the p300-deficient, butyrate-resistant HCT-R cell line suggests that ICG-001 may be particularly effective against butyrate-resistant CRCs that may be enriched among right-sided colonic neoplasms. The possibility that butyrate-resistance may be correlated to downregulation of p300 also needs to be explored.
Role of p300 in mediating the effects of butyrate on WNT signaling in CRC cells. (Left). In p300-expressing CRC cells (e.g., HCT-116 and SW620), p300-mediated WNT/catenin activity, which is repressed by p300 siRNA knockdown, contributes to WNT hyperactivation by butyrate. However, butyrate-mediated apoptosis, which is in part dependent on WNT signaling (29), is maintained in cells exposed to p300 siRNA. Exogenous (over)expression of p300 stimulates WNT/catenin activity and WNT hyperactivation in HCT-116, but not SW620, CRC cells. (Right). Previous reports have suggested that ICG-001, which represses CBP-mediated WNT signaling, works in part by inhibiting CBP/beta-catenin association and promoting p300/beta-catenin association (23-28). However, in the present study, we show that p300 expression is not required for the activity of ICG-001 on basal and butyrate-induced WNT/catenin activity, as demonstrated in the p300-deficient CRC cell lines HCT-R and HCT-15. Exogenous expression of p300 in these cell lines stimulates WNT/catenin activity and WNT hyperactivation. The effects of exogenous p300 expression on WNT signaling can in some cases be inhibited by the CBP-specific inhibitor ICG-001 by as yet unknown mechanisms. Arrows indicate positive interactions (activation); blocked lines indicate negative interactions (repression).
Further studies are required to fully evaluate the therapeutic potential of targeting p300-WNT activity; such studies will be facilitated by the development of specific pharmacological inhibitors of the association between p300 and beta-catenin.
Acknowledgements
We thank Dr. Michael Kahn for supplying ICG-001 and IQ-1. This work was supported by NIH NCI grant 1R15CA149589-01.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Korinek V, Barker N, Morin PJ, Van Wichen D, De Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997;275:1784-7
2. Morin J, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787-90
3. Bienz M, Clevers H. Linking colorectal cancer to WNT signaling. Cell. 2000;103:311-20
4. Lazarova DL, Bordonaro M, Carbone R, Sartorelli AC. Linear relationship between WNT activity levels and apoptosis in colorectal carcinoma cells exposed to butyrate. Internat J Cancer. 2004;110:523-31
5. Bordonaro M, Lazarova DL, Sartorelli AC. The activation of beta-catenin by WNT signaling mediates the effects of histone deacetylase inhibitors. Exp Cell Res. 2007;313:1652-66
6. Richter M, Jurek D, Wrba F, Kaserer K, Wurzer G, Karner-Hanusch J, Marian B. Cells obtained from colorectal microadenomas mirror early premalignant growth patterns in vitro. Euro J Cancer. 2002;38:1937-45
7. Kautenberger T, Beyer-Sehlmeyer G, Festag G, Haag N, Kuhler S, Kuchler A, Weise A, Marian B, Peters WHM, Liehr T, Claussen U, Pool-Zobel BL. The gut fermentation product butyrate, a chemopreventive agent, suppresses glutathione S-transferase theta (hGSTT1) and cell growth more in human colon adenoma cells (LT97) than tumor (HT29) cells. J Cancer Res Clin Oncol. 2005;131:692-700
8. Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Migrating cancer stem cells - an integrated concept of malignant tumour progression. Nature Reviews. 2005;5:744-9
9. Bingham SA, Day NE, Luben R, Ferrari P, Slimani N, Norat T, Clavel-Chapelon F, Kesse E, Nieters A, Boeing H, Tjonneland A, Overvad K, Martinzez C, Dorronsoro M, Gonzalez CA, Key TJ, Trichopolou A, Naska A, Vineis P, Tumino R, Krogh V, Bueno-de-Mesquita HB, Peeters PH, Berglund G, Hallmans G, Lund E, Skeie G, Kaaks R, Riboli E. Dietary fibre in food and protection against colorectal cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC): an observational study. Lancet. 2003;361:1496-501
10. Peters U, Sinha R, Chaterjee N, Subar AF, Ziegler RG, Kuldorff M, Bresalier R, Weissfeld JL, Flood A, Schtzkin A, Hayes RB. Dietary fibre and colorectal adenoma in a colorectal cancer early detection programme. Lancet. 2003;361:1491-5
11. Bingham SA, Norat T, Moskal A, Ferrari P, Slimani N, Chavel-Chapelon F, Kesse E, Nieters A, Boeing H, Tjonneland A, Overvad K, Martinzez C, Dorronsoro M, Gonzalez CA, Ardanaz E, Navarro C, Quiros JR, Key TJ, Day NE, Trichopoulo A, Naska A, Krogh V, Tumino R, Palli D, Panico S, Vineis P, Bueno-de-Mesquita HB, Ocke MC, Peeters PHM, Berglund G, Hallmans G, Lund E, Skeie G, Kaaks R, Riboli E. Is the association with fiber from foods in colorectal cancer confounded by folate intake? Cancer Epid Biom Prev. 2005;14:1552-6
12. Bingham S. Mechanisms and experimental evidence relating dietary fibre and starch to protection against large bowel cancer. Proc Nutr Soc. 1990;49:153-71
13. Cummings JH, Pomare EW, Branch WJ, Naylor CPE, MacFarlane GT. Short chain fatty acids in human large intestine, portal, hepatic, and venous blood. Gut. 1987;28:1221-7
14. Boffa LC, Luption JR, Mariani MR, Ceppi M, Newmark HL, Scalmati A, Lipkin M. Modulation of colonic epithelial cell proliferation, histone acetylation, and luminal short chain fatty acids by variation of dietary fiber (wheat bran) in rats. Cancer Res. 1992;52:5906-12
15. Heerdt BG, Houston MA, Augenlicht LH. Potentiation by specific short-chain fatty acids of differentiation and apoptosis in human colonic carcinoma cell lines. Cancer Res. 1994;54:3288-94
16. Hague A, Elder DJ, Hicks DJ, Paraskeva G. Apoptosis in colorectal tumor cells: induction by the short chain fatty acids butyrate, propionate and acetate and by the bile salt deoxycholate. Internat J Cancer. 1995;60:400-6
17. Medina V, Young GP, Edmonds B, James R, Appleton S, Zalewski PD. Induction of caspase-3 protease activity and apoptosis by butyrate and another inhibitor of histone deacetylase: dependence on protein synthesis and synergy with a mitochondrial/cytochrome c-dependent pathway. Cancer Res. 1997;57:3697-707
18. Marks PA, Dokmanovic M. Histone deacetylase inhibitors: discovery and development as anticancer agents. Expert Opin Investig Drugs. 2005;12:1497-511
19. Lin HY, Chen CS, Lin SP, Weng JR, Chen CS. Targeting histone deacetylase in cancer therapy. Med Res Rev. 2006;26:397-413
20. Bordonaro M, Mariadason JM, Aslam F, Heerdt BG, Augenlicht LH. Butyrate-induced apoptotic cascade in colonic carcinoma cells: modulation of the beta-catenin-Tcf pathway and concordance with effects of sulindac and trichostatin A but not curcumin. Cell Growth Differ. 1999;10:713-20
21. Albuquerque C, Breukel C, van der Luijt R, Fidalgo P, Lage P, Slors FGM, Leitao CN, Fodde R, Smits R. The just-right signaling model: APC somatic mutations are selected based on a special level of activation of the beta-catenin signaling cascade. Hum Mol Genet. 2002;11:1549-60
22. Bordonaro M, Lazarova D, Sartorelli AC. Hyperinduction of WNT signaling: a new paradigm for the treatment of colorectal cancer? Oncol Res. 2008;17:1-9
23. Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, Moon RT, Teo JL, Oh SW, Kim HY, Moon SH, Ha JR, Kahn M. A small molecule inhibitor of β-catenin/CREB-binding protein transcription. Proc Natl Acad Sci USA. 2004;101:12682-7
24. McMillan M, Kahn M. Investigating WNT signaling: a chemogenomic safari. Drug Disc Today. 2005;10:1467-74
25. Teo JL, Ma H, Nguyen C, lam C, Kahn M. Specific inhibition of CBP/β-catenin interaction rescues defects in neuronal differentiation caused by presenilin-1 mutayion. Proc Natl Acad Sci. 2005;102:12171-6
26. Ma H, Nguyen C, Lee KS, Kahn M. Differential roles for the coactivators CBP and p300 on TCF/β-catenin-mediated survivin gene expression. Oncogene. 2005;24:3619-31
27. Miyabayashi T, Teo JL, Yamamoto M, McMillian M, Nguyen C, Kahn M. WNT/β-catenin/CBP signaling maintains long-term murine stem cell pluripotency. Proc Natl Acad Sci USA. 2007;104:5668-73
28. Teo JL, Kahn M. The WNT signaling pathway in cellular proliferation and differentiation: a tale of two coactivators. Adv Drug Delivery Rev. 2010;62:1149-55
29. Lazarova DL, Chiaro C, Wong T, Drago E, Rainey A, O'Malley S, Bordonaro M. CBP activity mediates effects of the histone deacetylase inhibitor butyrate on WNT activity and apoptosis in colon cancer cells. J Cancer. 2013;4:481-490
30. Ionov Y, Matsui SI, Cowell JK. A role for p300/CREB binding protein genes in promoting cancer progression in colon cancer cell lines with microsatellite instability. Proc Natl Acad Sci. 2004;101:1273-8
31. Ionov Y, Nowak N, Perucho M, Markowitz S, Cowell JK. Manipulation of nonsense mediated decay identifies gene mutations in colon cancer cells with microsatellite instability. Oncogene. 2004;23:639-45
32. Iyer NG, Chin SF, Ozdag H, Daigo Y, Hu DE, Cariati M, Brindle K, Aparicio S, Caldas C. p300 regulates p53-dependent apoptosis after DNA damage in colorectal cancer cells by modulation of PUMA/p21 levels. Proc Natl Acad Sci. 2004;101:7386-91
33. Takahashi-Yanaga F, Kahn M. Targeting WNT signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. 2010;16:3153-62
34. Chiaro C, Lazarova DL, Bordonaro M. Tcf3 and cell cycle factors contribute to butyrate resistance in colorectal cancer cells. Biochem Biophys Res Commun. 2012;428:121-6
35. Waltzer L, Bienz M. Drosophila CBP represses the transcription factor TCF to antagonize Wingless signaling. Nature. 1998;395:521-5
36. Hecht A, Vleminckx K, Stemmler MP, van Roy F, Kremler R. The p300/CBP acetyltransferases function as transcriptional coactivators of beta-catenin in vertebrates. EMBO J. 2000;19:1839-50
37. Li J, Sutter C, Parker DS, Blauwkamp T, Fang M, Cadigan KM. CBP/p300 are bimodal regulators of WNT signaling. EMBO J. 2007;26:2284-94
38. Levy L, Wei Y, Labalette C, Wu Y, Renard CA, Buendia MA, Neuveut C. Acetylation of beta-catenin by p300 regulates beta-catenin-Tcf4 interaction. Mol Cell Biol. 2004;24:3404-14
39. Lazarova DL, Bordonaro M. Extreme fluctuations in Wnt/beta-catenin signaling as an approach for colon cancer prevention and therapy. Advanced Studies in Biology. 2012;4:351-62
Author contact
![]() Corresponding author: The Commonwealth Medical College, 525 Pine Street, Scranton, PA 18509. Tel: 570-504-9646 Fax: 570-504-9636 Email: mbordonaroorg.
Corresponding author: The Commonwealth Medical College, 525 Pine Street, Scranton, PA 18509. Tel: 570-504-9646 Fax: 570-504-9636 Email: mbordonaroorg.